

Background: RSK2-mediated prometastatic signaling mechanism remains unclear.
Results: RSK2-CREB pathway up-regulates Fascin-1 to promote filopodia formation, cancer cell invasion, and tumor metastasis.
Conclusion: RSK2-CREB-Fascin-1 pathway could be a promising therapeutic target in metastatic cancers.
Significance: These data will advance our understanding of signaling pathways that mediates RSK2-dependent prometastatic signals.
Keywords: Cell Invasion, Cell Migration, Metastasis, Serine Threonine Protein Kinase, Signal Transduction, Fascin-1, Ribosomal S6 Kinase 2, cAMP Response Element-binding, Filopodia
Abstract
Metastasis is the leading cause of death in patients with breast, lung, and head and neck cancers. However, the molecular mechanisms underlying metastases in these cancers remain unclear. We found that the p90 ribosomal S6 kinase 2 (RSK2)-cAMP response element-binding protein (CREB) pathway is commonly activated in diverse metastatic human cancer cells, leading to up-regulation of a CREB transcription target Fascin-1. We also observed that the protein expression patterns of RSK2 and Fascin-1 correlate in primary human tumor tissue samples from head and neck squamous cell carcinoma patients. Moreover, knockdown of RSK2 disrupts filopodia formation and bundling in highly invasive cancer cells, leading to attenuated cancer cell invasion in vitro and tumor metastasis in vivo, whereas expression of Fascin-1 significantly rescues these phenotypes. Furthermore, targeting RSK2 with the small molecule RSK inhibitor FMK-MEA effectively attenuated the invasive and metastatic potential of cancer cells in vitro and in vivo, respectively. Taken together, our findings for the first time link RSK2-CREB signaling to filopodia formation and bundling through the up-regulation of Fascin-1, providing a proinvasive and prometastatic advantage to human cancers. Therefore, protein effectors of the RSK2-CREB-Fascin-1 pathway represent promising biomarkers and therapeutic targets in the clinical prognosis and treatment of metastatic human cancers.
Introduction
More than 90% of mortality from cancer is not caused by the primary tumors but rather by metastases, which are a major prognostic indicator and cause it to be an incurable disease (1–5). Invasion is the process by which cancer cells leave the tumor and enter adjacent tissue (6). Cancer cells possess a broad spectrum of invasion mechanisms that are tightly regulated by a number of cell signaling proteins (1, 7, 8). We recently reported that p90 ribosomal S6 kinase 2 (RSK2)4 confers proinvasive and prometastatic potential to metastatic head and neck cancer cells (9). RSK2 is a Ser/Thr kinase that phosphorylates multiple signaling effectors, including transcription factors such as the cAMP response element-binding (CREB) protein (10), to regulate cell cycle, cell survival, and proliferation (reviewed in Refs. 11 and 12).
CREB is a transcription factor whose signaling is implicated in the promotion of tumor progression, growth stimulation, the conferring of apoptotic resistance, and the support of angiogenesis (13, 14). In human prostate cancer, CREB is associated with androgen-independent progression and promotes prostate cancer bone metastasis (15). RSK2 activates CREB by phosphorylating Ser-133, which then promotes CREB binding to the coactivator CBP (CREB-binding protein) and mediates functional contacts with the basal transcriptional machinery (16). Fascin-1 is a CREB transcription target that binds to actin and bundles F-actin into tightly packed parallel arrays (17–20). Fascin-1 is the only actin-bundling protein that localizes along the entire lengths of filopodia (21, 22), which are actin-containing spikes that aid in cell migration. Abundant filopodia formation is a critical hallmark of the invasive phenotype of cancer cells (23, 24). Elevated expression of Fascin-1 in cancer cells has been reported to correlate with an aggressive clinical outcome and poor prognosis in various types of cancers including liver cancer, melanoma, brain, and breast cancers (25–27). However, the mechanism by which proinvasive and prometastatic signals regulate CREB and Fascin-1 in metastatic human cancers remains largely unknown. Here, we report that the RSK2-CREB signaling pathway represents a common proinvasive and prometastatic pathway in diverse metastatic cancers that functions in part by up-regulating Fascin-1.
EXPERIMENTAL PROCEDURES
Antibodies
Antibodies include RSK2, vimentin, and GSK3α/β antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX); Fascin-1 antibodies (Santa Cruz Biotechnology, Inc. and Dako, Carpinteria, CA); phospho-CREB (Ser-133), CREB, phospho-RSK (Ser-380), and phospho-GSK3α/β (Ser-21/9) antibodies (Cell Signaling Technology, Inc., Danvers, MA); anti-FLAG and β-actin antibodies (Sigma Aldrich Corp.); Ki-67 antibody (Dako).
In Vitro Cell Invasion and Proliferation Assays
Cell invasion assays were performed as described (9). In brief, cells were seeded on Matrigel-coated chambers and incubated for 48 h. Invaded or non-invaded cells were fixed and stained in 25% methanol and 0.5% crystal violet. Total cell number was assessed by combining both non-invaded cells and invaded cells in the chambers. The relative invasion level was defined as the number of cells that had passed through the Matrigel-coated membranes normalized to the total cell number. Proliferation was determined by using the Celltiter96AQueous One solution proliferation kit (Promega, Madison, WI).
In Vitro Cell Migration Assay
Eight-μm pore Transwell inserts (BD Biosciences) bottoms were coated with collagen (40 μg/ml). Cells (3.3–6.6 × 103) were seeded in the upper chambers and were incubated for 24 h. Non-migrated cells on the upper surface or migrated cells on the lower surface of the membrane were removed with a cotton swab. The cells were fixed, stained, and counted as described above for the invasion assay.
Reagents
The RSK-specific inhibitor fmk has been described previously (28). SL0101 was purchased from Toronto Research Chemicals, Inc., Toronto, Ontario, Canada. shRNA constructs were purchased from Open Biosystems (Pittsburgh, PA). The sense strands of the RSK2, CREB, and Fascin-1 shRNAs were GCCTGAAGATACATTCTATTT, CAGTGGATAGTGTAACTGATT, and TTCTTGGAGGTCACAAACTTG, respectively. The RSK2 construct has been described previously (29). The image clone for Fascin-1 (GenBankTM accession no. BC000521) was purchased from Open Biosystems. The Fascin-1 gene was FLAG-tagged by PCR and subcloned into the pLHCX-derived gateway destination vector as described for expression in human cell lines (30). SKBR3 and A549 were from the American Type Culture Collection. 212LN and M4e have been described previously (9, 31). Cell line authentication was carried out using STR profiling by the RADIL CellCheckTM service.
Promoter Reporter Assay
The pmFascin-1-luc construct was kindly provided by Drs. D. Vignjevic and A. Reske-Kunz (Institut Curie and University of Mainz). The plasmid transfection and dual luciferase reporter assay were carried out according to the manufacturer's instructions (Promega, Madison, WI).
