

Background: Sphingosine kinases (Sphks) were proposed to be essential for inflammatory responses.
Results: Robust inflammatory responses were seen in macrophages that lack Sphks. However, intracellular sphingolipids and autophagic vesicles were induced.
Conclusion: Sphingosine kinases are not required for inflammation.
Significance: Attenuation of Sphk activity may not be critical for inflammation but could lead to altered sphingolipid levels and autophagy.
Keywords: Autophagy, Ceramide, Inflammation, Lysosomes, Macrophages, Sphingolipid, Sphingosine 1-Phosphate
Abstract
Sphingosine kinases (Sphks), which catalyze the formation of sphingosine 1-phosphate (S1P) from sphingosine, have been implicated as essential intracellular messengers in inflammatory responses. Specifically, intracellular Sphk1-derived S1P was reported to be required for NFκB induction during inflammatory cytokine action. To examine the role of intracellular S1P in the inflammatory response of innate immune cells, we derived murine macrophages that lack both Sphk1 and Sphk2 (MΦ Sphk dKO). Compared with WT counterparts, MΦ Sphk dKO cells showed marked suppression of intracellular S1P levels whereas sphingosine and ceramide levels were strongly up-regulated. Cellular proliferation and apoptosis were similar in MΦ Sphk dKO cells compared with WT counterparts. Treatment of WT and MΦ Sphk dKO with inflammatory mediators TNFα or Escherichia coli LPS resulted in similar NFκB activation and cytokine expression. Furthermore, LPS-induced inflammatory responses, mortality, and thioglycolate-induced macrophage recruitment to the peritoneum were indistinguishable between MΦ Sphk dKO and littermate control mice. Interestingly, autophagic markers were constitutively induced in bone marrow-derived macrophages from Sphk dKO mice. Treatment with exogenous sphingosine further enhanced intracellular sphingolipid levels and autophagosomes. Inhibition of autophagy resulted in caspase-dependent cell death. Together, these data suggest that attenuation of Sphk activity, particularly Sphk2, leads to increased intracellular sphingolipids and autophagy in macrophages.
Introduction
Sphingosine 1-phosphate (S1P)3 is a biologically active lipid that regulates many physiological processes, such as lymphocyte trafficking and vascular development (1, 2). S1P is generated by phosphorylation of free sphingosine (Sph) by two sphingosine kinases (Sphks) 1 and 2, which are highly conserved and ubiquitously expressed (3, 4). Cellular levels of S1P are regulated not only through its biosynthesis but also degradation by S1P lyase (5), S1P phosphatases (6), and intracellular lipid phosphate phosphatases (7). S1P exerts biological functions mostly through five cell surface G protein-coupled receptors S1P1–S1P5. In addition, it has recently been proposed that intracellular S1P binds directly to several proteins and regulates their functions. For example, S1P was proposed to bind to and activate TNFα receptor-associated factor 2 (TRAF2), histone deacetylase 1 (HDAC1), and HDAC2 to block their activity and mitochondrial protein prohibitin to modulate respiration (8–10).
Because Sphk enzymes are required to produce S1P, mice that lack both sphingosine kinases do not contain detectable levels of S1P (11). Because such global Sphk double KO mice are embryonic lethal due to a vascular defect, it was suggested that S1P signaling via its receptors constitutes an essential event in embryonic vascular development (11). However, single isoform knock-out, i.e. Sphk1−/− or Sphk2−/− mice, are phenotypically normal (12), suggesting that Sphk1 and Sphk2 have redundant functions and can compensate for each other to fulfill essential functions. Indeed, Sphk1 mRNA and activity were induced, and plasma S1P was elevated in Sphk2−/− mice (13–16), suggesting that lack of Sphk2 leads to a compensatory up-regulation of Sphk1 expression. The mechanism responsible for this compensation is not known.
