

Background: Stroma formation in pancreatic cancer is controlled via the activation of pancreatic stellate cells.
Results: Pancreatic stellate cells demonstrate different proteomic and functional attributes following specific stimuli.
Conclusion: The proteomic feature of pancreatic stellate cells shows TNF-α as a main contributor to activation.
Significance: The shift of proteomes in activated cells may help in understanding the processes of stromal formation and finding new drug targets.
Keywords: Microarray, Pancreatic Cancer, Proteomics, Stromal Cell, Tumor Necrosis Factor (TNF), Pancreatic Stellate Cell, Antibody Microarray
Abstract
Pancreatic stellate cells are key mediators in chronic pancreatitis and play a central role in the development of pancreatic fibrosis, stromal formation, and progression of pancreatic cancer. This study was aimed at investigating molecular changes at the level of the proteome that are associated with the activation of pancreatic stellate cells by proinflammatory factors, namely TNF-α, FGF2, IL6, and chemokine (C-C motif) ligand 4 (CCL4). They were added individually to cells growing in serum-free medium next to controls in medium supplemented with serum, thus containing a mixture of them all, or in serum-free medium alone. Variations were detected by means of a microarray of 810 antibodies targeting relevant proteins. All tested factors triggered increased proliferation and migration. Further analysis showed that TNF-α is the prime factor responsible for the activation of pancreatic stellate cells. CCL4 is associated with cellular neovascularization, whereas FGF2 and IL6 induction led to better cellular survival and decreased apoptotic activity of the stellate cells. The identified direct effects of individual cytokines on human pancreatic stellate cells provide new insights about their contribution to pancreatic cancer promotion.
Introduction
Every year, approximately 265,000 people die from pancreatic cancer worldwide. In the Western world, pancreatic cancer is ranked fourth in the list of cancer-related mortalities, although it is relatively rare in comparison with other tumor entities. However, mortality is close to incidence, and most patients die within a year after diagnosis (1). The disease develops without early symptoms and is usually diagnosed at very late stages. In addition, most patients have invasive and metastasized tumors at the time of diagnosis. This makes surgical intervention feasible only for about 20% of the patients (2). Although considerable advances have been made about understanding the molecular aspects of pancreatic cancer, there are still very few therapeutic modalities available (3, 4), and they usually have only a limited effect because the tumors develop resistance shortly after initiation of chemotherapy.
In pancreatic cancer, the desmoplastic microenvironment (stroma) surrounding tumor cells can contribute to as much as 90% of tumor mass (5). The stroma consists of pancreatic stellate cells (PSCs)3, endothelial, inflammatory, and dendritic cells and also a dense proteinaceous matter composed mainly of extracellular matrix proteins such as collagen, fibronectin, and proteoglycans (6). Although stromal cells lack the genetic alterations observed in malignant pancreatic cells, they communicate with inflamed and tumor cells, driven by cytokines, chemokines, growth factors, and other inflammatory mediators, and contribute to tumor invasiveness and aggressiveness, resistance to chemotherapy, and metastasis formation (7–11). The stellate cells are a prime contributor to stromal fibrosis formation in pancreatic cancer (12, 13) and key regulators in fibrogenesis during necroinflammation in chronic pancreatitis (14–16). The exocrine pancreas normally harbors PSCs in the preacinar and interlobular space as quiescent cells, containing vitamin A-rich lipid droplets in their cytoplasm. In the quiescent state, PSCs can hardly synthesize extracellular matrix (17). During inflammation, they are activated and mediate repair in pancreatic tissue as they migrate to the site of injury, contributing to cell proliferation and migration (18). The process of stellate cell activation involves loss of retinyl droplets, enlargement and proliferation, expression of a myofibroblast-like phenotype, and increased synthesis of extracellular matrix proteins (19, 20). During chronic inflammation, cytokines and growth factors are constantly released and, thus, activate PSCs continuously (21), which, in turn, act as an important factor to the course of pancreatic fibrogenesis and cancer (17, 23, 24). Although several growth factors, cytokines, and chemokines have been implicated in the process of activating PSCs and the transformation of cells to proliferative and fibrogenic myofibroblast, the contribution of each factor individually and the combination of the molecular events toward an activation have not been addressed in detail.
This study aims to define molecular events at the level of the proteome that occur as a consequence of PSC activation with individual proinflammatory mediators. Only few proteomic studies of PSCs have been conducted to date. They focus on the proteome variations between quiescent and activated PSCs in the presence of serum (25–27). However, serum does contain all kinds of growth factors, cytokines, and chemokines. Therefore, although these studies provide an overall picture, they obscure the individual effects. We performed analyses in the absence of serum, thus examining the direct effect of individual cytokines in vitro for a definition of the biological functions of each of them. For comparison, a serum-positive control was also investigated. The results provide new insights into the processes of pancreatic fibrogenesis during the course of inflammation and the contribution of PSCs to stroma formation in pancreatic cancer.
EXPERIMENTAL PROCEDURES
Materials
All materials used in this study were purchased from Sigma-Aldrich (Taufkirchen, Germany) unless stated otherwise and were of highest purity or protein grade. In the analysis, a set of 810 antibodies was used, as reported earlier (28). Their target proteins and origin are listed in supplemental Table S1.
Antibody Microarray Production
The microarrays were produced as described previously in detail (29, 30). In short, antibodies were spotted from 384-well plates on epoxysilane-coated slides (Nexterion-E, Schott, Jena, Germany) using contact printing (MicroGrid-2, BioRobotics, Cambridge, UK) with SMP3B pins (Telechem) at a humidity of 50–60%. The 15 μl of spotting buffer were made of Bicine buffer (50 mm, pH 8.5) containing 1.0 mm MgCl2, 5% trehalose, 0.005% Tween 20, and about 5 μg of the respective antibody. On each slide, the antibodies were printed in quadruplicates. After printing, the slides were equilibrated at a humidity of 50–60% overnight and subsequently stored under dry and dark conditions at 4 °C until use.
Cell Culture
Human immortalized PSCs were a gift from Ralf Jesnowski (Mannheim University Hospital, Germany) (31). Cells were maintained in Iscove's modified Dulbecco's medium (IMDM) supplemented with 10% heat-inactivated fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin at 37 °C and 5% CO2. The PSCs were cultured until 90% confluence and then washed twice with serum-free medium and once with Dulbecco's PBS. Before testing the effect of a supplemented factor, cells were incubated with serum-free IMDM for 24 h to synchronize cell growth. The medium was removed, and the cells were incubated with serum-free and phenol red-free IMDM under any of the six following conditions: 0% serum (negative control), 10% serum (positive control) and 10 ng/ml TNF-α, 10 ng/ml FGF2, 10 ng/ml IL6, or 5 ng/ml CCL4. After an incubation of 48 h, the cells were washed three times with ice-cold Dulbecco's PBS and subjected to protein extraction. Each treatment was performed independently in quintuplicate. To make sure that incubation for 48 h in serum-free medium was not affecting cell viability, cell growth was monitored at 24, 48, and 72 h for PSCs grown in serum-free medium. Cell viability was always 95% or higher.