SEM Experiments
Cells were seeded in silicon chips and fixed in 2.5% glutaraldehyde and 0.1 m cacodylate buffer, washed in the same buffer, and post fixed in 1% buffered osmium (2.5% glutaraldehyde in 0.1 m cacodylate buffer) for 1 h followed with a gentle wash in distilled water. The samples were dehydrated in ethanol and placed into the chilled chamber of a critical point dryer (Polaron, model E3000), which was filled with liquid CO2 under pressure. The ethanol was completely exchanged for liquid CO2 in the critical point dryer. The dried samples were attached to labeled aluminum stubs and sputter coated with ∼12–15 nm gold-palladium. The samples were imaged using a Topcon DS 130F field emission scanning electron microscope with an accelerating voltage of 10 kV.
Immunofluorescence Staining
A549 and SKBR3 cells were seeded on coverslips and fixed in PHEMO buffer as described previously (9). Cells were blocked in 10% goat serum and then stained with Alexa Fluor 555 conjugated phalloidin (5 units/ml, Invitrogen) to stain filamentous actin. The coverslips were washed in PBS, mounted, and imaged on a Zeiss LSM 510 META confocal microscope.
Xenograft Nude Mouse Assay
Animal experiments were carried out based on protocols approved by the Institutional Animal Care and Use Committee of Emory University. For xenograft experiments using three groups of mutant M4e cell lines, mice (athymic nu/nu, female, 4–6 weeks old, Taconic, Hudson, NY) were injected submandibular to the mylohyoid muscle with mutant cells as described previously (9). After 3 weeks, primary tumors and lymph nodes were collected, fixed, and paraffin-embedded. For FMK-MEA treatment, each of the nude mice (athymic nu/nu, female, 4–6 weeks old) were injected with 0.5 × 106 cells/100 μl of PBS submandibular to the mylohyoid muscle. On day 5 after injection, mice were divided into two groups with similar average weights with each group receiving either FMK-MEA or PBS. Each mouse was administered 80 mg/kg of FMK-MEA daily by intraperitoneal injection from 5 days after the xenograft for 16 days total. The control group received PBS alone on the same schedule. Tumor growth was recorded by measuring (every 2–3 days) two perpendicular diameters of the tumors using the formula 4π/3 × (width/2)2 × (length/2). Mice were sacrificed after 16 days post drug treatment. The lymph nodes and primary tumors were collected, fixed, and paraffin-embedded for histopathological analyses.
Immunohistochemical Staining (IHC)
The IHC was performed in a manner similar to a method described previously using the set of human HNSCC tissue samples used in Ref. 9 and lymph nodes or tumors obtained from xenograft mice. The anti-Fascin-1 antibody from Dako (1:1000), anti-phospho-CREB S133 antibody from Cell Signaling Technology (1:100), anti-vimentin antibody from Santa Cruz Biotechnology (1:1000), and anti-Ki-67 antibody from Dako (1:200) were used for staining.
RESULTS
The RSK2-CREB Pathway Is Commonly Activated in Diverse Metastatic Human Cancers, Leading to Up-regulated Gene Expression of the Prometastatic Protein Fascin-1
To determine whether RSK2-CREB is a common proinvasive and prometastatic signaling pathway, we tested the invasive and migration potential of diverse metastatic cell lines with either RSK2 or CREB knockdowns, including HNSCC 212LN, lung cancer cell line A549, and the breast cancer cell line SKBR3. Targeted down-regulation of CREB or RSK2 resulted in a significant reduction of cell invasion and migration in 212LN, A549, and SKBR3 cells, suggesting that the RSK-CREB signaling pathway confers an invasive potential in diverse metastatic human cancer cell lines (Fig. 1, A and B, respectively). Similar results were obtained using additional shRNA clones or siRNAs for RSK2 and CREB (data not shown). However, knockdowns of either RSK2 or CREB did not significantly affect the proliferation rate, suggesting that the resulting decrease in cell invasion and migration was not a consequence of reduced proliferation (data not shown).
FIGURE 1.

RSK2 activates CREB to promote cancer cell invasion and migration by up-regulating proinvasive Fascin-1. A, stable knockdown of RSK2 or CREB by shRNA (lower panels) results in significantly decreased invasion (upper panels) in the metastatic cell line HNSCC 212LN (left), the lung cancer cell line A549 (middle) and the breast cancer cell line SKBR3 (right). B, stable knockdown of RSK2 in 212LN, A549, and SKBR3 cells decreases cancer cell migration in a collagen-based migration assay. C, a summary of RT-PCR results detecting mRNA levels of a group of CREB transcription targets in metastatic lung cancer A549 and HNSCC M4e cells with a stable knockdown of RSK2 or CREB. One-step RT-PCR was performed using specific primers. The mRNA level of each transcription target was calculated as the signal intensity of each target band normalized to its relevant GAPDH band on agarose gels. The number indicates the mRNA ratio of each target in A549 and M4e cells with either RSK2 knockdown or CREB knockdown to the control cells with an empty vector. D, real-time RT-PCR results show that RSK2 knockdown results in decreased mRNA levels of the prometastatic CREB downstream target, Fascin-1 but not other CREB transcription targets in M4e cells. E, real-time RT-PCR results show that RSK2 knockdown results in decreased mRNA levels of Fascin-1 in various metastatic human cancer cells. F, knockdown of RSK2 resulted in attenuated Ser-133 phosphorylation of CREB, as well as attenuated protein expression levels of Fascin-1 in highly invasive 212LN, A549, and SKBR3 cells. G, RSK2 activates the fascin-1 gene promoter in a luciferase reporter assay. The Fascin-1 promoter-reporter construct and a plasmid containing the constitutively active mutant form of RSK2,Y707A (RSK2 CA) were co-transfected into 293T cells. After 24 h, cells were collected and assayed for luciferase expression. The results are shown as the firefly luciferase activity corrected for the Renilla luciferase activity in the cell lysate, in RSK2 CA-stimulated versus control cells and are expressed as the fold induction. All of the error bars shown in the figures represent the mean values ± S.D. from three replicates of each sample. p values were determined using Student's t test (*, p = 0.01–0.05; **, p < 0.01). The Western blot data presented are from one experiment that is representative of multiple independent experiments.