Recently, it was proposed that Sphk enzymes are essential for inflammatory responses. For example, intracellular S1P produced from Sphk1 was proposed to bind to the TRAF2 and stimulate its E3 ubiquitin ligase activity as a key mechanism for NFκB signaling (9). Because TRAF2 is a critical intermediate in the signal transduction of inflammatory cytokines (17–19), this work suggested an essential role of intracellular S1P in cytokine-induced inflammatory pathways. Other reports showed that inhibition of Sphk1 by its inhibitor and/or siRNA decreased expression of proinflammatory cytokines (20, 21). However, studies that attenuated Sphk1 activity or expression by either pharmacological inhibitors or Sphk1 siRNA rendered macrophages sensitive to Mycobacterium smegmatis infection (23), and deletion of Sphk1 exhibited disparate effects in mice against the inflammation and injury induced by LPS (24–26). In addition, we and others have reported that Sphk1−/− and Sphk2−/− mice do not exhibit attenuated inflammatory responses in several inflammatory models (25, 27, 28). Therefore, the role of Sphk enzymes in inflammation is unclear at present.
To examine in the issue of intracellular S1P as a critical mediator of cytokine-induced inflammatory responses in innate immune cells, we investigated murine macrophages that lack both Sphk isoenzymes. Our results suggest that intracellular S1P is not required for macrophages to respond to TNFα or LPS. Rather, Sphk2 is involved in an intracellular metabolic network that maintains sphingolipid homeostasis, which, when perturbed, leads to accumulation of sphingolipid metabolites and compensatory autophagy.
EXPERIMENTAL PROCEDURES
Animals
We obtained C57BL/6 mice and LysM-Cre mice, which express the Cre recombinase driven by the lysozyme M promoter (29), from the Jackson Laboratory. Sphk1flox/flox, Sphk2flox/flox, Sphk1−/−, and Sphk2−/− mice were described previously (11, 30). By cross-breeding, myeloid-specific Sphk1/Sphk2 knock-out mice (Sphk1flox/flox Sphk2−/− LysM-Cre+, or Sphk1flox/− Sphk2flox/− LysM-Cre+) were generated. All mice were >8 weeks old when used for the described experiments. Comparisons were done to Cre− littermate controls. All studies were performed under animal protocols approved by the IACUC of Weill Cornell Medical College, the University of California, San Francisco or the French Ministry of Agriculture.
Reagents and Cell Culture
C2-ceramide, Sph, and S1P were purchased from Avanti Polar Lipids. Doxorubicin, ammonium chloride (NH4Cl), and 3-methyladenine were purchased from Sigma. Bone marrow-derived macrophages (BMDMs) were isolated as described previously (31). The cells were cultured in DMEM containing 10% FBS and 20% L-cell conditioned media as a source of macrophage colony-stimulating factor (M-CSF). Macrophages were obtained as a homogeneous population of adherent cells after 5–7 days of culture. Thioglycolate-elicited peritoneal macrophages were isolated from mice 4 days after injection of 2 ml of sterile thioglycolate (Sigma). The cells were incubated at 37 °C in a humidified 5% CO2 atmosphere.
Western Blot Analysis
Cells were washed with ice-cold phosphate-buffered saline and lysed in radioimmuneprecipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% SDS, 1% Triton X-100, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 mm EDTA, 1 mm sodium orthovanadate, and 1 × protease inhibitor mixture). Protein concentrations of supernatants were determined by BCA protein assay kit (Pierce Chemical Co.). Equal amounts of protein were separated on 10% SDS-PAGE and blotted onto a nitrocellulose membrane. Immunoblot analysis was performed using the following antibodies: p-p65, p65, p-JNK, p-p38 (Cell Signaling); Sphk1, COX-2 (Cayman Chemical); Sphk2 and β-actin (Abcam). Blots were developed with the Western blot development kit from GE Healthcare.
Cell Viability Assay
The survival of macrophages under experiments was evaluated by the MTT dye reduction method. After each incubation time, the cells were incubated with yellow MTT dye (Sigma). The formazone crystals were dissolved in dimethyl sulfoxide, and absorbance was measured at 570 nm in a spectrometer Spectra max 250 (Bio-Rad).
