

Background: Protease-activated receptor 1 (PAR1) and PAR4 mediate thrombin signaling in platelets.
Results: Mutations in transmembrane helix 4 (TM4) of PAR1 or PAR4 disrupts α-thrombin-induced heterodimerization and PAR1-assisted PAR4 cleavage.
Conclusion: PAR1-PAR4 heterodimers are required for efficient PAR4 cleavage.
Significance: The dimerization of PAR1 and PAR4 may impact the effectiveness of PAR1 antagonists.
Keywords: Bioluminescence Resonance Energy Transfer (BRET), G-protein-coupled Receptors (GPCR), Protein-Protein Interactions, Receptor Regulation, Thrombin, PAR1, PAR4, Protease-activated Receptor
Abstract
Thrombin is a potent platelet agonist that activates platelets and other cells of the cardiovascular system by cleaving its G-protein-coupled receptors, protease-activated receptor 1 (PAR1), PAR4, or both. We now show that cleaving PAR1 and PAR4 with α-thrombin induces heterodimer formation. PAR1-PAR4 heterodimers were not detected when unstimulated; however, when the cells were stimulated with 10 nm α-thrombin, we were able to detect a strong interaction between PAR1 and PAR4 by bioluminescence resonance energy transfer. In contrast, activating the receptors without cleavage using PAR1 and PAR4 agonist peptides (TFLLRN and AYPGKF, respectively) did not enhance heterodimer formation. Preventing PAR1 or PAR4 cleavage with point mutations or hirugen also prevented the induction of heterodimers. To further characterize the PAR1-PAR4 interactions, we mapped the heterodimer interface by introducing point mutations in transmembrane helix 4 of PAR1 or PAR4 that prevented heterodimer formation. Finally, we show that mutations in PAR1 or PAR4 at the heterodimer interface prevented PAR1-assisted cleavage of PAR4. These data demonstrate that PAR1 and PAR4 require allosteric changes induced via receptor cleavage by α-thrombin to mediate heterodimer formation, and we have determined the PAR1-PAR4 heterodimer interface. Our findings show that PAR1 and PAR4 have dynamic interactions on the cell surface that should be taken into account when developing and characterizing PAR antagonists.
Introduction
Protease-activated receptors (PARs)2 are a unique class of G-protein-coupled receptors that are activated by proteolytic cleavage of the N terminus by serine proteases (1). There are four members of this family (PAR1-4). In the cardiovascular system, PARs are expressed in platelets, leukocytes, endothelial cells, and smooth muscle cells where they play an important role in hemostasis, thrombosis, inflammation, proliferation, and cell permeability (1, 2).
PAR1 has multiple roles on many cell types. One example of its diverse signaling is on endothelial cells where it can have barrier protective or barrier disruptive roles depending on the agonist and co-receptors present on the cell (3–6). The majority of research on PAR1 has been conducted in the context of human platelet activation by thrombin. A Phase III clinical trial with a PAR1 antagonist, vorapaxar, as an antiplatelet agent did not meet its primary end point due to bleeding complications (7). Recently, a crystal structure of PAR1 bound to vorapaxar was solved (8). Vorapaxar bound deep within the transmembrane helices explaining the essentially irreversible nature of its interaction with PAR1. However, the physical changes in PAR1 that occur after activation by thrombin have not been studied in detail.
PAR4 is the primary signaling receptor on platelets of many species. In human platelets, PAR1 and PAR4 have both overlapping and unique signaling functions (9, 10). For example, PAR4 activation produces a prolonged signal that is required for stable clot formation (11–14). Our laboratory has recently mapped the PAR4 homodimer interface to transmembrane helix 4 (15). Mutations that disrupt PAR4 homodimers also disrupt Ca2+ signaling. On human platelets, PAR4 also interacts with PAR1 to enhance PAR4 activation (16, 17). The details of this interaction are not known.
The activation of PAR1 and PAR4 requires the receptors to be cleaved by thrombin. PAR1 is an excellent thrombin substrate (18, 19). PAR1 has a hirudin-like sequence that binds tightly to thrombin exosite I (20). Based on biochemical and structural data from other tight exosite I binders, the PAR1 hirudin-like sequence likely induces thrombin into the protease conformation (21, 22). In contrast, PAR4 does not bind to exosite I and is a poor thrombin substrate when it is expressed on cells alone (16, 23). The inefficient activation of PAR4 is overcome by co-expression of PAR1 on human platelets and PAR3 on mouse platelets (17, 24). The proposed model for PAR1 or PAR3 enhancing PAR4 activation is that after cleavage of PAR1 or PAR3, thrombin remains bound to the hirudin-like sequence via exosite I and cleaves an adjacent PAR4. In addition to recruiting thrombin to the surface of cells, the exosite I interaction is likely holding thrombin in the protease conformation for efficient cleavage of PAR4 (16, 17, 25). The functional significance of PAR1 or PAR3 co-expression with PAR4 is a 10-fold reduction in the EC50 of thrombin activation of PAR4 (16, 17, 25).
In addition to PAR1 and PAR4 having important independent roles in platelet signaling, the two receptors act synergistically by PAR1 enhancing PAR4 activation. To fully understand the relationship between PAR1 and PAR4, it is essential to have a complete understanding of the molecular arrangement and dynamics of their interactions. The present study demonstrates that α-thrombin promotes efficient heterodimerization between PAR1 and PAR4. The individual activating peptides for PAR1 and PAR4, TFLLRN and AYPGKF, respectively, and mutating the cleavage site of PAR1 or PAR4 does not induce PAR1-PAR4 heterodimerization. The current study also defines the heterodimer interface and maps it to transmembrane helix 4 of both PAR1 and PAR4. Finally, mutations that disrupt PAR1-PAR4 heterodimerization also disrupt PAR1-assisted cleavage of PAR4 by α-thrombin. Taken together, the current study defines the PAR1-PAR4 interaction and demonstrates a functional significance of the PAR1-PAR4 heterodimer.
EXPERIMENTAL PROCEDURES
Reagents
Unless otherwise stated, all of the reagents were from Sigma. α-Thrombin was from Hematological Technologies (Essex Junction, VT).
Cell Culture
HEK293, HeLa, and COS-7 cells were from American Type Culture Collections and were cultured in DMEM supplemented with 10% bovine serum albumin (Hyclone, Logan, UT) and 1% penicillin/streptomycin (Invitrogen). Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Molecular Cloning
The human PAR4 and the PAR4 transmembrane helix 4 mutants have been described (see Table 1) (15). PAR4 with a mutation at the P1 residue of the thrombin cleavage site (PAR4-R47Q) was generated with overlapping PCR as previously described (18). The nomenclature is that of Schechter and Berger (26) where the residue preceding the scissile bond is designated as P1. Human PAR1 in pLK-neo has been described and was the starting material for subsequent PCR reactions (16). The HA tag (YPYDVPDYA) was added to the N terminus of the luciferase constructs, or V5 tag (GKPIPNPLLGLDST) was added to the N terminus of the GFP constructs at amino acid 26 such that the PAR1 signal sequence was removed. Alanine substitution mutants of PAR1 were generated with overlapping PCR using primers that mutated the relevant codon(s) to change Arg-41 (PAR1-R41A), Phe-2214.44, Leu-2244.47, Trp-2274.50, and Ile-2314.54 (PAR1-TM4-4A), Trp-2274.50 and Ile-2314.54 (PAR1-W227A/I231A), or Val-2354.58 and Leu-2394.62 (PAR1-V235A/L239A) (Table 1). The numbering system is that of Ballesteros and Weinstein (27). For co-immunoprecipitation studies, a T7 tag (MASMTGGQQMG) was added to the C terminus of wild type PAR4 using pcDNA3.1. HA-PAR1 wild type and HA-PAR1-W227A/I231A were expressed using pEF1α-IRES-neo (Addgene), referred to as pEF1. For cleavage assays, V5-PAR4-wt or V5-PAR4-TM4-4A was expressed using pIRES-puro (Addgene), referred to as pIRES. A detailed list of primers used for all cloning is available upon request.