To explore the molecular mechanism underlying RSK2-CREB pathway-enhanced metastasis, we surveyed potential links between RSK2 and a group of CREB transcription targets that are implicated in cell invasion and tumor metastasis, including heparanase-1 (32), insulin receptor substrate-2 (33), cysteine-rich protein 61 (Cyr61, CCN1) (34), matrix metalloproteinase 2 (35), and Fascin-1 (17, 18). To determine whether the RSK2-CREB pathway regulates these targets in metastatic cancer cells, we assessed the mRNA level of these potential targets in A549 and M4e cells with either RSK2 or CREB knockdown by quantitative RT-PCR. As shown in Fig. 1, C and D, the end point RT-PCR and real-time RT-PCR results show that knockdown of RSK2 did not result in a significant decrease in mRNA levels of heparanase-1, insulin receptor substrate-2, matrix metalloproteinase 2, and CCN1. In contrast, the mRNA levels of Fascin-1 resulted in a decrease in the presence of either RSK2 or CREB knockdown in metastatic lung cancer A549 and HNSCC M4e cells. These results suggest that Fascin-1, but not the other tested CREB targets, may be regulated by the RSK2-CREB signaling pathway in the tested metastatic cancer cells. Furthermore, shRNA-mediated knockdown of RSK2 resulted in a 30–90% reduction in Fascin-1 mRNA levels in invasive HNSCC 212LN cells, lung cancer A549 cells, and breast cancer SKBR3 cells, as determined by real-time RT-PCR (Fig. 1E). As shown in Fig. 1F, stable knockdown of RSK2 also resulted in reduced CREB activity, as assessed by S133 phosphorylation levels and the consequent reduction in Fascin-1 expression levels in these cell lines. Moreover, we examined whether RSK2 activity promoted Fascin-1 gene expression. We co-transfected 293T cells with a plasmid containing the fascin-1 promoter followed by a luciferase reporter gene and a constitutively active form of RSK2, Y707A. We observed that expression of RSK2 Y707A resulted in a significant increase in reporter activation driven by the fascin-1 promoter, as assessed by the luciferase activity, suggesting that RSK2 signaling promotes the gene expression of the Fascin-1 gene (Fig. 1G).
RSK2, Phospho-CREB, and Fascin-1 Levels Correlate in Primary Human Tumor Tissue Samples from HNSCC Patients
We have previously reported that the RSK2 expression pattern correlates with the metastatic progression of human head and neck cancer (9). Therefore, we sought to determine whether the levels of Fascin-1, phospho-CREB, and RSK2 exhibit a positive correlation with one another. The same set of human primary tumor tissue samples from HNSCC patients who were previously used to evaluate RSK2 expression patterns (9) was also used for Fascin-1 and phospho-CREB S133 IHC staining analysis. Primary tumors from patients with non-metastatic disease (TuMet−), primary tumors from patients with metastatic head and neck cancer (TuMet+) and paired metastatic lymph node samples from these same patients (LNMet+) were evaluated (Fig. 2A).
FIGURE 2.

The levels of RSK2, phospho-CREB, and Fascin-1 correlate with one another in primary human tumor tissue samples from HNSCC patients. A, IHC analysis was performed using 101 primary patient tissue specimens, including Tu(Met−), Tu(Met+), and LN(Met+) samples (Tu, tumor). Representative IHC staining images of RSK2, phospho-CREB S133, and Fascin-1 for each human HNSCC tissue specimen are shown. B, expressions were characterized by weighted index (WI = positive staining (%) × intensity score (0 ∼ 3+)). The correlation between RSK2, phospho-CREB, and Fascin-1 was assessed by the Pearson correlation coefficient using Graphpad Prism (version 6, Graphpad Software, Inc., La Jolla, CA). p values are two-tailed with significance considered to be p < 0.05.
As shown in Fig. 2B, positive staining of Fascin-1 and phospho-CREB at S133 in the tumor cells was identified using IHC signal intensity (scored as 0 to 3+). The data were compared with the results of RSK2 IHC analysis using the same set of samples. Weighted index (WI; staining intensity (0, 1+, 2+, 3+) × percent of cells stained (0 ∼ 100%)) of RSK2, phospho-CREB, and Fascin-1 were compared as a pair in each patient sample (n = 101). The correlation between the RSK2, phospho-CREB (p-CREB), and Fascin-1 expression was assessed by Pearson correlation coefficient. We found that the levels of RSK2 and p-CREB (left), p-CREB and Fascin-1 (middle), and RSK2 and Fascin-1 (right) are positively correlated with r values of 0.47, 0.40, and 0.38, respectively. Together, these results support our hypothesis that up-regulated RSK2 also up-regulates Fascin-1 through CREB signaling in metastatic human cancers.
RSK2-CREB Pathway Signals in Part through Fascin-1 to Promote Cancer Cell Invasion in Vitro and Tumor Metastasis in Vivo in a Xenograft Mouse Model
We also observed that stable knockdown of Fascin-1 by different shRNAs led to a significantly attenuated invasive potential without affecting the proliferation rate in diverse metastatic cancer cells, including 212LN, M4e, A549, and SKBR3 (Fig. 3A). As shown in Fig. 3B (upper panels), a stable knockdown of RSK2 significantly attenuated the invasive ability of 212LN, M4e, A549 and SKBR3 cells. In contrast, stable overexpression of FLAG-tagged Fascin-1 by retroviral infection in these cells partially rescued these phenotypes due to a lack of RSK2, resulting in increased cell invasion compared with the corresponding RSK2 knockdown control cells that did not exhibit any proliferative change (Fig. 3B). These data suggest that RSK2 signals through CREB to promote cancer cell invasion at least in part, by up-regulating prometastatic protein Fascin-1.
FIGURE 3.

RSK2-CREB pathway promotes cancer cell invasion and tumor metastasis in part by signaling through up-regulating Fascin-1. A, stable knockdown of Fascin-1 attenuates cell invasion of diverse metastatic cancer cells. B, expression of FLAG-tagged Fascin-1 significantly rescues the attenuated cell invasion potential in various metastatic cancer cells with stable knockdown of RSK2. Relative invasion was normalized to the invasion of control cells harboring empty vectors. p values were determined using Student's t test. C, stable overexpression of Fascin-1 significantly rescues the LN metastasis attenuated by RSK2 knockdown in an M4e xenograft mouse model (n = 7, mice injected with M4e cells harboring empty vectors; n = 8, mice injected with M4e cells with a RSK2 knockdown; n = 8, mice injected with M4e cells with RSK2 knockdown and overexpression of FLAG-Fascin-1). Western blot (WB) results show stable knockdown of RSK2 or expression of FLAG-tagged Fascin-1 in tumor lysates. D, xenograft experiment results show that RSK2 knockdown in M4e cells results in significantly reduced LN metastases, whereas overexpression of Fascin-1 partially rescues the reduced metastatic potential due to loss of RSK2. LN metastases were identified by H&E staining (upper panel) and human vimentin IHC staining (lower panel). Left, representative images of H&E stained or vimentin IHC stained LNs from each group are shown. Right, LN metastasis was determined by calculating the number of metastasized LNs of the total number of LN harvested by H&E staining (top). p values for H&E staining were determined by Fisher's exact test. The human vimentin IHC staining results are summarized by weighted index (bottom). p values for vimentin IHC staining were determined using Student's t test. E, RNAi-mediated down-regulation of RSK2 or overexpression of Fascin-1 did not affect tumor sizes or cell proliferation in tumors. Tumors were harvested and weighed at the experimental end point to determine tumor formation. Individual points represent differential weights of tumors from distinct mice (left). IHC staining of the proliferation marker protein Ki-67 was performed using primary tumor samples from each mouse (right). p values were determined using Student's t test (*, p = 0.01–0.05; **, p < 0.01; ns, not significant).