RNA Isolation and qRT-PCR Analysis
Total RNA was extracted from treated cells using RNA Stat-60 (Tel-Test Inc.) according to the manufacturer's instructions. Reverse transcription was carried out using a First Strand cDNA synthesis kit for RT-PCR (avian myeloblastosis virus) (Roche Applied Science) using random primers. Real-time PCR was performed on a 7500 real-time PCR system (Applied Biosystems) using Fast SYBR® Green Master Mix (Applied Biosystems), and relative RNA levels were calculated using the ΔΔCT method (31). Primer sets for qRT-PCR were Sphk1 (5′-AGGTGGTGAATGGGCTAATG-3′ and 5′-TGCTCGTACCCAGCATAGTG-3′), Sphk2 (5′-TGGTGCCAATGATCTCTGAA-3′ and 5′-CCAGACACAGTGACAATGCC-3′), IL-1 (5′-TTCTTTGGGTATTGCTTGGG-3′ and 5′-TTCTTTGGGTATTGCTTGGG-3′), IL-6 (5′-CCGGAGAGGAGACTTCACAG-3′ and 5′-TCCACGATTTCCCAGAGAAC-3′), TNFα (5′-CACTTGGTGGTTTGCTACGA-3′ and 5′-CATCGATGAGCTGATGCAGT-3′), and Gapdh (5′-AGAACATCATCCCTGCATCC-3′ and 5′-CACATTGGGGGTAGGAACAC-3′).
Intracellular Sphingolipid Analysis
Intracellular sphingolipid levels of ceramides, Sph, and S1P were analyzed by the Lipidomics Analytical Core at the Medical University of South Carolina using LC-MS/MS methods (32).
Lysosomal Staining with Dextran
For lysosome staining, BMDMs were plated on poly-d-lysine-coated coverslip glass-bottom dishes and incubated for 16 h with 2.2 mg/ml dextran conjugated to rhodamine (molecular weight, 70,000; Invitrogen) in complete growth medium. Cells were washed thoroughly in complete medium and then incubated in medium with or without 20 mm sphingosine for 4 h. Live cells were imaged in medium 2 and 0.2% (w/v) glucose on a Zeiss LSM510 laser scanning confocal microscope using a 63×, 1.4 numerical aperture plan Apochromat objective. Cell temperature was maintained at 37 °C with a heated stage and objective heater (33).
Flow Cytometry
BMDMs were harvested after 7 days of culture and then incubated on ice for 30 min with the indicated antibodies, washed, and processed using an LSR II flow cytometer (BD Biosciences). The antibodies used were R-phycoerythrin-conjugated anti-CD11b (BioLegend) and FITC-F4/80 (eBioscience). Stained cells were subsequently washed and analyzed using an LSR II flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (version 8; Tree Star, Inc.).
Immunofluorescence
Cells grown on glass coverslips were washed with PBS, fixed in 2% paraformaldehyde solution for 15 min at room temperature, and permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature. Immunofluorescence analysis was performed using anti LC3B antibody (Cell Signaling) and Alexa Fluor 594-conjugated secondary antibody (Invitrogen). Confocal laser scanning microscopy analysis was performed using a FluoView FV10i system (Olympus).
Endotoxemia
Female littermates 8 weeks or older were injected intraperitoneally with freshly prepared LPS (30 mg/kg; Escherichia coli 0111:B4; Sigma-Aldrich) in normal saline. Survival and righting reflexes were monitored every 12 h for 5 days, after which all surviving mice were sacrificed. Experimental animals without righting reflex at time of monitoring were considered moribund and were euthanized and included as nonsurvivors in survival curves generated by the Kaplan and Meier method using GraphPad Prism software.