TABLE 1.
PAR1 and PAR4 transmembrane point mutations
Sequences are shown with standard one-letter abbreviations. Wild type sequences are shown in plain font. Mutated residues are shown in bold font and underlined.

* Originally described in de la Fuente et al. (15).
Bioluminescence Resonance Energy Transfer (BRET)
Initial experiments determined the optimal expression of PAR1 for BRET experiments as previously described for PAR4 (15). HEK293 cells (1 × 105) were transfected with PAR1-GFP (0–1 μg) or PAR1-rLuc (0–0.5 μg) to determine the minimal amount of plasmid for sufficient GFP and luciferase signal. GFP was determined by excitation at 495 nm and emission at 515 nm. Luciferase activity was determined using coelenterazine H (5 μm) and measuring total light emission without a filter in a PerkinElmer Life Sciences Victor 3 plate reader. The conditions that gave optimal expression for BRET studies were used in subsequent experiments. BRET experiments were performed as previously described (15). To account for lower expression of PAR1, all PAR1-rLuc constructs were adjusted to 0.5 μg/transfection. PAR4-rLuc was used at 0.03 μg/transfection as previously described (15). The PAR4-GFP constructs were used at 0–0.25 μg/transfection, and the PAR1-GFP constructs were used at 0–5 μg/transfection. The BRET data from three or four independent experiments were pooled and analyzed by global fit to all of the data using Prism software (GraphPad). The best model (hyperbolic versus linear) for each data set was also determined using Prism. The BRET50 values were compared with an F test of the globally fit data with Prism; p < 0.05 was considered significant.
Co-immunoprecipitation and Western Blot Analysis
COS-7 cells in 10-cm plates were transiently transfected with PAR1-pEF1 (5 μg) and PAR4-T7-pcDNA3.1 (2 μg) or PAR1-pEF1 (5 μg) and PAR4-pcDNA3.1 (2 μg, no T7 tag). Transfected cells were removed from plates 48 h post-transfection and washed twice with PBS. Cells were stimulated with 10 nm α-thrombin for 20 min at 37 °C and lysed with lysis buffer (1% Triton X-100, 50 mm Tris, pH 7.4, 100 mm NaCl, 5 mm EDTA) containing protease inhibitors mixture (Roche Applied Science) rotating 30 min at 4 °C. Proteins were extracted by centrifugation of lysed cells at 12,000 × g for 30 min at 4 °C. The supernatant-containing 300 μg of protein was immediately incubated with T7 antibody-agarose (25 μl/sample, Novagen) and rotated at 4 °C overnight. Agarose beads were washed with 0.5× immunoprecipitation buffer three times, and the immunoprecipitated proteins were eluted with 0.1 m glycine, pH 2.6. Eluates were neutralized with Tris-HCl, pH 8.0, before adding 5× Laemmli reducing buffer. The samples were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Membranes were incubated with anti-PAR1 (1: 333 dilution; WEDE15 clone; Beckman Coulter) or anti-PAR4. Detection was performed with HRP-conjugated anti-mouse secondary antibody and an enhanced chemiluminescence system (Pierce).
Cleavage Assays
To determine the influence of PAR1 mutations on the rate of PAR4 cleavage, COS-7 cells in 10-cm plates were transiently transfected with HA-PAR1-pEF1 (0.5 μg) and V5-PAR4-pIRES (2 μg), HA-PAR1-W227A/I231A-pEF1 (5 μg) and V5-PAR4-pIRES (2 μg), or V5-PAR4-pIRES alone (2 μg). To determine the influence of PAR4 mutations, cells were transfected with HA-PAR1-pEF1 (5 μg) and V5-PAR4-TM4-4A-pIRES (2 μg) or V5-PAR4-TM4-4A-pIRES alone (2 μg). Transfected cells were removed from plates 24 h post-transfection with versene, washed twice with PBS, and resuspended at concentration of 106 cells/ml in PBS. Reactions were initiated at 37 °C by adding 10 nm α-thrombin or 100 nm α-thrombin. Reactions were stopped at different time points with hirudin (0.5 units/ml), washed once with PBS, and incubated with V5 tag antibody conjugated to Alexa Fluor 647 (AbD Serotec) with a 1:50 dilution. The decrease of mean fluorescence as a result of the loss of the V5 epitope was determined by flow cytometry on a BD LSRFortessa (Center for Aids Research, Immune Function Core, Case Western Reserve University). The mean fluorescence data were fit to an exponential decay with the equation fl = fl0e−kt to compare the rate of PAR4 cleavage. The initial cell surface expression of HA-PAR1 and V5-PAR4 was determined by quantitative flow cytometry before adding thrombin using HA (Cell Signaling Technology Inc) or V5 antibodies conjugated to Alexa Fluor 647 with a 1:50 dilution and performed essentially as described (15).
Calcium Mobilization Assays
HeLa cells were transfected with HA-PAR1-pEF1, HA-PAR1-W227A/I231A-pEF1, V5-PAR4-pIRES, or V5-PAR4-TM4-4A-pIRES, and stable clones were selected with puromycin (1 μg/ml) or Geneticin (1 mg/ml). Calcium mobilization was measured with Fura-2 as previously described (15, 28). Briefly, cells were removed from plates with versene, washed, and loaded with 5 μm Fura2-AM (Invitrogen) in Hepes-Tyrodes buffer supplemented with magnesium and calcium. Cells (2.5 × 105) were placed into 96-well plates, stimulated with α-thrombin, and read in a NOVOstar plate reader (BMG Labtech, Durham, NC). Fluorescence measurements were converted to intracellular calcium concentration by the formula of Grynkiewicz et al. (29).
RESULTS
Previously, we determined the optimal conditions to analyze PAR4 homodimers by BRET using the minimal expression levels necessary to avoid overexpression artifacts (15). Initial studies determined the minimal amount of plasmid to give sufficient PAR1-rLuc expression was 0.5 μg (data not shown) compared with 0.03 μg for PAR4 (15). The interaction between PAR1 and PAR4 was examined by BRET experiments that were designed to achieve similar levels of PAR1 and PAR4 expression. The initial experiments examined the PAR1-PAR4 heterodimer with BRET in unstimulated cells, which did not show a specific interaction, indicated by a linear relationship (Fig. 1A and Table 2). However, upon stimulation with 10 nm α-thrombin for 10 min there was a robust increase in the formation of PAR1-PAR4 heterodimers indicated by a hyperbolic curve (Fig. 1A and Table 2). The α-thrombin-induced interaction of PAR1 and PAR4 did not change when rLuc was fused to PAR1 and GFP was fused to PAR4 (Fig. 1B); the BRET50 (0.126 ± 0.08 versus 0.077 ± 0.03, p = 0.16) and BRETmax (0.17 ± 0.04 versus 0.26 ± 0.03, p = 0.26) were both unchanged (Table 2). The specificity of the BRET interaction was shown using the non-interacting GPCR rhodopsin (Fig. 1C). Similar to our previous studies with PAR4 (15), rhodopsin did not interact with PAR1. These data suggested that stimulation of the receptors with α-thrombin induced PAR1 and PAR4 to efficiently form heterodimers.