Protein Extraction and Labeling
Protein extraction was performed as described previously (32). In brief, after washing with ice-cold Dulbecco's PBS, the cells were layered with extraction solution composed of 50 mm Bicine buffer (pH 8.5) containing 20% glycerol, 1.0 mm MgCl2, 5.0 mm EDTA, 1.0 mm phenylmethanesulfonyl fluoride, 1.0 IU/ml benzonase (Merck Biosciences, Schwalbach, Germany), Halt protease and phosphatase inhibitor mixture (Thermo Scientific, Bonn, Germany), 0.5% Nonidet P-40 substitute, 1.0% cholic acid, 0.25% n-dodecyl-β-maltoside (GenaXXon Bioscience, Ulm, Germany), and 0.5% amidosulfobetaine-14. Flasks were kept on ice for 30 min with occasional mixing. The cells were scraped off and pipetted several times through a fine needle syringe, followed by centrifugation at 20,000 × g at 4 °C for 20 min. The supernatant was carefully collected not disturb the upper layer or the pellet. The protein concentration was determined with the bicinchoninic acid protein assay reagent kit (Thermo Scientific). The protein concentration was adjusted to 1.0 mg/ml using the lysis buffer as diluent. Samples were labeled with the DY-649 NHS-ester and DY-549 NHS-ester dyes (Dyomics, Jena, Germany) at a molar ratio of dye/protein of 7.5 as described earlier (29, 30).
Microarray Incubation, Scanning, and Image Processing
All steps were performed in the dark. Initially, the microarrays were washed twice for 10 and 5 min, respectively, with PBS containing 0.05% Tween 20 (PBST). Then the array surface was blocked with 10% nonfat dry milk (Bio-Rad) in PBST at room temperature for 3 h using Quadriperm chambers (Greiner Bio-One, Frickenhausen, Germany) on an orbital shaker. The incubation was done with 35 μg of dye-labeled sample in 5 ml of PBST supplemented with 10% milk at 4 °C overnight. Also, 35 μg of a common reference sample was added to all incubations and labeled with the second dye (33). The reference sample was made by pooling protein extracts prepared from 24 pancreatic cancer cell lines (28). Aliquots were stored frozen until use. In all experiments, the very same reference pool was used. After the incubation, the slides were washed four times (5 min each) with PBST, rinsed with deionized water, and dried in a ventilated oven at 37 °C. Scanning of the slides was performed with a Tecan Power Scanner (Tecan Austria GmbH, Grödig, Austria) at constant laser power and photomultiplier tube setting. Image analysis was performed with GenePix Pro 6.0 software (Molecular Devices).
Cell Proliferation Assay
For induction of cell proliferation, PSCs were seeded at 3000 cells/well in 96-well plates and cultured in IMDM supplemented with 10% heat-inactivated fetal bovine serum. Cells were allowed to adhere to the surface for 4 h and then washed twice with Dulbecco's PBS. Cell incubation with serum-free medium was performed for 24 h to synchronize cell growth. The serum-free medium was removed, and the cells were incubated for 48 h in serum-free and phenol red-free IMDM containing one of the following: 0% serum (negative control), 10% serum (positive control), 10 ng/ml TNF-α, 10 ng/ml FGF2, 10 ng/ml IL6, or 5 ng/ml CCL4. Subsequently, the medium was removed, and cell proliferation was determined using the CyQUANT NF cell proliferation assay kit (Invitrogen) according to the instructions of the manufacturer.
Cell Migration Assay
Cell migration was assayed using an in vitro wound healing assay. For this assay, cells were plated in 6-well plates. When the cells grew into full confluence, the medium was changed to serum-free IMEM, and the cells were incubated for another 24 h. A wound was created on the cell monolayer by scraping a gap using a sterile yellow micropipette tip and washing with PBS three times. Images of cells were taken using phase-contrast microscopy. The cells were incubated for 48 h with serum-free IMDM containing one of the following: 0% serum (negative control), 10% serum (positive control) and 10 ng/ml TNF-α, 10 ng/ml FGF2, 10 ng/ml IL6, or 5 ng/ml CCL4. At the end of the incubation time, images of the cells were taken.
Apoptosis Assay
The effect of the cytokines on PSC apoptosis was determined by measuring the mitochondrial membrane potential. To this end, the Mito-ID membrane potential kit from Enzo Life Sciences (Lörrach, Germany) was used according to the instructions of the manufacturer. A decrease in mitochondrial membrane potential is a function of accelerated apoptotic activity.
Assay of Cellular Reactive Oxygen Species
Cellular reactive oxygen species were measured using 2′,7′-dichlorofluorescein diacetate as described previously (34). 2′,7′-Dichlorofluorescein diacetate is a fluorescent dye that readily diffuses into cells and is hydrolyzed by esterases to the polar and non-fluorescent derivative 2′,7′-dichlorofluorescein that is trapped within cells. When present, the reactive oxygen species oxidize this compound to fluorescent 2′,7′-dichlorofluorescein. Briefly, PSCs were cultured in 96-well plates at 75% density and treated with various factors as detailed above. After a 48-h treatment, cells were loaded with 50 μm of 2′,7′-dichlorofluorescein diacetate for 30 min, and fluorescence intensities were measured at excitation/emission wavelengths of 485/530 nm, respectively.
Flow Cytometry
PSCs were cultured in 6-well plates at 75% density. After 48 h of incubation, cells were harvested and resuspended in Nicoletti buffer composed of 0.1% tri-sodium citrate, 0.1% Triton X-100, 50 μg/ml propidium iodide, and 50 μg/ml RNase A. Cells were mixed for a short time and incubated at 4 °C in the dark for 1.5 h. The cell cycle status was detected with a BD FACSCanto II flow cytometer (BD Biosciences) as recommended.
Immunohistochemistry Analysis
Immunohistochemistry analysis was performed as described previously (35). PDAC tissues were obtained from the Department of General, Visceral, and Transplantation Surgery at the University of Heidelberg. Informed written consent was obtained from patients, and the study was approved by the ethics committee of the University of Heidelberg. Samples were directly fixed with 5% paraformaldehyde solution and embedded in paraffin for immunohistochemistry analysis. Anti-filamin A (FLNA) (GeneTex, Irvine, CA), anti-matrix metalloproteinase 3 (MMP3) (BioCat GmbH, Heidelberg, Germany), anti-cytokeratin 19 (CK19) (Santa Cruz Biotechnology, Heidelberg, Germany), and anti-cortactin (BioCat GmbH), and anti-α-smooth muscle actin (α-SMA) antibodies were used at 1:100 dilution. Isotype-negative controls were used to correct for nonspecific binding. The imaging of section was performed using a Zeiss Axiocam 3.1 system (Jena, Germany).