Plasma Cytokines
Blood from the retro-orbital venous plexus was collected with EDTA-coated glass capillaries into EDTA tubes 8 h after intraperitoneal challenge with 30 mg/kg LPS. Plasma was removed after blood centrifugation at 500 × g for 10 min. Plasma levels of interleukin-6 (IL-6), interleukin-10 (IL-10), monocyte chemoattractant protein-1 (MCP-1), interferon-γ (IFN-γ), tumor necrosis factor α (TNFα), and interleukin-12p70 (IL-12p70) were determined using a cytometric bead array mouse inflammation kit (BD Biosciences) according to the manufacturer's instructions.
Statistical Analysis
All results are expressed as means ± S.E., except analysis of the thioglycolate-induced peritoneal macrophage (mean ± S.D.). Data were analyzed using Student's t test or one-way analysis of variance with Tukey's multiple comparison test, two-way analysis of variance (plasma cytokines), or log rank test (endotoxemia survival).
RESULTS
Sphks Are Not Essential for Myeloid Cell Survival, Proliferation, or Differentiation
To explore the role of sphingosine kinase isoenzymes in macrophages, Sphk1flox/flox Sphk2−/− mice (Sphk2−/−) were used as the source of Sphk2−/− macrophages, Sphk1flox/flox Sphk2−/− mice were crossed with lysozyme-Cre mice to generate the myeloid Sphk1 and Sphk2 double knock-out mice (MΦ Sphk dKO), which were the source of Sphk1- and Sphk2-null macrophages. The deletion efficiency of Sphk1 gene in isolated BMDMs from MΦ Sphk dKO mice was almost complete as determined by PCR of genomic DNA (Fig. 1A). RNA analysis by qRT-PCR showed that both Sphk1 and Sphk2 transcripts were down-regulated >90% in respective knock-out BMDMs (Fig. 1B). Immunoblot analysis of BMDM extracts showed complete loss of expression of Sphk2 and both Sphk1/Sphk2 in Sphk2−/− and MΦ Sphk dKO cells (Fig. 1C).
FIGURE 1.

Characterization of Sphk dKO macrophages. Efficiency of Sphk1 or Sphk2 deletion in isolated BMDMs was determined by PCR of genomic DNA (A), qRT-PCR of mRNA from BMDMs (B), and immunoblot analysis of BMDM extracts (C). Data are representative of at least three independent experiments. N/S, no signal detected. Data represent mean ± S.E. (error bars).
BMDMs from WT, Sphk2−/− and MΦ Sphk dKO mice were analyzed for the levels of S1P, sphingosine, and ceramides by LC-MS/MS. S1P levels were markedly diminished in both Sphk2−/− and MΦ Sphk dKO cells, suggesting that the activity of Sphk2 is important in the basal production of S1P (Fig. 2A). Interestingly, sphingosine levels were significantly elevated in Sphk2−/− and MΦ Sphk dKO. This suggests that lack of Sphk isoenzymes leads to the accumulation of the substrate sphingosine. Furthermore, ceramide levels were also elevated (Fig. 2B). Intracellular ceramide and sphingosine levels were at least 2 and 1 order of magnitude higher than S1P, respectively. These data suggest that lack of Sphk isoenzymes leads to metabolic “pileup” of sphingolipids.
FIGURE 2.

Sphk isoenzymes are not essential for myeloid cell survival or differentiation. A, intracellular sphingoid bases and ceramide level in untreated BMDMs were measured by LC-MS/MS. *, p < 0.05; **, p < 0.01 (compared with the WT group). n = 6 per group. Ceramide, total ceramide molecular species. B, the intracellular ceramide species in BMDMs were measured by LC-MS/MS. BMDMs were treated without or with 10 ng/ml TNFα or 20 μm Sph for 4 h. n = 4–6 per group. Data represent mean ± S.E. (error bars). C, flow cytometry detected CD11b and F4/80 expression on BMDMs after 7-day culture with 20% L929 conditioned medium. Note that Sphk2 or MΦ Sphk dKO cells show similar levels of differentiation. D, cell proliferation of BMDMs from 3 to 6 days after isolation was measured by the MTT assay. Data are representative of at least three independent experiments.