FIGURE 1.
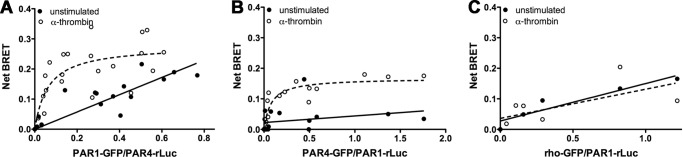
Thrombin modulates PAR1-PAR4 heterodimer formation. HEK293 cells were transfected with HA-PAR4-rLuc (0.03 μg) and V5-PAR1-GFP (0–2.5 μg) (A), HA-PAR1-rLuc (0.5 μg) and V5-PAR4-GFP (0–0.25 μg) (B), or HA-PAR1-rLuc (0.5 μg) and rhodopsin-GFP (0–1.5 μg) (C). Forty-eight hours post-transfection, the cells were unstimulated (solid circles) or treated with α-thrombin (10 nm) for 10 min (open circles). The cells were analyzed for GFP expression, luciferase activity, and BRET. The data are from three independent experiments in which all points were analyzed by global fit to hyperbolic or linear curve.
TABLE 2.
BRET50 and BRETmax values for PAR1-PAR4 heterodimers
PAR4-GFP and PAR1-Luc were stimulated with the indicated agonist for 10 min and examined in BRET assays. The data were fit to a hyperbolic curve, and the BRET50 and BRETmax for each pair were calculated. Nonspecific interactions are indicated as linear. Values are compared to PAR1-rLuc/PAR4-GFP and PAR4-rLuc/PAR1-GFP interactions with an F-test as described under “Experimental Procedures.”
| rLuc (donor) | GFP (acceptor) | 10 nM IIa | BRET50 | BRETmax |
|---|---|---|---|---|
| Heterodimers | ||||
| PAR4 | PAR1 | − | Linear | Linear |
| PAR4 | PAR1 | + | 0.077 ± 0.03 | 0.17 ± 0.01 |
| PAR1 | PAR4 | − | Linear | Linear |
| PAR1 | PAR4 | + | 0.126 ± 0.08 | 0.20 ± 0.04 |
| Homodimers | ||||
| PAR1 | PAR1 | − | 0.118 ± 0.07 | 0.57 ± 0.10 |
| PAR1 | PAR1 | + | 0.159 ± 0.07 | 0.52 ± 0.15 |
| PAR4 | PAR4 | − | 0.021 ± 0.01a | 0.26 ± 0.04 |
| PAR4 | PAR4 | + | 0.013 ± 0.01a | 0.29 ± 0.06 |
| PAR4 mutants | ||||
| PAR1 | PAR4-TM4-2A | + | 0.057 ± 0.023 | 0.21 ± 0.02 |
| PAR1 | PAR4-TM4-4A | + | Linear | Linear |
| PAR1 | PAR4-TM1-4A | + | 0.045 ± 0.040 | 0.17 ± 0.03 |
| PAR1 mutants | ||||
| PAR1-227/231 | PAR4 | + | Linear | Linear |
| PAR1-235/239 | PAR4 | + | 0.034 ± 0.024 | 0.42 ± 0.10 |
| PAR1-TM4-4A | PAR4 | + | Linear | Linear |
a Statistically different values are indicated (p ≤ 0.05).
Studies next determined if the induction of PAR1-PAR4 heterodimers was due to activation or cleavage of the receptor using PAR-activating peptides, PAR mutants, or the thrombin inhibitor hirugen. PAR1 and PAR4 activation peptides, TFLLRN or AYPGKF, respectively, can be used to activate the receptors in the absence of cleavage. Cells were prepared for BRET and stimulated with 50 μm TFLLRN and 500 μm AYPGKF simultaneously for 10 min. The PAR activation peptides did not induce PAR1-PAR4 heterodimers (Fig. 2A). Stimulation of the cells with TFLLRN or AYPGKF individually also did not induce PAR1-PAR4 heterodimer formation (data not shown). To examine the requirement of PAR1 and PAR4 cleavage for the induction of heterodimers, the Arg in the P1 position of PAR1 or PAR4 was mutated and tested for its ability to form heterodimers. PAR1-R41A did not interact with PAR4 wild type after stimulation with 10 nm α-thrombin for 10 min (Fig. 2B). Similarly, the cleavage-deficient PAR4, PAR4-R47Q, did not interact with wild type PAR1 (Fig. 2C). Furthermore, blocking thrombin exosite I with hirugen also inhibited the induction of PAR1-PAR4 heterodimers (Fig. 2D). Flow cytometry with antibodies to the N-terminal tags HA or V5 on PAR1 or PAR4, respectively, confirmed that hirugen blocked the cleavage of PAR1 and PAR4 by thrombin (Fig. 2E).
FIGURE 2.

PAR1 and PAR4 cleavage by thrombin is required to modulate dimerization. A, BRET assays were performed in HEK293 cells expressing HA-PAR1-rLuc (0.5 μg) and V5-PAR4-GFP (0–0.25 μg) after stimulation with TFLLRN (50 μm) and AYPGKF (500 μm) simultaneously for 10 min. HA-PAR4-rLuc (0.03 μg) and V5-PAR1-R41A-GFP (0–2.5 μg) (B) and HA-PAR4-R47Q-Luc (0.03 μg) and V5-PAR1-GFP (0–0.5 μg) (C) were analyzed by BRET assays after treatment with α-thrombin (10 nm) for 10 min. D, BRET assays were performed in cells expressing HA-PAR1-rLuc and V5-PAR4-GFP stimulated with α-thrombin (10 nm) pretreated with hirugen (10 μm). For all BRET assays, GFP expression and luciferase activity were determined to calculate the GFP/rLuc ratio. The data are from three independent experiments in which all points were analyzed by global fit to hyperbolic or linear curve. E, the cleavage of HA-PAR1-rLuc or V5-PAR4-GFP by 10 nm α-thrombin pretreated with hirugen was analyzed by flow cytometry; error bars indicate S.D. of three independent experiments.
PAR1 and PAR4 also form homodimers (15, 30). In contrast to the PAR1-PAR4 heterodimers, thrombin did not influence the BRET50 or BRETmax for PAR1 or PAR4 homodimers (Fig. 3, A and B; Table 2). The BRET50 values were used to compare the relative affinities of α-thrombin-stimulated PAR1 homodimers, PAR4 homodimers, and PAR1-PAR4 heterodimers (Table 2). PAR4 homodimers had a lower BRET50 (higher relative affinity) (0.013 ± 0.01) compared with PAR1-PAR4 heterodimers regardless if the rLuc was fused to PAR4 (0.077 ± 0.026, p = 0.02) or PAR1 (0.126 ± 0.08, p = 0.01). PAR4 homodimers also had a lower BRET50 compared with PAR1 homodimers (0.159 ± 0.077, p = 0.01). Finally, the BRET50 for PAR1 homodimers and PAR1-PAR4 heterodimers were not statistically different regardless if the pLuc was fused to PAR1 or PAR4 (p = 0.80 or 0.14, respectively).