We next assessed the effect of rescuing the expression of Fascin-1 in cells with a stable knockdown of RSK2 on metastatic potential in vivo using a xenograft mouse model. Three groups of nude mice were injected with either highly metastatic M4e cells stably transduced with empty vectors, cells with a stable knockdown of RSK2, or cells with a stable knockdown of RSK2 and overexpressing Fascin-1 (Fig. 3C). The mice were sacrificed at the experimental end point at day 21 when M4e cells were observed to induce detectable lymph node (LN) metastasis. We characterized metastases in the tissue samples of LNs using H&E staining (Fig. 3D, upper panels) as well as IHC staining of vimentin, a mesenchymal cell marker that is expressed only in metastatic cancer cells (Fig. 3D lower panels). The staining was quantified using weighted index (WI = positive staining (%) × intensity score (0 ∼ 3+)). We found that overexpression of Fascin-1 in RSK2 knockdown cells significantly rescued the reduction in LN metastases due to RSK2 deficiency in vivo in xenograft mice (Fig. 3D). Moreover, IHC staining studies revealed reduced CREB phosphorylation and Fascin-1 expression levels in lymph nodes infiltrated by RSK2 knockdown cells compared with lymph nodes infiltrated by control cells, whereas overexpression of FLAG-Fascin-1 in RSK2 knockdown cells rescues decreased Fascin-1 level as well as attenuated LN metastasis (data not shown). We next determined whether altered protein expression levels of RSK2 and/or Fascin-1 might affect the metastatic potential of cancer cells by altering cell proliferation. As shown in Fig. 3E left, no significant difference in the primary tumor size was detected among the three groups of mice injected with M4e cells with empty vectors, a stable knockdown of RSK2, or RSK2 knockdown and overexpression of FLAG-Fascin-1. Stable knockdown of RSK2 and/or overexpression of Fascin-1 in M4e cells did not affect cell proliferation, as assessed by the percentage of cells with positive staining of Ki-67 in each primary tumor (Fig. 3E, right). These results suggest that the RSK2-CREB pathway signals through Fascin-1 to promote tumor metastasis by a proliferation-independent mechanism.
RSK2 Signals through Fascin-1 to Promote Filopodia Formation
Fascin-1 is the main actin filament-bundling protein in filopodia, the finger-like cellular extensions that play an important role in cell migration and cancer cell invasion. To determine whether RSK2 is required for filopodia formation, we performed scanning electron microscopy to observe the filopodia on the surface of metastatic human cancer cells with or without stable knockdown of RSK2. As shown in Fig. 4, A and B, stable knockdown of RSK2 by shRNA in A549 cells resulted in a loss of filopodia (Fig. 4A, middle), compared with control cells with empty vectors (Fig. 4A, left). Overexpression of FLAG-Fascin-1 partially rescued the formation of filopodia in cells with a stable knockdown of RSK2 (Fig. 4A, right). Similar results were obtained using breast cancer SKBR3 cells. As shown in Fig. 4, C and D, SKBR3 cells with a knockdown of RSK2 exhibited an altered cellular morphology, with a disruption of the tips of filopodial bundles and a reduced number of filopodia on the cell surface (Fig. 4C, middle) compared with control cells harboring empty vectors (Fig. 4C, left). Fascin-1 overexpression in SKBR3 cells with RSK2 knockdown partially rescued filopodia formation (Fig. 4C, right).
FIGURE 4.

RSK2 signals through CREB → Fascin-1 to promote filopodia formation in metastatic cancer cells. A549 and SKBR3 cells were seeded on silicon chips and evaluated with SEM. A, representative SEM images show the filopodia formed on the surface of A549 cells. Stable knockdown of RSK2 results in a decreased number of filopodia on the surface of A549 cells (middle) compared with control A549 cells harboring empty vectors (left). Overexpression of Fascin-1 partially rescues attenuated filopodia formation due to RSK2 knockdown in A549 cells (right). Representative filopodia are indicated by arrows. Scale bars, 2 μm. B, the relative average number of filopodial tips per field in each A549 cell line was counted from eight random fields of view and plotted. Statistical significance was determined using Student's t test. C, representative SEM imaging results show filopodia formed on the surface of SKBR3 cells. Left, control SKBR3 cells with empty vectors. Middle and right, RSK2 knockdown leads to a reduced number of filopodial tips on the surface of SKBR3 cells and morphological change (middle), whereas stable expression of Fascin-1 rescues the reduced filopodia formation and altered morphology upon RSK2 knockdown (right). Scale bars, 5 μm. D, the relative average number of filopodial tips per eight random fields in each SKBR3 cell line was counted and plotted. Statistical significance was determined using Student's t test (**, p < 0.01).
In support of these observations, we observed that actin filaments were well organized and distributed evenly throughout the control A549 and SKBR3 cells transduced with empty vectors (Fig. 5, A and B, left, respectively). In contrast, knockdown of RSK2 in A549 and SKBR3 cells induced the disruption of actin filaments and the redistribution of actin to the cell membrane (Fig. 5, A and B, middle, respectively). However, stable expression of FLAG-Fascin-1 rescued the formation of actin filaments in A549 and SKBR3 cells with a stable knockdown of RSK2 (Fig. 5, A and B, right, respectively). Moreover, stable overexpression of FLAG-Fascin-1 significantly rescued the attenuated invasive ability of A549, SKBR3, and 212LN cells caused by RSK2 knockdown, whereas treatment with the actin disrupting compound, latrunculin A reversed this rescue effect of Fascin-1 overexpression (Fig. 5C). Thus, these results together suggest a transcription-dependent mechanism by which RSK2 signals through CREB to promote actin bundling in filopodia, and consequently, cancer cell invasion, which is mediated, in part, by upregulating prometastatic Fascin-1.
FIGURE 5.

RSK2 requires Fascin-1 to promote filopodia bundling. A, immunofluorescence staining results show the filopodia bundling detected by staining filopodia-associated actin filaments using Alexa Fluor 555-conjugated phalloidin in A549 cells. Stable RSK2 knockdown decreased filopodia bundling in A549 cells (middle) compared with control cells with empty vectors (left). Overexpression of Fascin-1 rescues the filopodia bundling attenuated by RSK2 knockdown in A549 cells (right). B, filopodia bundle formation in SKBR3 cells detected by immunofluorescence staining. RSK2 knockdown abolishes filopodia bundle formation in SKBR3 cells (middle) compared with control cells (left). Overexpression of Fascin-1 partially rescues the formation of filopodia bundling attenuated by RSK2 knockdown in SKBR3 cells (right). Arrows indicate actin bundling associated with phalloidin. Original magnification, 1000×. C, overexpression of Fascin-1 significantly rescued the decreased invasion of A549, SKBR3, and 212LN cells due to knockdown of RSK2, while treatment with latrunculin A (Lat A), which disrupts actin bundling abolished the rescue effect of Fascin-1 overexpression. p values were determined using Student's t test (*, p = 0.01–0.05; **, p < 0.01).