Immunoblotting Analysis
To test for the expression of α-SMA in stimulated cells, PSCs were cultured and treated, and the cellular protein was extracted as above. Protein was loaded in Laemmli buffer, run on SDS-PAGE, and transferred onto a nitrocellulose membrane. α-SMA bands were probed with anti-α-SMA antibodies diluted to 1:1000.
Data Analysis
The microarray data were analyzed with the Chipster software package (v1.4.6, CSC, Finland). The median of the signal intensities in the red (DY-649) and green (DY-549) channels, respectively, was used. The data were normalized using the Loess method with background correction offset (0, 50) of the normexp method (36). A test for significance between the control and treatment groups was performed using the empirical Bayes test with a Bonferroni-Hochberg adjustment (37). The empirical Bayes test makes use of a moderated t statistics in which posterior residual standard deviations are applied rather than normal standard deviations. This results in a far more stable inference when the number of arrays is small (37). A p value of 0.05 or less was considered significant. Multiple-set Venn diagrams were generated using the open source software VENNTURE (38). The biofunctional annotation of the differentially expressed proteins was performed with Ingenuity Pathways Analysis software (version 6.3, Ingenuity Systems, Redwood City, CA). The prediction of variations in biological functions was performed using a z-score of +2 or −2, respectively, as threshold for significance. Protein functional interaction networks were evaluated using the open source software STRING 9.0 (39). Unpaired Student's t test (two-tailed) was used to compare the control, serum-free incubations with each of the other treatments in the proliferation and reactive oxygen species production assays. The inter- and intra-assay coefficient of variance was less than 20%.
RESULTS
For the analysis of the effects of particular cytokines, human immortalized PSCs (31) were grown in serum-free medium to avoid the effects of the various growth factors contained in serum. TNF-α, CCL4, IL6, or FGF2 were added to the medium separately. Also, control experiments were conducted in the absence of any supplement or in the presence of serum. Two of the four studied growth factors were selected on the basis of the results from analyzing protein expression variations in 650 pancreatic cancer tissue samples.4 In pancreatic ductal adenocarcinoma, the amount of TNF-α was found to be directly correlated with the percentage of tumor cells. Conversely, the same was true for CCL4 and the percentage of stroma cells. The third factor, IL6, was present at substantial concentrations in the secretome of 16 pancreatic cancer cell lines (data not shown). FGF2, finally, was included because it has frequently been reported as a PSC activator (20, 24).
After cell growth, the cellular proteins were isolated and analyzed on a complex microarray of 810 antibodies that permit the detection of 741 proteins (supplemental Table S1) that are closely associated with cancer, and particularly pancreatic tumors (33). The same format was used for a recent comparative analysis of 24 pancreatic cancer cell lines (28). All measurements were done on five biological replicates for each condition. In comparison to cells grown in serum-free medium, a substantial number of proteins, ranging from 174 to 283, were regulated in the presence of the four cytokines (supplemental Tables S2–S5). Fig. 1 shows the number of proteins that were uniquely or commonly regulated under each of the four growth conditions. Only the expression of 42 proteins was similarly changed under all four conditions, although differences in the degree of variation were observed (Fig. 2). Another three proteins also always showed a change of expression, but of opposing directions. Examples of proteins that were similarly regulated under all treatment conditions are FLNA and MMP3. Identical to the cell culture experiments, the expression of FLNA and MMP3 was also found up-regulated in stromal cells of PDAC tissue sections (Fig. 3).
FIGURE 1.
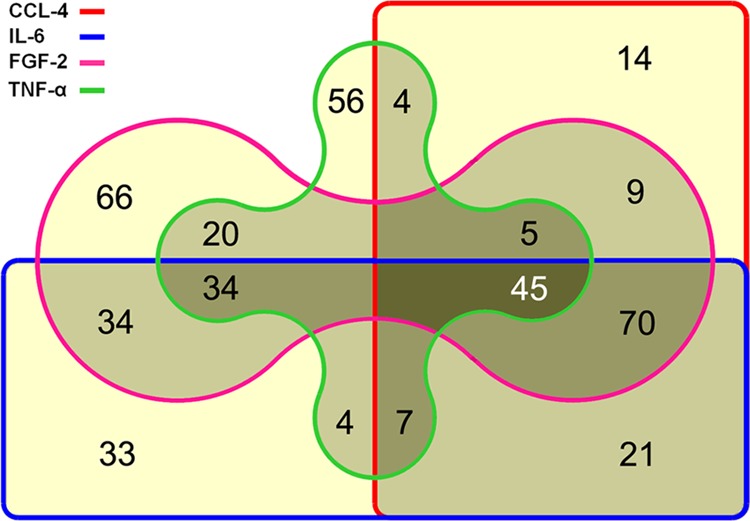
Venn diagram showing the number of proteins that were found regulated upon growth of PSCs in the presence of TNF-α, CCL-4, IL-6, and FGF-2. A yellow background indicates the number of proteins uniquely regulated at one condition only. Light gray and medium gray stand for molecules that were regulated at two or three growth conditions, respectively. The number of proteins that were differentially expressed under all four conditions is shown in the area with the darkest background.
FIGURE 2.

Expression level variations of the 45 commonly regulated proteins, presented as the logarithm of fold change (Log-FC).
FIGURE 3.