FIGURE 3.

Thrombin does not modulate PAR1 or PAR4 homodimers. HEK293 cells were transfected with HA-PAR1-Luc (0.5 μg) and V5-PAR1-GFP (0–2.5 μg) (A) or HA-PAR4-Luc (0.03 μg) and V5-PAR4-GFP (0–0.24 μg) (B). Forty-eight hours post-transfection, the cells were unstimulated or treated with α-thrombin (10 nm) and analyzed for GFP expression, luciferase activity, and BRET. The data are from three independent experiments in which all points were analyzed by global fit to hyperbolic or linear curve.
We next wanted to determine the PAR1-PAR4 heterodimer interface. We previously showed that the PAR4 homodimer interface is in transmembrane helix 4 (TM4) (15). Therefore, we tested our panel of PAR4-TM4 alanine substitution mutants (Table 1) for the ability to associate with PAR1. The location of the mutations is shown in the molecular model of PAR4 that was generated by Swiss Modeler (Fig. 4A) (31–33). Initial studies examined the surface expression with quantitative flow cytometry to ensure that each of the PAR4 mutants was expressed on the cell surface to the same degree as PAR4-wt (Fig. 4B). HEK293 cells were prepared for BRET assays and stimulated with 10 nm α-thrombin for 10 min. PAR4-TM4-2A (PAR4-L209A/L213A) was able to interact with PAR1 similar to PAR4 wild type with a BRET50 of 0.057 ± 0.023, p = 0.30 (Fig. 4C, Table 2). In contrast, PAR4-TM4-4A (PAR4-L192A/L194A/M198A/L202A) did not interact with PAR1, indicated by the linear relationship (Fig. 4D, Table 2). Residues in TM1 have also been reported to mediate GPCR dimers/oligomers (34, 35). Therefore, we mutated four conserved residues in TM1 of PAR4 (Table 1) and tested their role in mediating the PAR1 interaction. PAR4-TM1-4A (PAR4-L79A/V80A/L83A/L86A) interacted with PAR1 with the same BRET50 as PAR4 wild type (0.045 ± 0.040, p = 0.40) (Fig. 4E, Table 2).
FIGURE 4.

Residues in transmembrane helix 4 of PAR4 mediate interaction with PAR1. A, structural model of PAR4 based on rhodopsin generated with Swiss Modeler. TM1 is indicated in red, and TM4 is in blue. Residues that were analyzed in this study are shown as sticks. The residues in which alanine substitution disrupted α-thrombin-induced heterodimerization are shown in orange. The nomenclature is that of Ballesteros and Weinstein (27). B, the expression of PAR4 wild type and mutants in HEK293 was analyzed by flow cytometry with an anti-V5 antibody. C–E, BRET assays were performed in HEK293 cells expressing HA-PAR1-rLuc (0.5 μg) and V5-PAR4-TM4-2A-GFP (C), V5-PAR4-TM4-4A -GFP (D), or V5-PAR4-TM1-4M-GFP (0–0.25 μg) (E). Forty-eight hours post-transfection, the cells were treated with α-thrombin (10 nm) for 10 min and analyzed for GFP expression, luciferase activity, and BRET. The data are from three independent experiments in which all points were analyzed by global fit to hyperbolic or linear curve.
Next, we wanted to determine the interface on PAR1 for the PAR1-PAR4 heterodimer. We generated a model of PAR1 using Swiss Modeler to guide our mutagenesis studies (15, 31–33). This PAR1 model revealed residues in TM4 that were directed away from the body of the receptor (Fig. 5A). A series of alanine substitution mutants in TM4 of PAR1 were generated to determine their association with PAR4 (Table 1). The surface expression of each of the PAR1 mutants was verified by quantitative flow cytometry (Fig. 5B). The PAR1 mutants were tested in BRET assays for their ability to interact with PAR4 wild type. Similar to PAR4, mutations in residues near the outer membrane, PAR1-V235A/L239A, did not disrupt the ability of α-thrombin to mediate PAR1-PAR4 heterodimerization; the BRET50 (0.034 ± 0.024) was not statistically different from PAR1-wt (p = 0.16) (Fig. 5C, Table 2). However, mutating two residues near the center of TM4, PAR1-W227A/I231A, disrupted the interaction with PAR4 indicated by the linear BRET curve (Fig. 5D). Mutating two additional residues also disrupted PAR1-PAR4 heterodimers (Fig. 5E). These data suggest that, like PAR4 homodimers, several residues in TM4 of both receptors mediate the interaction between PAR1 and PAR4. Importantly, the residues in TM4 that mediate the PAR1-PAR4 interactions on both receptors (shown in orange in Figs. 4A and 5A) are in the same region along TM4 and align in a manner that would be capable of forming the interface.
FIGURE 5.

Residues in transmembrane helix 4 of PAR1 mediate interaction with PAR4. A, structural model of PAR1 based on rhodopsin generated with Swiss Modeler. TM1 is indicated in red, and TM4 is in blue. Residues that were analyzed in this study are shown as sticks. The residues in which alanine substitution disrupted α-thrombin-induced heterodimerization are shown in orange. The nomenclature is that of Ballesteros and Weinstein (27). B, the expression of PAR1 wild type and mutants in HEK293 was analyzed by flow cytometry with an anti-HA antibody. C–E, BRET assays were performed in HEK293 cells expressing V5-PAR4-GFP (0-.24 μg) and HA-PAR1-V235A/L239A-rLuc (C), HA-PAR1-W227A/I231A-rLuc (D), or HA-PAR1-TM4-4A-rLuc (0.5 μg) (E). Forty-eight hours post-transfection the cells were treated with α-thrombin (10 nm) for 10 min and analyzed for GFP expression, luciferase activity, and BRET. The data are from two or three independent experiments in which all points were analyzed by global fit to hyperbolic or linear curve.
We next examined the ability of PAR1 and PAR4 mutants to induce intracellular signaling by measuring the calcium mobilization in response to α-thrombin (Fig. 6). Consistent with our previous results, HeLa cells do not respond to α-thrombin in calcium mobilization (Fig. 6C) (16). When expressed on HeLa cells, PAR4-TM4-4A retained the ability to respond to 30 nm α-thrombin, however, with reduced efficiency compared with PAR4 (Fig. 6A). These data are consistent with our previous results (15). Similarly, PAR1-W227A/I231A also retained the ability to induce intracellular calcium mobilization but at a reduced level compared with the PAR1 (Fig. 6C). We verified by flow cytometry that HA-PAR1-W227A/I231A and V5-PAR4-TM4-4A PAR4 mutants were expressed to the same level as HA-PAR1-wt and V5-PAR4-wt, respectively, on the surface of HeLa cells (Fig. 6, B and D). These data show that the PAR1 and PAR4 mutants that disrupt the PAR1-PAR4 interface are expressed on the cell surface and have partial activity.
FIGURE 6.