Targeting RSK2 with a Derivative of fmk, FMK-MEA Attenuates Cancer Cell Invasive Potential in Vitro and Metastatic Potential in Vivo
We previously demonstrated that treatment with an RSK-specific small molecule inhibitor, fmk inhibits RSK2 kinase activity and attenuates HNSCC cell invasion in vitro (9). To better evaluate the potential efficacy of targeting RSK2 in diverse metastatic human cancers, we evaluated a second generation RSK inhibitor FMK-MEA, which is a water-soluble derivative of fmk, in the treatment of diverse metastatic human cancer cell lines in vitro and xenograft nude mice with LN metastasis in vivo. FMK-MEA treatment inhibited RSK2 kinase activity as assessed by Ser-386 phosphorylation, in diverse, highly invasive human cancer cell lines including 212LN, M4e, A549, and SKBR3 cells (Fig. 6A). Treatment with the RSK-specific inhibitor FMK-MEA significantly attenuated RSK2 activity, as assessed by the phosphorylation levels of Ser-386 and the consequent invasive ability of A549 cells (Fig. 6B, left). A similar result was obtained using an alternative specific RSK inhibitor, SL0101 (Fig. 6B, right) (36). Stable overexpression of FLAG-Fascin-1 rescued the reduced invasive potential of cells treated with FMK-MEA or SL0101, although Fascin-1 overexpression did not rescue the inhibition of RSK2 activity by FMK-MEA or SL0101 (Fig. 6B). These data, together with those reported in Fig. 3B in which we targeted RSK2 with shRNA, suggest that RSK2 may signal through CREB to promote cancer cell invasion by up-regulating the prometastatic protein Fascin-1. In an in vitro cell invasion assay using highly invasive cells, we observed that targeting RSK2 by FMK-MEA treatment inhibited cell invasion (Fig. 6C). We also observed that FMK-MEA treatment inhibits RSK2 kinase activity and cancer cell invasion with comparable effectiveness as fmk (data not shown). However, treatment with FMK-MEA did not significantly affect the proliferation rate of these cells (Fig. 6D).
FIGURE 6.

The RSK inhibitor FMK-MEA effectively attenuates cancer cell invasion in diverse metastatic human cancer cells. A, treatment with FMK-MEA (6 μm) effectively inhibits RSK2 kinase activity, as was assessed by the phosphorylation levels of S386 of RSK2 detected in a Western blot using a specific phospho-RSK antibody (p-S380) to probe various metastatic human cancer cells, including 212LN, M4e, A549, and SKBR3. B, inhibition of RSK2 by the specific RSK inhibitors, FMK-MEA (6 μm) or SL0101 (25 μm), results in a reduction in the invasive ability of A549 cells, whereas Fascin-1 overexpression rescues the reduced invasive potential of cells treated with FMK-MEA or SL0101. Relative invasiveness was normalized to the invasion of control cells harboring empty vectors. C, targeting RSK2 with FMK-MEA (6 μm) results in reduced invasiveness of metastatic human cancer cells in an in vitro Matrigel invasion assay. Relative invasiveness was normalized to the control cells without drug treatment. D, treatment with FMK-MEA (6 μm) did not attenuate the cell proliferation rate. Cell number was determined using a proliferation assay kit, and the cell viability was normalized to a standard curve of cell number. The RSK2 activity assay (A), invasion assay (B and C), and proliferation assay (D) were performed in parallel after 48 h of drug treatment. p values were determined using Student's t test (ns, not significant; *, p = 0.01–0.05; **, p < 0.01).
In agreement with these observations, FMK-MEA treatment (80 mg/kg/day for 16 days by intraperitoneal injection) in highly metastatic M4e cell xenograft nude mice resulted in a significant attenuation of LN metastasis (Fig. 7A). FMK-MEA treatment had no effect on the tumor size (Fig. 7B), and the proliferation rate of the primary tumor (Fig. 7C) compared with the control mice treated with PBS. Efficacy of FMK-MEA in xenograft HNSCC tumor and metastasis mouse models was evaluated by decreased phosphorylation levels of RSK2 and CREB as well as reduced Fascin-1 expression levels in LNs and tumors (Fig. 8). These results are consistent with our observations that targeting RSK2 with shRNA attenuated LN metastasis but not primary tumor growth in xenograft mice (Fig. 3, C–E).
FIGURE 7.
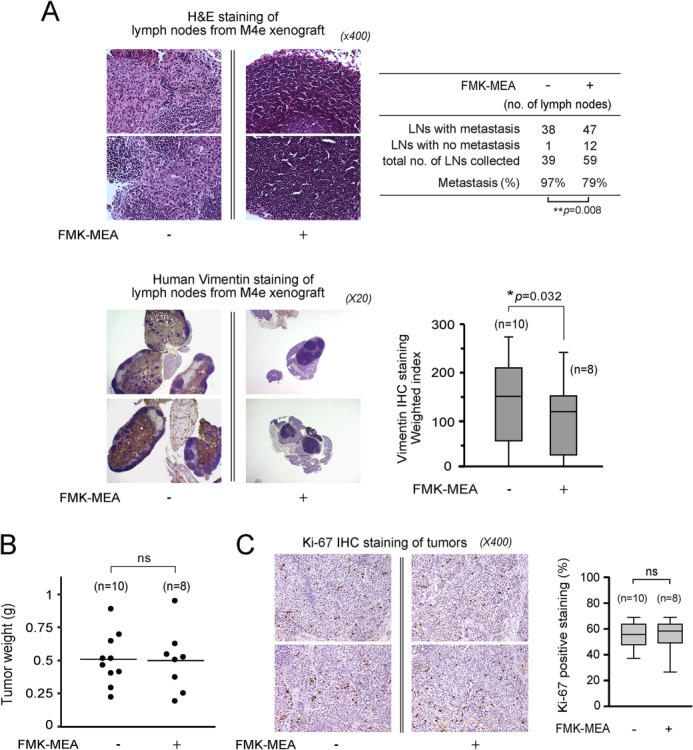
Treatment with the RSK inhibitor, FMK-MEA, results in significantly reduced LN metastasis but does not reduce tumor cell proliferation and growth in xenograft nude mice injected with highly metastatic M4e cells. A, left: representative images of H&E staining and human vimentin IHC staining of LN samples from M4e xenograft nude mice treated with PBS or FMK-MEA are shown. Right, LN metastasis was determined by calculating the number of metastasized LNs out of the total number of LN harvested by H&E staining (top). p values for H&E staining were determined by Fisher's exact test. The weighted index of the human vimentin IHC results shows that FMK-MEA treatment significantly reduces the LN metastasis in M4e xenograft mice compared with control mice treated with PBS (bottom). p values for vimentin IHC staining were determined using Student's t test (n = 10, mice injected with PBS; n = 8, mice injected with FMK-MEA). B and C, targeting RSK2 by FMK-MEA did not affect tumor size or cell proliferation within tumors. Primary tumors were weighed at the experimental end point from xenograft nude mice. Individual points represent different weights of tumors from different mice (B). IHC staining of the proliferation marker protein Ki-67 was performed using primary tumor samples from each mouse. Representative images of IHC staining of Ki-67 are shown (C). p values were determined using Student's t test.