Sphk2−/− and MΦ Sphk dKO mice were grossly indistinguishable from WT mice, and no obvious abnormality was detected. To examine whether the loss of Sphk isoenzymes affects BMDM differentiation, macrophage makers (CD11b and F4/80) were analyzed by flow cytometry in BMDMs. More than 98% cells expressed both of these macrophage markers (Fig. 2C), suggesting that lack of Sphk isoenzymes does not impair differentiation of hematopoietic stem cells into macrophages. Moreover, kinetics of cell proliferation were similar among BMDMs derived from WT, Sphk2−/−, and MΦ Sphk dKO mice (Fig. 2D), suggesting that Sphk isoenzymes are not required for myeloid cell survival or proliferation in vitro.
Sphk1 and 2 Are Not Necessary for Macrophage Inflammatory Responses in Vitro
Intracellular levels of sphingoid bases (Sph, dihydrosphingosine), sphingoid base 1-phosphates (S1P, dihydro-S1P), and ceramide molecular species were quantified in BMDMs after TNFα treatment. Surprisingly, TNFα treatment (4 h) did not result in significant alterations in intracellular sphingoid bases or the phosphorylated derivatives in BMDMs isolated from Sphk2−/−, MΦ Sphk dKO, and WT mice (Fig. 3A).
FIGURE 3.

Sphk isoenzymes are not necessary for inflammatory responses. A, intracellular sphingoid bases and total ceramides in TNFα-treated BMDMs quantified by LC-MS/MS. *, p < 0.05; **, p < 0.01 (compared with the WT group). n = 4 per group. Data represent mean ± S.E. (error bars). B, immunodetection of total p65 and phosphorylated forms of p65, p38, and JNK in whole cell extracts of BMDMs. Cells were either stimulated or not with 10 ng/ml TNFα for 15 min or 100 ng/ml LPS for 30 min. C, immunodetection of COX-2 in whole cell extracts of BMDMs stimulated or not with 100 ng/ml LPS for 24 h. D, qRT-PCR detection of TNFα, IL-1, and IL-6 mRNA in BMDMs cultured with 100 ng/ml LPS for 0, 2, and 6 h. Bar graphs present data as -fold changes relative to untreated (0 h) cultures (n = 9). Data represent mean ± S.E. (error bars).
Inflammatory responses in macrophages are regulated by key signal transduction intermediates, such as NFκB and stress-activated protein kinase (SAPK) members, p38 and JNK. To examine the role of Sphk isoenzymes and intracellular S1P in macrophage inflammatory signal transduction, we examined the activation of NFκB p65, p38 SAPK, and JNK phosphorylation. Because BMDMs showed strong activation of p-p65 and p-p38 after a 15-min treatment with TNFα and 30-min treatment with LPS (data not shown), we treated BMDMs with TNFα for 30 min and LPS for 15 min. BMDMs from Sphk2−/−, MΦ Sphk dKO, and WT mice showed equivalent activation of p-p65, p-JNK and p-p38 after treatment with LPS or TNFα (Fig. 3B). Further, cyclooxygenase-2 (COX-2), a key downstream inflammatory gene, was also induced to a similar extent in BMDMs from Sphk2−/−, MΦ Sphk dKO, and WT mice 24 h after LPS treatment (Fig. 3C). Similar findings were seen in thioglycolate-elicited peritoneal macrophages (data not shown).
We also treated BMDMs with LPS for 2 and 6 h, and mRNAs involved in the inflammatory response were detected by qPCR. The results indicate that IL-1, IL-6, and TNFα mRNA were induced by LPS treatment, but no significant differences were observed among BMDMs from Sphk2−/−, MΦ Sphk dKO, and WT mice (Fig. 3D).