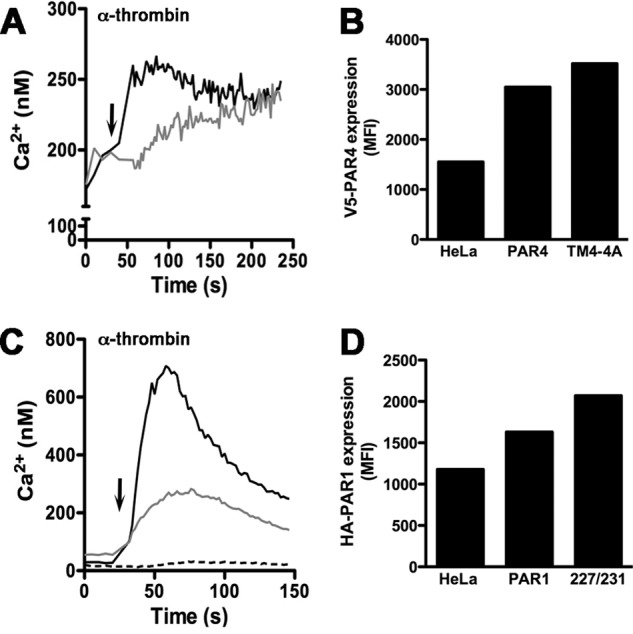
Thrombin-mediated intracellular calcium mobilization. HeLa cells expressing V5-PAR4 (black line) or V5-PAR4-TM4-4A (gray line) were loaded with Fura-2 in HEPES-Tyrode buffer, pH 7.4, containing 2 mm CaCl2 (A). Calcium mobilization was measured over time in response to 30 nm α-thrombin (indicated by the arrow) (A). The surface expression of V5-PAR4 or V5-PAR4-TM4-4A in the HeLa cells was determined by flow cytometry with a V5 tag antibody conjugated to AlexaFluor 647 (B). Calcium mobilization was determined in HeLa cells expressing HA-PAR1 (black line), HA-PAR1-W227A/I231A (gray line), or parental HeLa cells (dashed line) as in panel A (C). MFI, mean fluorescence intensity. The surface expression of HA-PAR1 or HA-PAR1-W227A/I231A was determined by flow cytometry with an HA antibody conjugated to AlexaFluor 647 (D).
To examine the PAR1-PAR4 interaction with a second technique, we co-expressed PAR1 and PAR4 in COS-7 cells for co-immunoprecipitation studies. A T7 epitope was added to the C terminus of PAR4 to facilitate co-immunoprecipitation with anti-T7 agarose beads. PAR4 without a T7-epitope was used as a negative control. Cells expressing PAR1 and PAR4-cT7 were unstimulated or stimulated with 10 nm α-thrombin before lysis. We were able to detect a specific interaction by co-immunoprecipitation with and without stimulation by α-thrombin (Fig. 7). To reliably detect the interaction with the co-immunoprecipitation experiments, a 10-fold higher expression of PAR1 and PAR4 was required than for the BRET experiments. This may have contributed to the interactions in the unstimulated cells. Finally, the PAR4 lacking the T7 epitope also failed to co-immunoprecipitate PAR1, indicating that PAR1 was not binding to the T7-agarose non-specifically.
FIGURE 7.
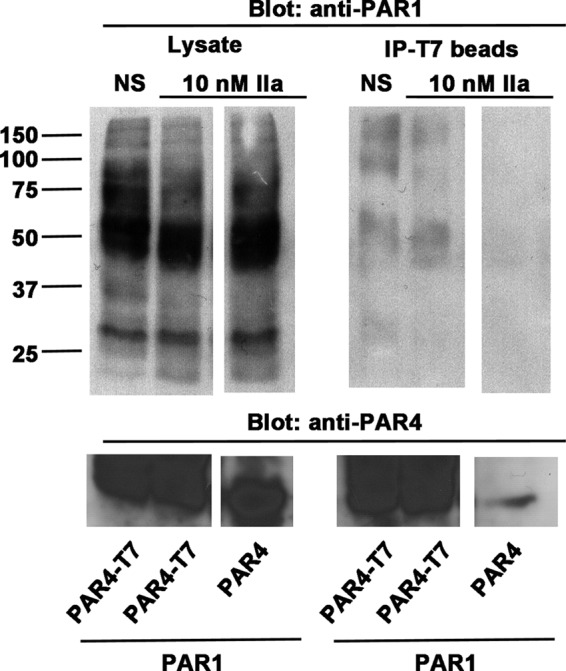
PAR1 and PAR4 co-immunoprecipitation studies. COS-7 cells were co-transfected with PAR1 (5 μg) and PAR4-cT7 (2 μg) or with PAR4 (no cT7, 2 μg); cT7 is a C-terminal T7 epitope. Forty-eight hours post transfection, cells were harvested, stimulated with α-thrombin (10 nm), lysed, and incubated with T7-agarose overnight at 4 °C. Co-immunoprecipitations (IP) were separated on SDS-PAGE and immunoblotted with anti-PAR1 or anti-PAR4. The space between lanes indicates samples run on the same gel in non-contiguous lanes, NS is nonstimulated.
In human platelets PAR1 serves as a cofactor for cleavage and activation of PAR4 at low thrombin concentrations (≤10 nm) (16, 17). In the previous studies we showed that PAR1 lowered the EC50 of PAR4 cleavage by α-thrombin 6-fold when coexpressed on HeLa cells (16). Therefore, we wanted to determine if PAR1-PAR4 heterodimers were required for PAR1-assisted PAR4 cleavage. To mimic the conditions that were used in the BRET assays, COS-7 cells expressing PAR4 alone or with PAR1 were treated with 10 nm α-thrombin. Consistent with previous studies, PAR4 was not efficiently cleaved with 10 nm α-thrombin when expressed on cells alone (Fig. 8A, open circles). Coexpression of PAR1 increased the rate of cleavage (Fig. 8A, black circles). The rate constant (k) for PAR4 alone (0.006 ± 0.001) was 2.3-fold greater in the presence of PAR1 (0.014 ± 0.007 p = 0.03) (Table 3). Mutations in transmembrane helix 4 of PAR1 that disrupt PAR1-PAR4 heterodimers did not enhance PAR4 cleavage (Fig. 8A, gray circles and Table 3). Quantitative flow cytometry was used to ensure that the initial PAR4 expression was the same in each of the experiments (Fig. 8E, black bars). The initial expression of PAR1 and PAR1-W227A/I231A was also verified by flow cytometry (Fig. 8D, black bars). Disrupting PAR1-PAR4 heterodimers with PAR4 mutations also abolished PAR1-assisted PAR4 cleavage (Fig. 8B). Coexpression of PAR1 did not influence the expression of PAR4-TM4-4A (Fig. 8E, dark gray bars). PAR1 was deliberately expressed at higher levels to confirm that the failure to increase the rate of PAR4 cleavage was not due to low expression of PAR1 (Fig. 8E, gray bars). Finally, we verified that PAR4-TM4-4A did not have intrinsic properties that made it less efficiently cleaved than PAR4 by increasing the α-thrombin concentration to 100 nm (Fig. 8C and Table 3). The disappearance of the V5 epitope was not due to internalization as PAR4-R47Q remained on the surface after stimulation with 100 nm α-thrombin (data no shown). Taken together, these data suggest that PAR1-PAR4 heterodimers are required for PAR1-assisted cleavage of PAR4.
FIGURE 8.