FIGURE 8.
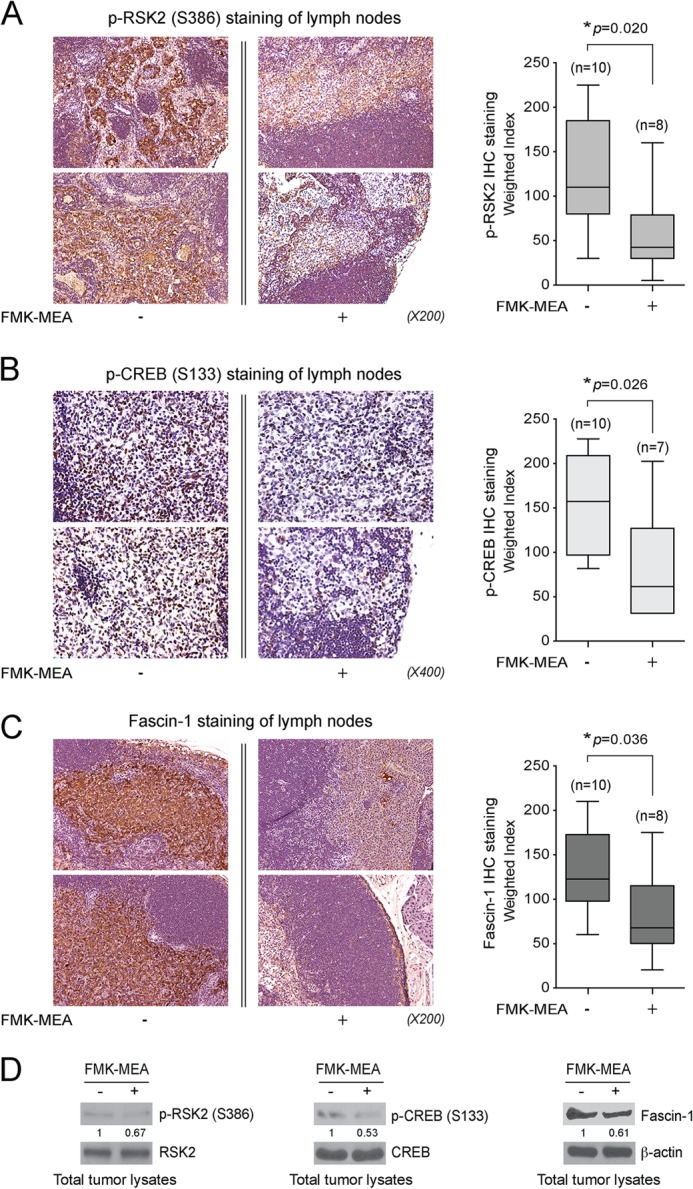
Treatment with RSK inhibitor FMK-MEA significantly attenuates activation of RSK2-CREB-Fascin-1 pathway in metastatic cells in vivo. A–C, IHC staining of phospho-RSK2 Ser-386 (A), phospho-CREB Ser-133 (B), and Fascin-1 (C) using LN samples from M4e xenograft nude mice treated with PBS or FMK-MEA. Representative IHC staining images for each group are shown on the left. Expression was characterized by weighted index (WI = positive staining (%) × intensity score (0∼3+)). p values were determined by Student's t test. D, Western blot results show a decrease of RSK2 and CREB activity as well as Fascin-1 expression upon FMK-MEA treatment in total tumor lysates.
DISCUSSION
Our findings suggest that the RSK2-CREB pathway is commonly activated in diverse metastatic human cancers and mediates proinvasive and prometastatic signals at least in part by up-regulating gene expression of the transcription target Fascin-1. Moreover, we observed that the levels of RSK2, phospho-CREB, and Fascin-1 positively correlate in primary human tumor tissue samples from HNSCC patients and also correlate with tumor progression. Furthermore, we provide evidence that treatment with a newly developed, potent RSK inhibitor, FMK-MEA, effectively reduced the metastatic potential of cancer cells in xenograft mice, whereas overexpression of Fascin-1 partially rescued the reduced metastasis due to attenuation of RSK2 in vivo. Together, these findings have for the first time linked the RSK2-CREB signaling to filopodia formation and bundling by up-regulating Fascin-1, providing a proinvasive and prometastatic advantage to human cancers. Thus, protein effectors of the RSK2-CREB-Fascin-1 pathway may represent promising biomarkers and therapeutic targets in the prognosis and treatment of metastatic human cancers.
Fascin-1 is an emerging anti-cancer target due to the important role it plays in filopodia formation and consequent cell migration (37). CREB is known to regulate and activate Fascin-1 transcription in carcinoma cells (17, 18). Knockdown of RSK2 results in decreased cancer cell invasion and tumor metastasis, which correlate with reduced filopodia formation. Fascin-1 expression rescues these phenotypes in RSK2 knockdown cells, suggesting that RSK2 phosphorylates and activates CREB to up-regulate Fascin-1, which then promotes filopodia formation and consequently contributes to cancer cell migration, invasion, and tumor metastasis. We previously reported a novel RSK2 phosphorylation target Hsp27, which regulates actin dynamics. RSK2 activates Hsp27 to promote formation of actin filaments and cell invasion via direct phosphorylation of Hsp27 at both Ser-78 and Ser-82 (9). This is a short-term “transcription-independent” mechanism underlying RSK2-mediated, proinvasive and prometastatic signaling that controls the turnover of actin filaments and bundles in cancer cells. Our new findings demonstrate that the RSK2-CREB pathway up-regulates Fascin-1 to promote filopodia formation and bundling, providing an additional long term “transcription-dependent” mechanism. Further research is warranted to explore how RSK2 signaling coordinates the activation of Hsp27 and the expression of Fascin-1 to control actin bundling during filopodia formation and how this promotes cancer cell migration, invasion, and tumor metastasis.
It is worth noting that residual filopodia formation occurs on the cell surface of RSK2 stable knockdown cells. This may be due to basal levels of RSK2 expression despite shRNA-mediated knockdown in the cells or RSK2-CREB-Fascin-1 and/or RSK2-Hsp27-independent pathways that may contribute to filopodia formation such as β-catenin/TCF signaling (38). β-catenin is believed to induce the expression of genes involved in invasion and metastasis such as matrix metalloproteinases and the cell adhesion molecule L1 (39–41). Previous studies have shown that the fascin-1 gene is a direct transcriptional target of β-catenin/TCF signaling in colon cancer (38). GSK3β, which is a downstream substrate of RSK2, binds and phosphorylates β-catenin, resulting in ubiquitin-mediated degradation of β-catenin (42). Therefore, future studies exploring how RSK2 and β-catenin coordinate signaling pathways to promote cancer cell invasion and tumor metastasis are warranted. In addition, previous reports suggest that other CREB transcription targets, including plasminogen activator inhibitor-1 and osteopontin, may also be important in the regulation of cell invasion and migration (43–45). Future studies to examine whether these effectors also participate in RSK2-CREB mediated proinvasive and prometastatic signaling are also warranted.