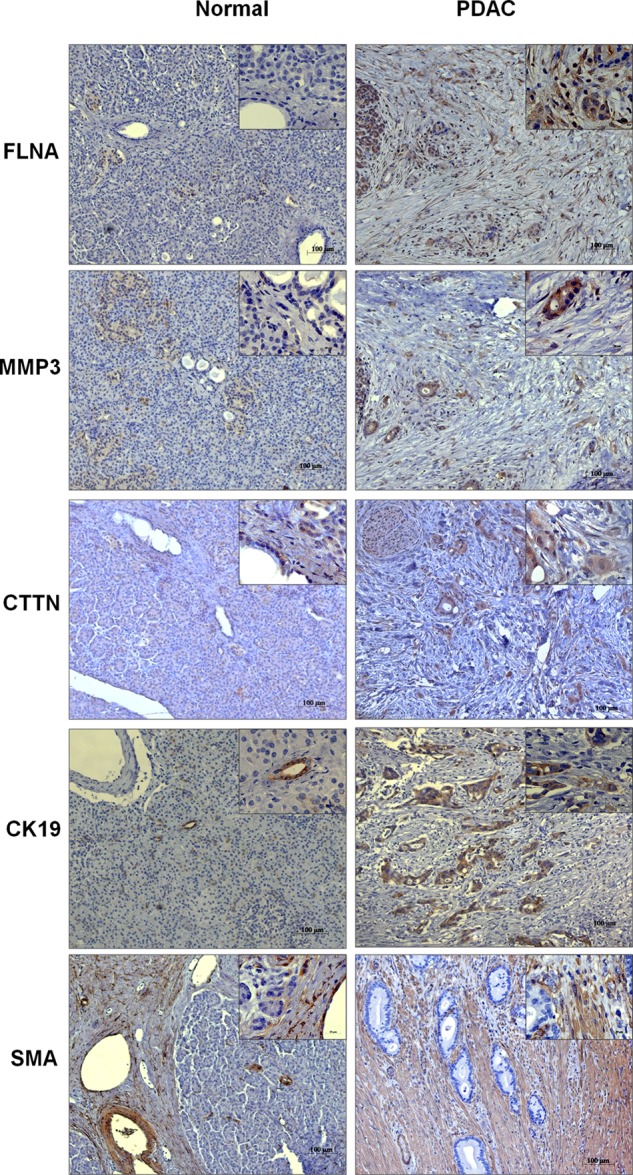
Immunohistochemical analysis of normal and PDAC tissues. Tissue sections were analyzed with antibodies binding FLNA, MMP3, and cortactin (CTTN) in normal versus PDAC tissue sections. Cytokeratin 19 (CK19) and SMA were used to stain ductal and stellate cells, respectively. Original magnification was 100- and 663-fold for the insets.
A functional annotation of the proteins was performed that exhibited expression level variations, yielding results that were common to all four or some of the conditions or unique for only one of them (supplemental Tables S6–S9). The common functions were related to cancer, cell death, cell growth, and proliferation. Under each condition, at least 30% of the proteins with different expression levels belonged to these functional classes. Another feature common to all four factors was the development of blood vessels and vasculogenesis.
TNF-α treatment resulted in a significant regulation of 174 proteins, of which 56 were unique to TNF-α (supplemental Table S2). The expression pattern for some of these unique proteins, like cortactin (up-regulated), was found to have a similar regulation in PDAC tissue sections (Fig. 3). Many of the unique proteins belong to the hepatic fibrosis and hepatic stellate cell activation signaling pathway, underlined by an overexpression of IGF1 and IL4. Also, an activation of cell-to-cell signaling and interaction, fibrosis, proliferation of fibroblasts, and differentiation of progenitor cells could be seen (supplemental Table S6). Other functions were chemotaxis and homing of monocytes and development of T cells and lymphocytes. The data also predicted an increased synthesis of reactive oxygen species upon TNF-α treatment and changes at the level of the cell cycle with a significantly increased S phase activity. All of these results indicate a direct implication of TNF-α in the activation of PSCs, their differentiation to myofibroblast-like cells, and their regulatory interaction with immune cells. Additionally, the contribution of TNF-α to PSC activation was further confirmed by the finding of increased α-SMA expression in these cells (Fig. 4).
FIGURE 4.
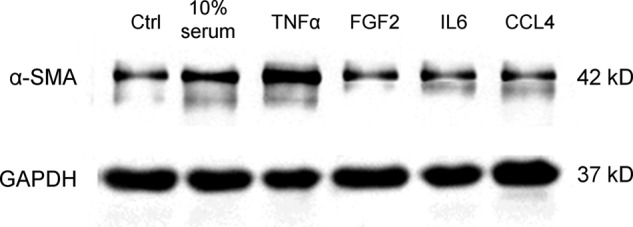
Immunoblot analysis showing the expression of α-SMA in PSCs under various treatment conditions. Ctrl, control.
Treatment of PSCs with FGF2 resulted in changes of 283 proteins. Similar to TNF-α, most of these proteins were related to hepatic fibrosis and hepatic stellate cell activation signaling. Also, functions related to gene expression were substantially triggered by FGF2 (supplemental Table S7). The elevated expression of endogenous TGFb1 and IL4 suggest a potential mechanism toward an acceleration of gene expression in PSCs.
Upon IL6 treatment, 247 regulated proteins were detected. Further analysis of these proteins showed that 33 were significantly expressed proteins only in IL6 (supplemental Table S4). Generally, a decrease in several functions was observed with significant prediction from the expression pattern of the regulated proteins upon IL6 treatment of PSCs (supplemental Table S8). There were decreases in activation of neutrophils, adhesion of monocytes, apoptosis of epithelial cells, cell death, a quantity of calcium ions, and binding of vascular endothelial cells.
In the CCL4-treated cells (supplemental Table S5), the expression of 174 proteins was changed significantly. A unique set of 14 proteins was differentially expressed only with CCL4, whose functions indicated increased neovascularization (supplemental Table S9).
We performed several functional analyses to confirm the predictions that were made on the basis of the proteome data. The cellular response to TNF-α treatment indicated an increase in S phase activity. This was confirmed by FACS analysis (Fig. 5). Also, the study of the proliferation of PSCs treated with the four cytokines led to results that are in agreement with the proteome data. A growth of stellate cells in the presence of TNF-α, FGF2, and CCL4 resulted in a significant increase in proliferation (Fig. 6). The same trend was seen for IL6, although to a lesser extent. Further, a wound healing scratch assay was applied to investigate the influence of the proinflammatory factors on the migration ability of PSCs compared with cells grown in serum-free medium. Exposure to TNF-α, FGF2, and CCL4 caused a strong increase in the number of PSCs that migrated into the empty space created by scratching (Fig. 7). Migration was also observed in an experiment with IL6, but the migration effect was delayed compared with the other proinflammatory cytokines (data not shown). All this is in agreement with the proteome analysis. Even the smaller effect observed for incubations with IL6 concurs directly with the degree of proteome variations detected under this growth condition.
FIGURE 5.

FACS sorting analysis revealed the percentage of PSCs (% of events) in S phase. For each condition, three independent measurements were performed.
FIGURE 6.

Proliferation of PSCs under various incubation conditions. The assay measured the incorporation of a fluorescent dye to the DNA that was proportional to the number of cells in the system. The fluorescence intensities are shown in arbitrary units (AU). The p values were calculated in comparison with the control cells grown in serum-free medium.
FIGURE 7.