Dimerization is required for PAR1-assisted PAR4 cleavage by α-thrombin. V5-PAR4 was expressed on COS-7 cells alone (open circles), with PAR1 (black circles), or with PAR1-W227A/I231A (gray circles). PAR4 cleavage by 10 nm α-thrombin was determined by measuring the loss of the V5 epitope over time by flow cytometry (A). V5-PAR4-TM4-4A was expressed on COS-7 cells alone (open circles) or with PAR1 (black circles), and the loss of the V5-epitope in response to 10 nm α-thrombin was measured over time (B). V5-PAR4 (black circles) or V5-PAR4-TM4-4A (open circles) were expressed on COS-7 cells and treated with 100 nm α-thrombin (C). Quantitative flow cytometry was used to determine the expression of HA-PAR1 (black bar) or HA-PAR1-W227A/I231A (227/231) (dark gray bar) in the presence of V5-PAR4 and HA-PAR1 expression in the presence of V5-PAR4-TM4-4A (P4-4A) (light gray bar) (D). Quantitative flow cytometry to determine V5-PAR4 expression in the absence or presence of PAR1 or PAR1-W227A/I231A (227/231) (black bars) and V5-PAR4-TM4-4A (P4-4A) expression in the absence or presence of PAR1 (gray bars) (E).
TABLE 3.
Rate PAR4 cleavage on cells
The rate of PAR4 cleavage by 10 or 100 nm α-thrombin was determined with COS7 cells expressing V5-PAR4 or V5-PAR4-TM4-4A by measuring the disappearance of an N-terminal V5 epitope. The influence of PAR1 was determined by co-expressing PAR1 or PAR1-W227A/I231A.
| Rate constant |
|||
|---|---|---|---|
| 10 nM IIa | 100 nM IIa | ||
| k | |||
| PAR4 | 0.006 ± 0.001 | 1.07 ± 0.71 | |
| PAR4 | PAR1 | 0.014 ± 0.007a | |
| PAR4 | PAR1-W227A/I231A | 0.006 ± 0.005 | |
| PAR4-TM4-4A | 0.007 ± 0.003 | 1.04 ± 0.41 | |
| PAR4-TM4-4A | PAR1 | 0.007 ± 0.0003 | |
a The rates constants were compared using a t test and were considered statistically different from PAR4 alone at p < 0.05.
DISCUSSION
The current report shows that PAR1 and PAR4 form heterodimers that are modulated by α-thrombin. In contrast, the PAR1 and PAR4 activation peptides, TFLLRN and AYPGKP, respectively, were unable to induce heterodimers. We also show that cleavage-deficient mutants of PAR1 or PAR4 do not form heterodimers when stimulated with α-thrombin, confirming the requirement of receptor cleavage for heterodimerization. Additional experiments mapped the PAR1-PAR4 heterodimer interface to four residues in TM4 of both PAR1 and PAR4. Finally, mutations that disrupt PAR1-PAR4 heterodimers also disrupt PAR1-assisted cleavage of PAR4 by α-thrombin. Taken together, our studies have defined the PAR1-PAR4 heterodimer and have linked these physical interactions to enhanced PAR4 cleavage by PAR1.
Our studies show that thrombin induces PAR1-PAR4 heterodimers with BRET. We have focused on BRET because it allows us to examine the PAR1-PAR4 interactions in living cells at near physiologic levels of expression (Figs. 4 and 5). Importantly, the levels of expression required for the BRET studies were sufficient to detect an enhanced rate of cleavage of PAR4 by PAR1 (Fig. 8). We were also able to show the PAR1-PAR4 interactions with co-immunoprecipitation experiments (Fig. 7), which agree with Leger et al. (17). However, we interpret our co-immunoprecipitation experiments with caution because the level of expression required for us to reliably detect PAR1 and PAR4 interactions with this technique was 10-fold higher than required for the BRET and cleavage studies. Our interpretation is that at high expression levels, PAR1 and PAR4 can specifically interact in a thrombin-independent manner. Leger et al. (17) also showed PAR1 and PAR4 co-immunoprecipitation from human platelets that were stimulated with thrombin, which are in agreement with our BRET studies. However, we have not been able to co-immunoprecipitate PAR1 and PAR4 from human platelets using the antibodies available to us.
The heterodimerization is specific to α-thrombin. This suggests that PAR1 and PAR4 undergo specific allosteric rearrangement when stimulated with thrombin that is distinct from the activation peptides. This may be due to the conformational changes that must occur for the tethered ligand to activate the receptor after cleavage. The movement of the N terminus likely causes long-range conformational changes through the receptor that would not be required when the receptors are stimulated by the peptides that activate PAR1 and PAR4 independently of receptor cleavage. An alternative explanation is that PAR1 and PAR4 form constitutive heterodimers and cleavage of the receptors results in the rearrangement of the C termini, which facilitates the detection of the BRET signal. The evidence against this interpretation is mutations that disrupt dimerization also disrupt PAR-1-assisted PAR4 cleavage. This will best be addressed by high resolution time-resolved imaging techniques that are currently being developed (36). A third possibility is that α-thrombin mediated the interaction by forming a trimolecular complex with PAR1 and PAR4. Our data in which α-thrombin pretreated with hirugen does not induce PAR1-PAR4 heterodimers suggests that the induction of PAR1-PAR4 heterodimers may be an exosite I-driven process (Fig. 2D). However, blocking exosite I also prevented cleavage of PAR1 and PAR4, which makes it difficult to separate the role of thrombin exosite I in efficient PAR1 cleavage and the potential cross-linking of PAR1 and PAR4. Finally, PAR1 and PAR4 may be differentially localized in the plasma membrane regions such as lipid rafts. Activation of the receptors may alter the interactions that PAR1 and PAR4 have with the membrane such that they are then able to interact with one another. Future studies will need to focus on the lipid environment and how it influences the protein-protein interactions.
There are many studies that have examined the arrangement of GPCRs in the membranes of cells and synthetic membranes (37–40). From these studies there is compelling evidence for both monomeric and dimeric/oligomeric receptors having functional significance. Recent studies using a minimal system of purified proteins and high density lipoprotein discs show that rhodopsin and β2-adrenergic receptors can functional as monomers (37, 38). For example, Whorton et al. (37) demonstrated that monomeric β2-adrenergic receptor was able to mediate agonist-dependent nucleotide exchange from the Gs heterotrimeric G-protein. However, recent studies by Jastrzebska et al. (41, 42) demonstrated that bovine rhodopsin purified from native tissues is functional as a heteropentamer existing of two receptors in complex with a heterotrimeric G-protein. There is also structural evidence for GPCRs existing as dimers (43, 44). Recently, a crystal structure of PAR1 bound to vorapaxar was solved (8). The structure provides key details of the vorapaxar binding site on PAR1. However, the structural changes that occur upon PAR1 cleavage by thrombin are still unknown. Furthermore, the influence of other membrane proteins, such as PAR4, are also unknown.
Many GPCR dimers are noncovalent, and the receptors are in equilibrium between monomers and dimers. An elegant study by Kasai et al. (45) has determined the two-dimensional KD of the monomer to dimer transition for the N-formyl peptide receptor. In this study using single molecule fluorescence the authors determined that N-formyl peptide receptor monomers convert to dimers every 150 ms and the dimers dissociate to monomers every 91 ms with an overall average of 41% of the receptors in a dimer at any given time. These studies highlight the dynamic nature of membrane receptor dimers. Our results show that PAR1-PAR4 heterodimers are also dynamic in nature and can be induced by α-thrombin (Fig. 1). In contrast, PAR1 and PAR4 homodimers were not affected by stimulation with α-thrombin (Fig. 3). The question remains whether PAR1 and PAR4 form dimers or oligomers. Our studies map the heterodimer interface to TM4; the same region that we have determined as the PAR4 homodimer interface (15). Therefore, our data suggest that PAR1 and PAR4 are an equilibrium between monomers and homodimers, and stimulating with α-thrombin induces the PAR1 and PAR4 monomers to form heterodimers. The current study describes the conditions in which PAR1-PAR4 heterodimers efficiently form the interaction interface. These data will be essential for the development of quantitative studies to examine the stability of the homodimers compared with the heterodimers. An alternative hypothesis is that PAR1 homodimers may interact with PAR4 homodimers to form higher order oligomers as has been described for other GPCRs (36).