We previously demonstrated that fmk is a potent RSK inhibitor and treatment with fmk attenuates HNSCC cell invasion, but not proliferation (9). Here we are the first to test a bioavailable derivative of fmk, FMK-MEA, in animals using a xenograft mouse model. FMK-MEA is a fluoromethylketone molecule that was designed to specifically exploit two selectivity filters for RSK. Fmk potently inactivates the C-terminal auto-kinase domain activity of RSK1 and RSK2 with high specificity in mammalian cells (28). FMK-MEA significantly attenuated the invasive ability of all the metastatic human cancer cell lines tested. Our data supports the notion that targeting RSK2 with specific small molecule drugs may represent a potential therapeutic strategy to attenuate metastatic cancer cell invasion and tumor metastasis. We previously demonstrated that targeting RSK2 effectively induces apoptotic cell death and attenuates cell proliferation in t(4;14) FGFR3-positive multiple myeloma cells or FLT3-ITD-positive acute myeloid leukemia cells (29, 46). We also showed that RSK specific inhibitors such as fmk might have minimal nonspecific cytotoxicity in human cells (29).
However, in contrast to the proproliferative and prosurvival role of RSK2 in hematopoietic malignancies, targeting RSK2 with FMK-MEA does not effectively inhibit cell proliferation in diverse human tumor cells, including the head and neck cancer cell lines 212LN and M4e, the lung cancer cell line A549 or the breast cancer cell line SKBR3. Although FMK-MEA effectively attenuates LN metastasis in xenograft nude mice injected with M4e cells, the drug treatment did not affect tumor cell proliferation or tumor growth in vivo. This is consistent with our previous findings that stable a knockdown of RSK2 significantly inhibits the metastatic potential of cancer cells in xenograft mice, whereas a lack of RSK2 has no significant impact on tumor cell proliferation and tumor growth (9). Such differential reliance on RSK2 signaling in cell proliferation among different human cancer cells may be due to different oncogenic events and/or the genetic backgrounds of different types of cancers. Thus, our findings warrant the further development of combined therapeutic strategies to treat metastatic cancers using RSK2 antagonists and other anti-cancer reagents to abrogate both tumor growth and metastasis.
Acknowledgments
We thank Jeannette Taylor at Emory University Electron Microscopy Core for assistance with scanning electron microscopy. We thank Jong Seok Lee for technical support. We acknowledge the microscope core facility at the Winship Cancer Institute of Emory University.
This work was supported in part by the American Cancer Society Grant RSG-11-081-01 (to S. K.), the National Institutes of Health/NCI SPORE in Head and Neck Cancer P50CA128613 Career Development Program Award (to S. K.), and a Robbins Scholar Award (to S. K.). M. T. and T.-L. G. are employees of Cell Signaling Technology, Inc. J. T. is as an inventor on a patent application filed by the University of California that describes the RSK inhibitor FMK.
- RSK2
- p90 ribosomal S6 kinase 2
- CREB
- cAMP response element-binding protein
- HNSCC
- head and neck squamous cell carcinoma
- IHC
- immunohistochemical staining
- LN
- lymph node
- fmk
- fluoromethyl ketone.
REFERENCES
- 1. Fidler I. J. (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 3, 453–458 [DOI] [PubMed] [Google Scholar]
- 2. Nguyen D. X., Massagué J. (2007) Genetic determinants of cancer metastasis. Nat. Rev. Genet. 8, 341–352 [DOI] [PubMed] [Google Scholar]
- 3. Gupta G. P., Massagué J. (2006) Cancer metastasis: building a framework. Cell 127, 679–695 [DOI] [PubMed] [Google Scholar]
- 4. Talmadge J. E., Fidler I. J. (2010) AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 70, 5649–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valastyan S., Weinberg R. A. (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedl P., Alexander S. (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 [DOI] [PubMed] [Google Scholar]
- 7. Sahai E. (2005) Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 15, 87–96 [DOI] [PubMed] [Google Scholar]
- 8. Friedl P., Wolf K. (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374 [DOI] [PubMed] [Google Scholar]
- 9. Kang S., Elf S., Lythgoe K., Hitosugi T., Taunton J., Zhou W., Xiong L., Wang D., Muller S., Fan S., Sun S. Y., Marcus A. I., Gu T. L., Polakiewicz R. D., Chen Z. G., Khuri F. R., Shin D. M., Chen J. (2010) p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J. Clin. Invest. 120, 1165–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buck M., Poli V., Hunter T., Chojkier M. (2001) C/EBPβ phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol. Cell 8, 807–816 [DOI] [PubMed] [Google Scholar]
- 11. Kang S., Chen J. (2011) Targeting RSK2 in human malignancies. Expert Opin. Ther. Targets 15, 11–20 [DOI] [PubMed] [Google Scholar]
- 12. Anjum R., Blenis J. (2008) The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 9, 747–758 [DOI] [PubMed] [Google Scholar]
- 13. Abramovitch R., Tavor E., Jacob-Hirsch J., Zeira E., Amariglio N., Pappo O., Rechavi G., Galun E., Honigman A. (2004) A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer Res. 64, 1338–1346 [DOI] [PubMed] [Google Scholar]
- 14. Jiang H., Chen S. S., Yang J., Chen J., He B., Zhu L. H., Wang L. (2012) CREB-binding protein silencing inhibits thrombin-induced endothelial progenitor cells angiogenesis. Mol. Biol. Rep. 39, 2773–2779 [DOI] [PubMed] [Google Scholar]
- 15. Wu D., Zhau H. E., Huang W. C., Iqbal S., Habib F. K., Sartor O., Cvitanovic L., Marshall F. F., Xu Z., Chung L. W. (2007) cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene 26, 5070–5077 [DOI] [PubMed] [Google Scholar]
- 16. Xing J., Ginty D. D., Greenberg M. E. (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273, 959–963 [DOI] [PubMed] [Google Scholar]
- 17. Kim S. J., Choi I. J., Cheong T. C., Lee S. J., Lotan R., Park S. H., Chun K. H. (2010) Galectin-3 increases gastric cancer cell motility by up-regulating fascin-1 expression. Gastroenterology 138, 1035–45.e1–2 [DOI] [PubMed] [Google Scholar]
- 18. Hashimoto Y., Loftis D. W., Adams J. C. (2009) Fascin-1 promoter activity is regulated by CREB and the aryl hydrocarbon receptor in human carcinoma cells. PLoS One 4, e5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishikawa R., Sakamoto T., Ando T., Higashi-Fujime S., Kohama K. (2003) Polarized actin bundles formed by human fascin-1: their sliding and disassembly on myosin II and myosin V in vitro. J. Neurochem. 87, 676–685 [DOI] [PubMed] [Google Scholar]