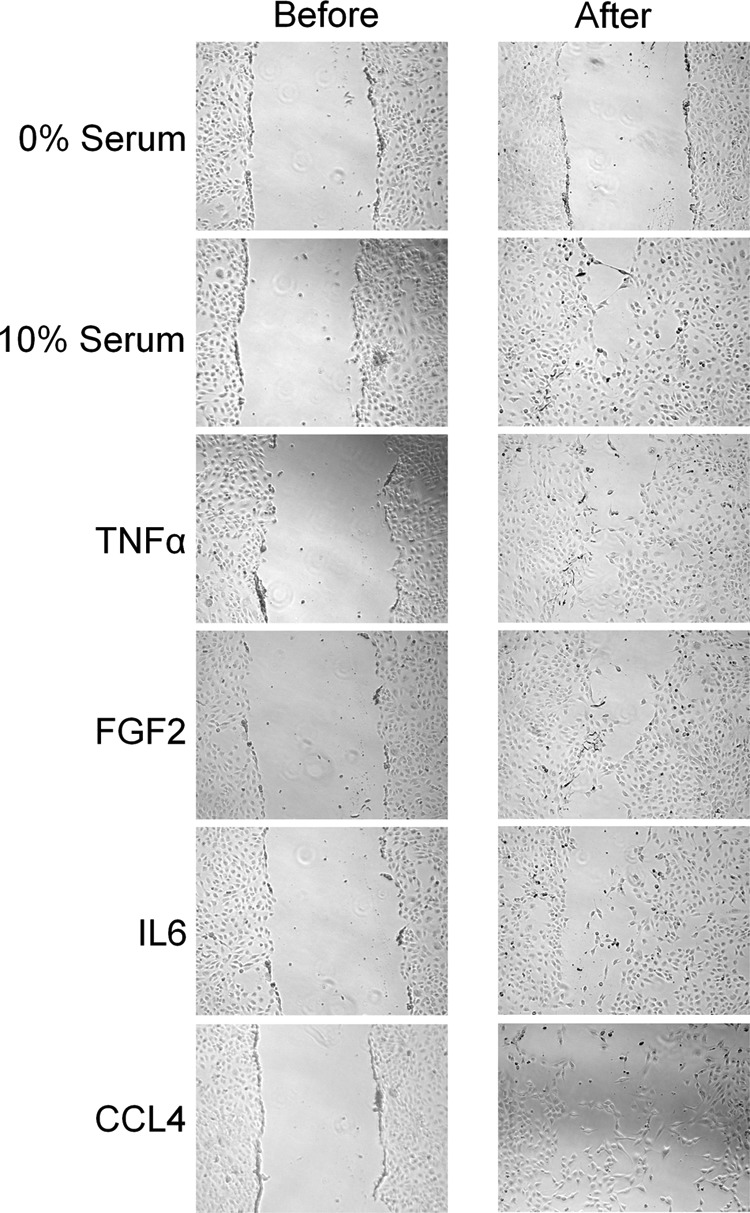
Migration assay. PSCs were grown to confluence. A gap was generated by physically scraping off cells. The number of cells was determined before and after 48 h of growth under the indicated incubation conditions.
A result common to all four conditions was the effect of TNF-α, FGF2, IL6, and CCL4 on apoptosis of PSCs. As compared with serum-free medium, PSCs treated with either one of the factors and also cells growing in 10% serum showed decreased apoptotic activity, as determined by the enhanced mitochondrial membrane potential (Fig. 8), which was measured as described under “Experimental Procedures.” The findings are comparable with the functional annotation results that are on the basis of the observed variations in the protein content of the cells. TNF-α showed the most anti-apoptosis effect in PSCs, followed by CCL4.
FIGURE 8.

Apoptosis assay. The fluorescence signal is representative for the mitochondrial membrane potential, which, in turn, is a function of reduced apoptotic activity. The higher the signal, the smaller the apoptosis activity. The measurements were done relative to analyses in serum-free medium. AU, arbitrary units.
The proteome data also indicated a strongly increased reactive oxygen species synthesis upon incubation with TNF-α. Therefore, the level of cellular reactive oxygen species was determined under the four conditions. As expected, the level of reactive oxygen species increased more than 5-fold when the cells were incubated with TNF-α, whereas no change could be observed in presence of the other cytokines (Fig. 9). Only cells grown in 10% serum exhibited an increased level as well, but the effect was much less pronounced.
FIGURE 9.

Analysis of the level of reactive oxygen species. The cellular level of reactive oxygen species was determined as a function of the conversion of 2′,7′-dichlorofluorescein to its oxidized and then fluorescent version. AU, arbitrary units; Ctrl, growth in serum-free medium.
DISCUSSION
PSCs play a central role in pancreatic fibrogenesis during inflammation and cancer. Several mediators are involved in the communication process between inflammatory cells, tumor cells, and PSCs. This study evaluates the contribution of TNF-α, FGF2, IL6, and CCL4 to the activation and proliferation of PSCs from a proteomics point of view. Analysis of the expression patterns indicates distinct functional characteristics in response to each of the cytokines under investigation.
Of all four factors, TNF-α exhibited changes at the proteome level that are associated with the most distinct functional variations. Most noticeable, cell activation was observed only with TNF-α but with none of the other three factors. The overexpression of α-SMA is described as a hallmark of PSC activation (40). Our data showed that TNF-α caused the highest α-SMA level among all treatment conditions, even that of 10% serum. This finding suggests a potentially strong link between the TNF-α level in PDAC and stromal cell activation. Besides cellular activation, there was a strong increase in proteins that are relevant for the synthesis of reactive oxygen species. TNF-α is a well known stimulant of reactive oxygen species synthesis and oxidative stress (41), and reactive oxygen species have been reported to induce PSC activation (42). Upon TNF-α treatment, an overexpression of principal proliferation and profibrogenic cytokines was observed, such as IGFBP2, TGFB1, IL15, SERPIN1, TNF-αSF10, CFLAR (CASP8- and FADD-like apoptosis regulator), and MMP11. It has been shown earlier that IGFBP2 participates in regulating cell proliferation, differentiation, and apoptosis (43). Increased levels of TNF-α are well documented in pancreatic cancer patients (44–46) and have been implicated in playing an important role in tumor microenvironment remodeling (47). The high level of TNF-α in pancreatic cancer could be the reason of the constant priming of PSCs and their sustained proliferation and fibrogenesis.
CCL4 is a strong stimulant to fibrosis in vitro and in vivo (48) and has been shown to exert angiogenic effects in animal models (49). High levels of CCL4 have been reported in chronic pancreatitis (50). In this study, CCL4 stimulated a similar but less intensive effect than TNF-α. The effect of CCL4 on PSCs could be due to an induction of the endogenous levels of TNF-α in the cells, as observed in this study. There is also a potential synergistic effect between TNF-α and CCL4, affecting the cellular proteome of PSCs toward monitoring the endothelial system. Although TNF-α gave rise to changes in the levels of PSC proteins that increase cellular development and movement of endothelial cells, CCL4 seems to increase neovascularization. The expression of proteins that regulate neovascularization, such as IL2 (51), IL1B (52), TGFB1 (53), and matrix metalloproteinases (54), was found significantly altered by treatment of PSCs with CCL4. In addition to endothelial promoting effects, CCL4-treated PSCs, along with cells grown with FGF2, also exhibited functions that are related to tumor development.