The molecular arrangement of GPCRs on the cell surface has been studied for several receptors (40, 46). The specific region that is involved in the dimer interface varies, but there are common themes that emerge. TM4 is a common interface for GPCRs to interact with one another (15, 35, 47, 48). Our recent studies have mapped the homodimer interface for PAR4 to a region on TM4 (15). Mutating Leu-2094.58 and Leu-2134.62 to alanine reduced PAR4 homodimer formation. Furthermore, mutating Leu-1924.41, Leu-1944.43, Met-1984.47, and Leu-2024.51 to alanine completely disrupted the formation of PAR4 homodimers. In these studies, mutating single amino acids was not sufficient to disrupt PAR4 homodimers. A molecular model of PAR4 has this series of residues directed away from the body of the receptor and potentially available to mediate receptor dimers. We examined if these residues with outward facing side chains in TM4 of PAR4 had a role in mediating PAR1-PAR4 heterodimers (Fig. 4A). In contrast to PAR4 homodimers, mutating Leu-2094.54 and Leu-2134.62 did not disrupt PAR1-PAR4 heterodimer formation in response to 10 nm α-thrombin (Fig. 4C). However, mutating Leu-1924.41, Leu-1944.43, Met-1984.47, and Leu-2024.51 to alanine dramatically reduced the ability of PAR1 and PAR4 to interact in response to α-thrombin (Fig. 4D). These four residues are located near the cytoplasmic side to TM4 (Fig. 4A) and are also at the PAR4 homodimer interface (15). Importantly, each of our mutants that disrupts heterodimer formation is expressed on the cell surface (Figs. 4B and 5B) and is capable of mediated Ca2+ mobilization (Fig. 6). The dopamine D2 receptor and α1β-adrenoreceptor have been reported to form dimers/oligomers using residues in TM1 and TM4 (34, 35). In our studies, mutating conserved residues in TM1 of PAR4 did not disrupt the formation of PAR1-PAR4 heterodimers (Fig. 4E). The residues in PAR1 that mediate the PAR1-PAR4 heterodimer are also in TM4 (Fig. 5). These residues are also found near the cytoplasmic side of TM4 and align with the critical residues identified in TM4 of PAR4. In summary, our data suggest that PAR1-PAR4 heterodimer are arranged in the membrane at a TM4-TM4 interface.
There are several GPCRs that have important signaling functions in platelets. We have described the dynamics of PAR1 and PAR4 heterodimers and have mapped the region for the interactions. Importantly, we have linked heterodimerization to PAR1-assisted PAR4 cleavage. These data are in agreement with earlier studies by Nakanishi-Matsui et al. (25) that demonstrated that the exodomain of PAR1 was not able to enhance PAR4 cleavage when fused to CD8 as it is unlikely that the PAR1-CD8 chimera is interacting with PAR4. PAR3 is well known to be a required cofactor of PAR4 activation on mouse platelets. We have recently shown that mouse PAR3 and mouse PAR4 form constitutive heterodimers and PAR3 negatively influences PAR4 signaling (28). Previous studies by Covic et al. (12) demonstrated that sequential activation of PAR1 and PAR4 on human platelets results in an increase in ADP response, which is dependent on PAR4. The sequential activation of PAR1 and PAR4 by thrombin may allow the receptors to rearrange in the platelet membrane and alter the signaling response. A recent study by Li et al. (49) described how PAR4 cooperates with the ADP receptor, P2Y12. In these studies platelets from arrestin-2-deficient mice had a reduction in Akt phosphorylation and fibrinogen binding in response to PAR4 activation. Li et al. (49) also showed that PAR4 physically interacts with the P2Y12 receptor in an agonist-dependent manner and may recruit arrestin-2 to PAR4. There is a complex arrangement of receptors and signaling molecules at the platelet surface in which PAR4 appears to be a common player. Future studies will need to examine how these and other GPCRs interact on platelets to mediate their full range of signaling in vivo.
The current data show that the induction of heterodimers is specific to α-thrombin. PAR1 and PAR4 both have distinct roles in platelet signaling. A recent Phase III clinical trial with the PAR1 antagonist vorapaxar that did not meet its primary end point underscores the need to fully understand the interactions between platelet receptors and how these interactions influence receptor function (7). Future studies will need to generate models that can differentiate between heterodimer and homodimer signaling in platelets. The current work provides a framework for building platelet-specific models to examine the dynamics and signaling consequences of these interactions. In addition, understanding the molecular arrangement of PAR1 and PAR4 will provide insight for the development of antiplatelet therapies.
This work was supported, in whole or in part, by National Institutes of Health Grant HL098217 (to M. N.). This work was also supported by American Heart Association (AHA) Beginning Grant-in-aid 0865441D, AHA Scientist Development Grant 10SDG2600021, an American Society for Hematology Scholar Award (to M. N.), and by the Cytometry and Imaging Microscopy Core Facility of the Case Comprehensive Cancer Center Grant P30CA043703.
- PAR
- protease-activated receptor
- BRET
- bioluminescence resonance energy transfer
- GPCR
- G-protein-coupled receptor
- TM
- transmembrane helix.