- 20. Hayashi Y., Toda K., Saibara T., Okamoto S., Osanai M., Enzan H., Lee G. H. (2008) Expression of fascin-1, an actin-bundling protein, in migrating hepatoblasts during rat liver development. Cell Tissue Res. 334, 219–226 [DOI] [PubMed] [Google Scholar]
- 21. Small J. V. (1988) The actin cytoskeleton. Electron Microsc. Rev. 1, 155–174 [DOI] [PubMed] [Google Scholar]
- 22. Small J. V., Stradal T., Vignal E., Rottner K. (2002) The lamellipodium: where motility begins. Trends Cell Biol. 12, 112–120 [DOI] [PubMed] [Google Scholar]
- 23. Vignjevic D., Kojima S., Aratyn Y., Danciu O., Svitkina T., Borisy G. G. (2006) Role of fascin in filopodial protrusion. J. Cell Biol. 174, 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vignjevic D., Yarar D., Welch M. D., Peloquin J., Svitkina T., Borisy G. G. (2003) Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 160, 951–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Machesky L. M., Li A. (2010) Fascin: Invasive filopodia promoting metastasis. Commun Integr. Biol. 3, 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams J. C. (2004) Roles of fascin in cell adhesion and motility. Curr. Opin. Cell Biol. 16, 590–596 [DOI] [PubMed] [Google Scholar]
- 27. Baldassarre M., Ayala I., Beznoussenko G., Giacchetti G., Machesky L. M., Luini A., Buccione R. (2006) Actin dynamics at sites of extracellular matrix degradation. Eur. J. Cell Biol. 85, 1217–1231 [DOI] [PubMed] [Google Scholar]
- 28. Cohen M. S., Zhang C., Shokat K. M., Taunton J. (2005) Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308, 1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang S., Dong S., Gu T. L., Guo A., Cohen M. S., Lonial S., Khoury H. J., Fabbro D., Gilliland D. G., Bergsagel P. L., Taunton J., Polakiewicz R. D., Chen J. (2007) FGFR3 activates RSK2 to mediate hematopoietic transformation through tyrosine phosphorylation of RSK2 and activation of the MEK/ERK pathway. Cancer Cell 12, 201–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hitosugi T., Fan J., Chung T. W., Lythgoe K., Wang X., Xie J., Ge Q., Gu T. L., Polakiewicz R. D., Roesel J. L., Chen G. Z., Boggon T. J., Lonial S., Fu H., Khuri F. R., Kang S., Chen J. (2011) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X., Liu Y., Gilcrease M. Z., Yuan X. H., Clayman G. L., Adler-Storthz K., Chen Z. (2002) A lymph node metastatic mouse model reveals alterations of metastasis-related gene expression in metastatic human oral carcinoma sublines selected from a poorly metastatic parental cell line. Cancer 95, 1663–1672 [DOI] [PubMed] [Google Scholar]
- 32. Aucoin R., Reiland J., Roy M., Marchetti D. (2004) Dominant-negative CREB inhibits heparanase functionality and melanoma cell invasion. J. Cell. Biochem. 93, 215–223 [DOI] [PubMed] [Google Scholar]
- 33. Nagle J. A., Ma Z., Byrne M. A., White M. F., Shaw L. M. (2004) Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol. Cell. Biol. 24, 9726–9735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun Z. J., Wang Y., Cai Z., Chen P. P., Tong X. J., Xie D. (2008) Involvement of Cyr61 in growth, migration, and metastasis of prostate cancer cells. Br. J. Cancer 99, 1656–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie S., Price J. E., Luca M., Jean D., Ronai Z., Bar-Eli M. (1997) Dominant-negative CREB inhibits tumor growth and metastasis of human melanoma cells. Oncogene 15, 2069–2075 [DOI] [PubMed] [Google Scholar]
- 36. Smith J. A., Poteet-Smith C. E., Xu Y., Errington T. M., Hecht S. M., Lannigan D. A. (2005) Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 65, 1027–1034 [PubMed] [Google Scholar]
- 37. Chen L., Yang S., Jakoncic J., Zhang J. J., Huang X. Y. (2010) Migrastatin analogues target fascin to block tumour metastasis. Nature 464, 1062–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vignjevic D., Schoumacher M., Gavert N., Janssen K. P., Jih G., Laé M., Louvard D., Ben-Ze'ev A., Robine S. (2007) Fascin, a novel target of β-catenin-TCF signaling, is expressed at the invasive front of human colon cancer. Cancer Res. 67, 6844–6853 [DOI] [PubMed] [Google Scholar]
- 39. Gavert N., Conacci-Sorrell M., Gast D., Schneider A., Altevogt P., Brabletz T., Ben-Ze'ev A. (2005) L1, a novel target of β-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J. Cell Biol. 168, 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takahashi M., Tsunoda T., Seiki M., Nakamura Y., Furukawa Y. (2002) Identification of membrane-type matrix metalloproteinase-1 as a target of the beta-catenin/Tcf4 complex in human colorectal cancers. Oncogene 21, 5861–5867 [DOI] [PubMed] [Google Scholar]
- 41. Stein U., Arlt F., Walther W., Smith J., Waldman T., Harris E. D., Mertins S. D., Heizmann C. W., Allard D., Birchmeier W., Schlag P. M., Shoemaker R. H. (2006) The metastasis-associated gene S100A4 is a novel target of β-catenin/T-cell factor signaling in colon cancer. Gastroenterology 131, 1486–1500 [DOI] [PubMed] [Google Scholar]
- 42. Ding Q., Xia W., Liu J. C., Yang J. Y., Lee D. F., Xia J., Bartholomeusz G., Li Y., Pan Y., Li Z., Bargou R. C., Qin J., Lai C. C., Tsai F. J., Tsai C. H., Hung M. C. (2005) Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Mol. Cell 19, 159–170 [DOI] [PubMed] [Google Scholar]
- 43. Jalvy S., Renault M. A., Lam Shang Leen L., Belloc I., Reynaud A., Gadeau A. P., Desgranges C. (2007) CREB mediates UTP-directed arterial smooth muscle cell migration and expression of the chemotactic protein osteopontin via its interaction with activator protein-1 sites. Circ. Res. 100, 1292–1299 [DOI] [PubMed] [Google Scholar]
- 44. Gutierrez L. S., Schulman A., Brito-Robinson T., Noria F., Ploplis V. A., Castellino F. J. (2000) Tumor development is retarded in mice lacking the gene for urokinase-type plasminogen activator or its inhibitor, plasminogen activator inhibitor-1. Cancer Res. 60, 5839–5847 [PubMed] [Google Scholar]
- 45. Dimova E. Y., Samoylenko A., Kietzmann T. (2010) FOXO4 induces human plasminogen activator inhibitor-1 gene expression via an indirect mechanism by modulating HIF-1α and CREB levels. Antioxid. Redox Signal. 13, 413–424 [DOI] [PubMed] [Google Scholar]
- 46. Elf S., Blevins D., Jin L., Chung T. W., Williams I. R., Lee B. H., Lin J. X., Leonard W. J., Taunton J., Khoury H. J., Kang S. (2011) p90RSK2 is essential for FLT3-ITD- but dispensable for BCR-ABL-induced myeloid leukemia. Blood 117, 6885–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]