Pancreatic tumor cells have been shown to express and secrete FGF2, which contributes to enhancing the malignant phenotype of pancreatic cancer (55). The most prominent effect of FGF2 observed in this study was a change in the cellular proteome toward a decreased level of cell death and simultaneously improved cellular survival, the latter associated with a higher overall degree of gene expression. In total, 45 DNA binding proteins were regulated by FGF2, including the molecules TP53, CDKN1A, MYCN, (N-Myc proto-oncogene protein) IL1A, IGF1, IL4 and TGFB1, which are known for their regulatory effect on gene expression and cell survival. Similarly to TNF-α and CCL4, FGF2 influences gene expression also through binding of protein binding sites. FGF2 regulated the expression of 30 proteins, which are involved in modulating human gene expression via binding to the promoter E-box motif of many genes. However, network analyses indicate that FGF2 actually triggers an alternative regulative pathway via an endogenous rise of IGF1 and INS expression levels (data not shown).
As compared with measurements with TNF-α and CCL4, PSC growth in presence of IL6 exhibited reduced cellular movement, required for migration and invasion, but still higher values than those measured in serum-free medium. Significantly elevated levels of IL6 have been reported in pancreatic cancer cell lines (28) and medium of these cells (56) as well as in sera of pancreatic cancer patients (57). The anti-apoptotic effect of IL6 is well documented (58, 59). From the expression variation of 23 proteins, it can be concluded that in IL6-treated cells there is a decrease in the level of ionized cellular calcium. Some cytokines are known to increase the cellular calcium level, as reported for IL1B (60), INFG (61) and IL8 (62). The protein levels of these three cytokines were actually found repressed by IL6. This result fits well to the fact that IL6 causes a reduction in the cytosolic level of calcium (63). Under certain pathological stimuli, calcium can play a detrimental role in cell death and apoptosis (64). The anti-apoptotic effect triggered by IL6 is presumably attributable to its effect on calcium modulatory proteins. It is of interest that a treatment of PSCs with FGF2 caused an increase in endogenous levels of IL6, which may explain the similar effect of the two on cell survival. Expression of several proteins was reduced that control cell death, such as BCL2, TP53, AIFM1 and MYCN, when PSCs were grown with FGF2 or IL6. However, the anti-apoptotic effect was more prominent with IL6, probably as a result of down-regulation of CASP3 (22), which was not observed with FGF2.
From the above, an overview of the influence and combinatorial effect of the four pro-inflammatory factors on PSCs can be generated that is on the basis of an analysis at the protein level and the functional characteristics that are associated to them (Fig. 10). TNF-α seems to be the prime activator of these cells. Along with CCL4, it also contributes to changing the PSC proteome toward an endothelial system. FGF2 and IL6 increase cellular survival, presumably via an accelerated cellular gene expression mechanism by the former and a decreasing apoptotic activity through modulation of ionized calcium by the latter. However, in vivo, PSCs are not only influenced by these four factors. Indeed, we found that cell growth with 10% serum caused a significant change in the expression level of 230 proteins. 47 of them were not observed with the four cytokines alone and have unique functional characteristics (supplemental Table S10). This indicates that the presence of other effectors is very likely to have a pathophysiological influence on PSCs in health and disease.
FIGURE 10.

Schematic summary of the different effects of TNF-α, FGF2, IL6, and CCL4 on PSCs.
Acknowledgments
The technical assistance of Brunhilde Bentzinger is gratefully acknowledged.
This work was supported in part by the European Union as part of the Affinomics consortium.

This article contains supplemental Tables S1–S10.
A. J. Marzoq, N. Giese, J. D. Hoheisel, and M. S. S. Alhamdani, unpublished results.
- PSC
- pancreatic stellate cell
- IMDM
- Iscove's modified Dulbecco's medium
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- FLNA
- filamin A
- SMA
- smooth muscle actin
- PDAC
- pancreatic ductal adenocarcinoma.
REFERENCES
- 1. Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M. J. (2009) Cancer statistics, 2009. CA-Cancer J. Clin. 59, 225–249 [DOI] [PubMed] [Google Scholar]
- 2. Li D., Xie K., Wolff R., Abbruzzese J. L. (2004) Pancreatic cancer. Lancet 363, 1049–1057 [DOI] [PubMed] [Google Scholar]
- 3. Matsubara J., Ono M., Honda K., Negishi A., Ueno H., Okusaka T., Furuse J., Furuta K., Sugiyama E., Saito Y., Kaniwa N., Sawada J., Shoji A., Sakuma T., Chiba T., Saijo N., Hirohashi S., Yamada T. (2010) Survival prediction for pancreatic cancer patients receiving gemcitabine treatment. Mol. Cell. Proteomics 9, 695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yokoyama Y., Nimura Y., Nagino M. (2009) Advances in the treatment of pancreatic cancer. Limitations of surgery and evaluation of new therapeutic strategies. Surg. Today 39, 466–475 [DOI] [PubMed] [Google Scholar]
- 5. Fidler I. J. (2002) The organ microenvironment and cancer metastasis. Differentiation 70, 498–505 [DOI] [PubMed] [Google Scholar]
- 6. Erkan M., Reiser-Erkan C., Michalski C. W., Kleeff J. (2010) Tumor microenvironment and progression of pancreatic cancer. Exp. Oncol. 32, 128–131 [PubMed] [Google Scholar]
- 7. Edmonds C., Cengel K. A. (2008) Tumor-stroma interactions in pancreatic cancer. Will this SPARC prove a raging fire? Cancer Biol. Ther. 7, 1816–1817 [DOI] [PubMed] [Google Scholar]
- 8. Hwang R. F., Moore T., Arumugam T., Ramachandran V., Amos K. D., Rivera A., Ji B., Evans D. B., Logsdon C. D. (2008) Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Infante J. R., Matsubayashi H., Sato N., Tonascia J., Klein A. P., Riall T. A., Yeo C., Iacobuzio-Donahue C., Goggins M. (2007) Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 25, 319–325 [DOI] [PubMed] [Google Scholar]
- 10. Liotta L. A., Kohn E. C. (2001) The microenvironment of the tumour-host interface. Nature 411, 375–379 [DOI] [PubMed] [Google Scholar]
- 11. Mahadevan D., Von Hoff D. D. (2007) Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 6, 1186–1197 [DOI] [PubMed] [Google Scholar]