REFERENCES
- 1. Coughlin S. R. (2005) Protease-activated receptors in hemostasis, thrombosis, and vascular biology. J. Thromb. Haemost. 3, 1800–1814 [DOI] [PubMed] [Google Scholar]
- 2. Leger A. J., Covic L., Kuliopulos A. (2006) Protease-activated receptors in cardiovascular diseases. Circulation 114, 1070–1077 [DOI] [PubMed] [Google Scholar]
- 3. Riewald M., Ruf W. (2005) Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J. Biol. Chem. 280, 19808–19814 [DOI] [PubMed] [Google Scholar]
- 4. Russo A., Soh U. J., Paing M. M., Arora P., Trejo J. (2009) Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc. Natl. Acad. Sci. U.S.A. 106, 6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Soh U. J., Trejo J. (2011) Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. U.S.A. 108, E1372–E1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bae J. S., Yang L., Manithody C., Rezaie A. R. (2007) The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood 110, 3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tricoci P., Huang Z., Held C., Moliterno D. J., Armstrong P. W., Van de Werf F., White H. D., Aylward P. E., Wallentin L., Chen E., Lokhnygina Y., Pei J., Leonardi S., Rorick T. L., Kilian A. M., Jennings L. H., Ambrosio G., Bode C., Cequier A., Cornel J. H., Diaz R., Erkan A., Huber K., Hudson M. P., Jiang L., Jukema J. W., Lewis B. S., Lincoff A. M., Montalescot G., Nicolau J. C., Ogawa H., Pfisterer M., Prieto J. C., Ruzyllo W., Sinnaeve P. R., Storey R. F., Valgimigli M., Whellan D. J., Widimsky P., Strony J., Harrington R. A., Mahaffey K. W. (2012) Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes. N. Engl. J. Med. 366, 20–33 [DOI] [PubMed] [Google Scholar]
- 8. Zhang C., Srinivasan Y., Arlow D. H., Fung J. J., Palmer D., Zheng Y., Green H. F., Pandey A., Dror R. O., Shaw D. E., Weis W. I., Coughlin S. R., Kobilka B. K. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492, 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holinstat M., Voss B., Bilodeau M. L., McLaughlin J. N., Cleator J., Hamm H. E. (2006) PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J. Biol. Chem. 281, 26665–26674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voss B., McLaughlin J. N., Holinstat M., Zent R., Hamm H. E. (2007) PAR1, but not PAR4, activates human platelets through a Gi/o/phosphoinositide-3 kinase signaling axis. Mol. Pharmacol. 71, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 11. Kahn M. L., Nakanishi-Matsui M., Shapiro M. J., Ishihara H., Coughlin S. R. (1999) Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest. 103, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Covic L., Gresser A. L., Kuliopulos A. (2000) Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39, 5458–5467 [DOI] [PubMed] [Google Scholar]
- 13. Covic L., Singh C., Smith H., Kuliopulos A. (2002) Role of the PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky-Pudlak syndrome. Thromb. Haemost. 87, 722–727 [PubMed] [Google Scholar]
- 14. Mazharian A., Roger S., Berrou E., Adam F., Kauskot A., Nurden P., Jandrot-Perrus M., Bryckaert M. (2007) Protease-activating receptor-4 induces full platelet spreading on a fibrinogen matrix. Involvement of ERK2 and p38 and Ca2+ mobilization. J. Biol. Chem. 282, 5478–5487 [DOI] [PubMed] [Google Scholar]
- 15. de la Fuente M., Noble D. N., Verma S., Nieman M. T. (2012) Mapping human protease-activated receptor 4 (PAR4) homodimer interface to transmembrane helix 4. J. Biol. Chem. 287, 10414–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nieman M. T. (2008) Protease-activated receptor 4 uses anionic residues to interact with α-thrombin in the absence or presence of protease-activated receptor 1. Biochemistry 47, 13279–13286 [DOI] [PubMed] [Google Scholar]
- 17. Leger A. J., Jacques S. L., Badar J., Kaneider N. C., Derian C. K., Andrade-Gordon P., Covic L., Kuliopulos A. (2006) Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation 113, 1244–1254 [DOI] [PubMed] [Google Scholar]
- 18. Nieman M. T., Schmaier A. H. (2007) Interaction of thrombin with PAR1 and PAR4 at the thrombin cleavage site. Biochemistry 46, 8603–8610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jacques S. L., LeMasurier M., Sheridan P. J., Seeley S. K., Kuliopulos A. (2000) Substrate-assisted catalysis of the PAR1 thrombin receptor. Enhancement of macromolecular association and cleavage. J. Biol. Chem. 275, 40671–40678 [DOI] [PubMed] [Google Scholar]
- 20. Liu L. W., Vu T. K., Esmon C. T., Coughlin S. R. (1991) The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J. Biol. Chem. 266, 16977–16980 [PubMed] [Google Scholar]
- 21. Kamath P., Huntington J. A., Krishnaswamy S. (2010) Ligand binding shuttles thrombin along a continuum of zymogen- and proteinase-like states. J. Biol. Chem. 285, 28651–28658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huntington J. A. (2012) Thrombin plasticity. Biochim. Biophys. Acta 1824, 246–252 [DOI] [PubMed] [Google Scholar]
- 23. Bah A., Chen Z., Bush-Pelc L. A., Mathews F. S., Di Cera E. (2007) Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc. Natl. Acad. Sci. U.S.A. 104, 11603–11608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kahn M. L., Zheng Y. W., Huang W., Bigornia V., Zeng D., Moff S., Farese R. V., Jr., Tam C., Coughlin S. R. (1998) A dual thrombin receptor system for platelet activation. Nature 394, 690–694 [DOI] [PubMed] [Google Scholar]
- 25. Nakanishi-Matsui M., Zheng Y. W., Sulciner D. J., Weiss E. J., Ludeman M. J., Coughlin S. R. (2000) PAR3 is a cofactor for PAR4 activation by thrombin. Nature 404, 609–613 [DOI] [PubMed] [Google Scholar]
- 26. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 27. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 28. Arachiche A., de la Fuente M., Nieman M. T. (2013) Calcium mobilization and protein kinase C activation downstream of protease-activated receptor 4 (PAR4) is negatively regulated by PAR3 in mouse platelets. PLoS ONE 8, e55740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 30. McLaughlin J. N., Patterson M. M., Malik A. B. (2007) Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 104, 5662–5667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL. An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace. A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 33. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 34. Lopez-Gimenez J. F., Canals M., Pediani J. D., Milligan G. (2007) The α1b-adrenoceptor exists as a higher-order oligomer. Effective oligomerization is required for receptor maturation, surface delivery, and function. Mol. Pharmacol. 71, 1015–1029 [DOI] [PubMed] [Google Scholar]
- 35. Guo W., Urizar E., Kralikova M., Mobarec J. C., Shi L., Filizola M., Javitch J. A. (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 27, 2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patowary S., Alvarez-Curto E., Xu T. R., Holz J. D., Oliver J. A., Milligan G., Raicu V. (2013) The muscarinic M3 acetylcholine receptor exists as two differently sized complexes at the plasma membrane. Biochem. J. 452, 303–312 [DOI] [PubMed] [Google Scholar]
- 37. Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., Sunahara R. K. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whorton M. R., Jastrzebska B., Park P. S., Fotiadis D., Engel A., Palczewski K., Sunahara R. K. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 283, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park P. S., Filipek S., Wells J. W., Palczewski K. (2004) Oligomerization of G protein-coupled receptors. Past, present, and future. Biochemistry 43, 15643–15656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Birdsall N. J. (2010) Class A GPCR heterodimers. Evidence from binding studies. Trends Pharmacol. Sci. 31, 499–508 [DOI] [PubMed] [Google Scholar]
- 41. Jastrzebska B., Ringler P., Lodowski D. T., Moiseenkova-Bell V., Golczak M., Müller S. A., Palczewski K., Engel A. (2011) Rhodopsin-transducin heteropentamer. Three-dimensional structure and biochemical characterization. J. Struct. Biol. 176, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jastrzebska B., Orban T., Golczak M., Engel A., Palczewski K. (2013) Asymmetry of the rhodopsin dimer in complex with transducin. FASEB J. 27, 1572–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lodowski D. T., Salom D., Le Trong I., Teller D. C., Ballesteros J. A., Palczewski K., Stenkamp R. E. (2007) Crystal packing analysis of rhodopsin crystals. J. Struct. Biol. 158, 455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang J., Chen S., Zhang J. J., Huang X. Y. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kasai R. S., Suzuki K. G., Prossnitz E. R., Koyama-Honda I., Nakada C., Fujiwara T. K., Kusumi A. (2011) Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 192, 463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prinster S. C., Hague C., Hall R. A. (2005) Heterodimerization of g protein-coupled receptors. Specificity and functional significance. Pharmacol. Rev. 57, 289–298 [DOI] [PubMed] [Google Scholar]
- 47. Wang H. X., Konopka J. B. (2009) Identification of amino acids at two dimer interface regions of the α-factor receptor (Ste2). Biochemistry 48, 7132–7139 [DOI] [PubMed] [Google Scholar]
- 48. McMillin S. M., Heusel M., Liu T., Costanzi S., Wess J. (2011) Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 286, 28584–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li D., D'Angelo L., Chavez M., Woulfe D. S. (2011) Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J. Biol. Chem. 286, 3805–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]