- 12. Omary M. B., Lugea A., Lowe A. W., Pandol S. J. (2007) The pancreatic stellate cell. A star on the rise in pancreatic diseases. J. Clin. Invest. 117, 50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Apte M. V., Haber P. S., Applegate T. L., Norton I. D., McCaughan G. W., Korsten M. A., Pirola R. C., Wilson J. S. (1998) Periacinar stellate shaped cells in rat pancreas. Identification, isolation, and culture. Gut 43, 128–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bachem M. G., Schünemann M., Ramadani M., Siech M., Beger H., Buck A., Zhou S., Schmid-Kotsas A., Adler G. (2005) Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921 [DOI] [PubMed] [Google Scholar]
- 15. Masamune A., Watanabe T., Kikuta K., Shimosegawa T. (2009) Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin. Gastroenterol. Hepatol. 7, S48–54 [DOI] [PubMed] [Google Scholar]
- 16. Mews P., Phillips P., Fahmy R., Korsten M., Pirola R., Wilson J., Apte M. (2002) Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut 50, 535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bachem M. G., Zhou S., Buck K., Schneiderhan W., Siech M. (2008) Pancreatic stellate cells. Role in pancreas cancer. Langenbecks Arch. Surg. 393, 891–900 [DOI] [PubMed] [Google Scholar]
- 18. Shimizu K. (2008) Mechanisms of pancreatic fibrosis and applications to the treatment of chronic pancreatitis. J. Gastroenterol. 43, 823–832 [DOI] [PubMed] [Google Scholar]
- 19. Bachem M. G., Schneider E., Gross H., Weidenbach H., Schmid R. M., Menke A., Siech M., Beger H., Grünert A., Adler G. (1998) Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115, 421–432 [DOI] [PubMed] [Google Scholar]
- 20. Masamune A., Sakai Y., Kikuta K., Satoh M., Satoh A., Shimosegawa T. (2002) Activated rat pancreatic stellate cells express intercellular adhesion molecule-1 (ICAM-1) in vitro. Pancreas 25, 78–85 [DOI] [PubMed] [Google Scholar]
- 21. Dunér S., Lopatko Lindman J., Ansari D., Gundewar C., Andersson R. (2010) Pancreatic cancer. The role of pancreatic stellate cells in tumor progression. Pancreatology 10, 673–681 [DOI] [PubMed] [Google Scholar]
- 22. Caruso J. A., Hunt K. K., Keyomarsi K. (2010) The neutrophil elastase inhibitor elafin triggers rb-mediated growth arrest and caspase-dependent apoptosis in breast cancer. Cancer Res. 70, 7125–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vonlaufen A., Joshi S., Qu C., Phillips P. A., Xu Z., Parker N. R., Toi C. S., Pirola R. C., Wilson J. S., Goldstein D., Apte M. V. (2008) Pancreatic stellate cells. Partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093 [DOI] [PubMed] [Google Scholar]
- 24. Schneider E., Schmid-Kotsas A., Zhao J., Weidenbach H., Schmid R. M., Menke A., Adler G., Waltenberger J., Grünert A., Bachem M. G. (2001) Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am. J. Physiol. Cell Physiol. 281, C532-C543 [DOI] [PubMed] [Google Scholar]
- 25. Wehr A. Y., Furth E. E., Sangar V., Blair I. A., Yu K. H. (2011) Analysis of the human pancreatic stellate cell secreted proteome. Pancreas 40, 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paulo J. A., Urrutia R., Banks P. A., Conwell D. L., Steen H. (2011) Proteomic analysis of a rat pancreatic stellate cell line using liquid chromatography tandem mass spectrometry (LC-MS/MS). J. Proteomics 75, 708–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paulo J. A., Urrutia R., Banks P. A., Conwell D. L., Steen H. (2011) Proteomic analysis of an immortalized mouse pancreatic stellate cell line identifies differentially-expressed proteins in activated vs. nonproliferating cell states. J. Proteome Res. 10, 4835–4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alhamdani M. S., Youns M., Buchholz M., Gress T. M., Beckers M. C., Maréchal D., Bauer A., Schröder C., Hoheisel J. D. (2012) Immunoassay-based proteome profiling of 24 pancreatic cancer cell lines. J. Proteomics 75, 3747–3759 [DOI] [PubMed] [Google Scholar]
- 29. Alhamdani M. S., Schröder C., Hoheisel J. D. (2010) Analysis conditions for proteomic profiling of mammalian tissue and cell extracts with antibody microarrays. Proteomics 10, 3203–3207 [DOI] [PubMed] [Google Scholar]
- 30. Schröder C., Alhamdani M. S., Fellenberg K., Bauer A., Jacob A., Hoheisel J. D. (2011) Robust protein profiling with complex antibody microarrays in a dual-colour mode. Methods Mol. Biol. 785, 203–221 [DOI] [PubMed] [Google Scholar]
- 31. Jesnowski R., Fürst D., Ringel J., Chen Y., Schrödel A., Kleeff J., Kolb A., Schareck W. D., Löhr M. (2005) Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis. Deactivation is induced by Matrigel and N-acetylcysteine. Lab. Invest. 85, 1276–1291 [DOI] [PubMed] [Google Scholar]
- 32. Alhamdani M. S., Schröder C., Werner J., Giese N., Bauer A., Hoheisel J. D. (2010) Single-step procedure for the isolation of proteins at near-native conditions from mammalian tissue for proteomic analysis on antibody microarrays. J. Proteome Res. 9, 963–971 [DOI] [PubMed] [Google Scholar]
- 33. Schröder C., Jacob A., Tonack S., Radon T. P., Sill M., Zucknick M., Rüffer S., Costello E., Neoptolemos J. P., Crnogorac-Jurcevic T., Bauer A., Fellenberg K., Hoheisel J. D. (2010) Dual-color proteomic profiling of complex samples with a microarray of 810 cancer-related antibodies. Mol. Cell. Proteomics 9, 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cathcart R., Schwiers E., Ames B. N. (1984) Detection of picomole levels of lipid hydroperoxides using a dichlorofluorescein fluorescent assay. Methods Enzymol. 105, 352–358 [DOI] [PubMed] [Google Scholar]
- 35. Erkan M., Kleeff J., Esposito I., Giese T., Ketterer K., Büchler M. W., Giese N. A., Friess H. (2005) Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 24, 4421–4432 [DOI] [PubMed] [Google Scholar]
- 36. Ritchie M. E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G. K. (2007) A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 [DOI] [PubMed] [Google Scholar]
- 37. Smyth G. K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 38. Martin B., Chadwick W., Yi T., Park S. S., Lu D., Ni B., Gadkaree S., Farhang K., Becker K. G., Maudsley S. (2012) VENNTURE. A novel Venn diagram investigational tool for multiple pharmacological dataset analysis. PloS ONE 7, e36911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011) The STRING database in 2011. Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erkan M., Adler G., Apte M. V., Bachem M. G., Buchholz M., Detlefsen S., Esposito I., Friess H., Gress T. M., Habisch H. J., Hwang R. F., Jaster R., Kleeff J., Klöppel G., Kordes C., Logsdon C. D., Masamune A., Michalski C. W., Oh J., Phillips P. A., Pinzani M., Reiser-Erkan C., Tsukamoto H., Wilson J. (2012) StellaTUM. Current consensus and discussion on pancreatic stellate cell research. Gut 61, 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adamson G. M., Billings R. E. (1992) Tumor necrosis factor induced oxidative stress in isolated mouse hepatocytes. Arch. Biochem. Biophys. 294, 223–229 [DOI] [PubMed] [Google Scholar]
- 42. Masamune A., Watanabe T., Kikuta K., Satoh K., Shimosegawa T. (2008) NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G99-G108 [DOI] [PubMed] [Google Scholar]
- 43. Lowenfels A. B., Maisonneuve P. (2005) Risk factors for pancreatic cancer. J. Cell. Biochem. 95, 649–656 [DOI] [PubMed] [Google Scholar]
- 44. Poch B., Lotspeich E., Ramadani M., Gansauge S., Beger H. G., Gansauge F. (2007) Systemic immune dysfunction in pancreatic cancer patients. Langenbecks Arch. Surg. 392, 353–358 [DOI] [PubMed] [Google Scholar]
- 45. Zhou G., Niu L., Chiu D., He L., Xu K. (2012) Changes in the expression of serum markers CA242, CA199, CA125, CEA, TNF-α and TSGF after cryosurgery in pancreatic cancer patients. Biotechnol. Lett. 34, 1235–1241 [DOI] [PubMed] [Google Scholar]
- 46. Karayiannakis A. J., Syrigos K. N., Polychronidis A., Pitiakoudis M., Bounovas A., Simopoulos K. (2001) Serum levels of tumor necrosis factor-α and nutritional status in pancreatic cancer patients. Anticancer Res. 21, 1355–1358 [PubMed] [Google Scholar]
- 47. Balkwill F. (2002) Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 13, 135–141 [DOI] [PubMed] [Google Scholar]
- 48. Reichenbach V., Ros J., Fernández-Varo G., Casals G., Melgar-Lesmes P., Campos T., Makriyannis A., Morales-Ruiz M., Jiménez W. (2012) Prevention of fibrosis progression in CCl4-treated rats. Role of the hepatic endocannabinoid and apelin systems. J. Pharmacol. Exp. Ther. 340, 629–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin Y. J., Lai M. D., Lei H. Y., Wing L. Y. (2006) Neutrophils and macrophages promote angiogenesis in the early stage of endometriosis in a mouse model. Endocrinology 147, 1278–1286 [DOI] [PubMed] [Google Scholar]
- 50. Zeh H. J., Winikoff S., Landsittel D. P., Gorelik E., Marrangoni A. M., Velikokhatnaya L., Winans M. T., Lee K., Moser A., Bartlett D., Lotze M. T., Siegfried J. M., Whitcomb D., Papacristou G., Slivka A., Bigbee W. L., Lokshin A. E. (2005) Multianalyte profiling of serum cytokines for detection of pancreatic cancer. Cancer Biomark. 1, 259–269 [DOI] [PubMed] [Google Scholar]
- 51. Epstein R. J., Hendricks R. L., Stulting R. D. (1990) Interleukin-2 induces corneal neovascularization in A/J mice. Cornea 9, 318–323 [PubMed] [Google Scholar]
- 52. Yang F., Dou H. L., Ma Z., Li Y. L., Lu X. R., Wang X., He P. P. (2010) Serum inflammatory factors in patients with idiopathic choroidal neovascularization. Ocular Immunol. Inflamm. 18, 390–394 [DOI] [PubMed] [Google Scholar]
- 53. Wara A. K., Foo S., Croce K., Sun X., Icli B., Tesmenitsky Y., Esen F., Lee J. S., Subramaniam M., Spelsberg T. C., Lev E. I., Leshem-Lev D., Pande R. L., Creager M. A., Rosenzweig A., Feinberg M. W. (2011) TGF-β1 signaling and Kruppel-like factor 10 regulate bone marrow-derived proangiogenic cell differentiation, function, and neovascularization. Blood 118, 6450–6460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi W., Liu J., Li M., Gao H., Wang T. (2010) Expression of MMP, HPSE, and FAP in stroma promoted corneal neovascularization induced by different etiological factors. Curr. Eye Res. 35, 967–977 [DOI] [PubMed] [Google Scholar]
- 55. Tian X., Chen G., Zhou S., Henne-Bruns D., Bachem M., Kornmann M. (2012) Interactions of pancreatic cancer and stellate cells are mediated by FGFR1-III isoform expression. Hepatogastroenterology 59, 1604–1608 [DOI] [PubMed] [Google Scholar]
- 56. Li R., Hou J., Xu Q., Liu Q. J., Shen Y. J., Rodin G., Li M. (2012) High level interleukin-6 in the medium of human pancreatic cancer cell culture suppresses production of neurotransmitters by PC12 cell line. Metab. Brain Dis. 27, 91–100 [DOI] [PubMed] [Google Scholar]
- 57. Mroczko B., Groblewska M., Gryko M., Kedra B., Szmitkowski M. (2010) Diagnostic usefulness of serum interleukin 6 (IL-6) and C-reactive protein (CRP) in the differentiation between pancreatic cancer and chronic pancreatitis. J. Clin. Lab. Anal. 24, 256–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tomlins C., Storey A. (2010) Cutaneous HPV5 E6 causes increased expression of osteoprotegerin and interleukin 6 which contribute to evasion of UV-induced apoptosis. Carcinogenesis 31, 2155–2164 [DOI] [PubMed] [Google Scholar]
- 59. Law H. K., Tu W., Liu E., Lau Y. L. (2008) Insulin-like growth factor I promotes cord blood T cell maturation through monocytes and inhibits their apoptosis in part through interleukin-6. BMC Immunol. 9, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Campbell V., Lynch M. A. (2000) The role of ceramide in the modulation of intracellular Ca2+ levels by interleukin 1 β in rat cortical synaptosomes. Cytokine 12, 487–490 [DOI] [PubMed] [Google Scholar]
- 61. Hill H. R., Augustine N. H., Jaffe H. S. (1991) Human recombinant interferon γ enhances neonatal polymorphonuclear leukocyte activation and movement, and increases free intracellular calcium. J. Exp. Med. 173, 767–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mahali S., Raviprakash N., Raghavendra P. B., Manna S. K. (2011) Advanced glycation end products (AGEs) induce apoptosis via a novel pathway. Involvement of Ca2+ mediated by interleukin-8 protein. J. Biol. Chem. 286, 34903–34913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Adebanjo O. A., Moonga B. S., Yamate T., Sun L., Minkin C., Abe E., Zaidi M. (1998) Mode of action of interleukin-6 on mature osteoclasts. Novel interactions with extracellular Ca2+ sensing in the regulation of osteoclastic bone resorption. J. Cell Biol. 142, 1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hajnóczky G., Davies E., Madesh M. (2003) Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 304, 445–454 [DOI] [PubMed] [Google Scholar]