Macrophage Sphk1 and 2 Are Not Necessary for Inflammatory Responses in Vivo
Next, to investigate the role of myeloid Sphk isoenzymes in inflammatory responses in vivo, we employed LPS to model the systemic inflammatory response characteristic of septic shock. We first observed that knocking out Sphk1 in all cells did not protect mice from LPS-induced mortality (Fig. 4A). To exclude compensation by Sphk2, myeloid cell-specific Sphk1/Sphk2 double knock-out mice (Sphk1flox/− Sphk2flox/− LysM-Cre+) were challenged with 30 mg/kg LPS. No significant difference was observed between MΦ Sphk dKO and littermate controls (Sphk1flox/− Sphk2flox/− LysM-Cre−) in the plasma levels of inflammatory cytokines TNFα, INF-γ, MCP-1, IL-6, IL-12, and IL-12 or mortality (Fig. 4, B and C). We employed the thioglycolate-induced peritonitis model to explore whether macrophage recruitment is altered in Sphk2−/− and MΦ Sphk dKO mice in response to inflammatory insult. The mice were injected with 3% thioglycolate, and peritoneal macrophages were isolated by lavage 4 days later. Recruited peritoneal macrophage numbers were similar in WT, Sphk2−/−, and MΦ Sphk dKO mice (Fig. 4D). These results suggest that macrophage Sphk isoenzymes are not necessary for inflammatory responses in vivo.
FIGURE 4.

Myeloid Sphk is not critical for inflammatory responses in vivo. A and B, survival curves of Sphk1−/− and Sphk1+/+ mice (A), Sphk1flox/− Sphk2flox/− LysM-Cre+ and Sphk1flox/− Sphkflox/− LysM-Cre− littermate (B) mice after the injection of LPS (30 mg/kg intraperitoneally). The number of mice in each group is indicated. C, plasma levels of cytokines 8 h after the injection of LPS (30 mg/kg intraperitoneally). The number of mice in each group is indicated. D, total macrophage numbers in peritoneal lavage samples quantified 4 days after injection of 2 ml of 3% thioglycolate (n = 3). Data represent mean ± S.D. (error bars).
Sphk2 Is Critical for Intracellular Sphingoid Base Homeostasis in BMDMs
As shown above, Sphk2−/− and MΦ Sphk dKO BMDMs contained higher levels of sphingosine and ceramides than WT BMDMs. When treated with exogenous sphingosine, further increases in intracellular sphingosine and ceramides were observed in MΦ Sphk dKO BMDMs (Fig. 5). In particular, C16 ceramide was greatly enhanced (Fig. 2B). This was also observed, albeit to a slightly lesser extent in Sphk2−/− BMDMs. S1P was increased in Sphk2−/− BMDMs, which was expected because these cells express Sphk1. These data suggest that lack of Sphk isoenzymes leads to intracellular accumulation of sphingosine and ceramide in BMDMs.
FIGURE 5.
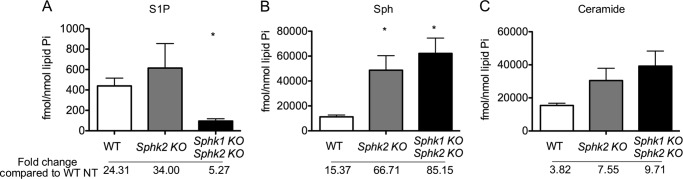
Accumulation of intracellular ceramide and Sph induced by exogenous sphingosine treatment. The intracellular levels of S1P (A), Sph (B), and total ceramides (C) in BMDMs treated with 20 μm Sph for 4 h were measured by LC-MS/MS. *, p < 0.05 (compared with the WT group); n = 4 per group. Data represent mean ± S.E. (error bars).
Treatment of WT BMDMs with exogenous sphingosine led to strong induction of mRNA and protein for Sphk2 (Fig. 6). This was also seen in HEK293 cells. In contrast, Sphk1 was not induced. These data suggest that accumulation of intracellular sphingosine leads to the compensatory increase in Sphk2 expression and the formation of S1P.
FIGURE 6.
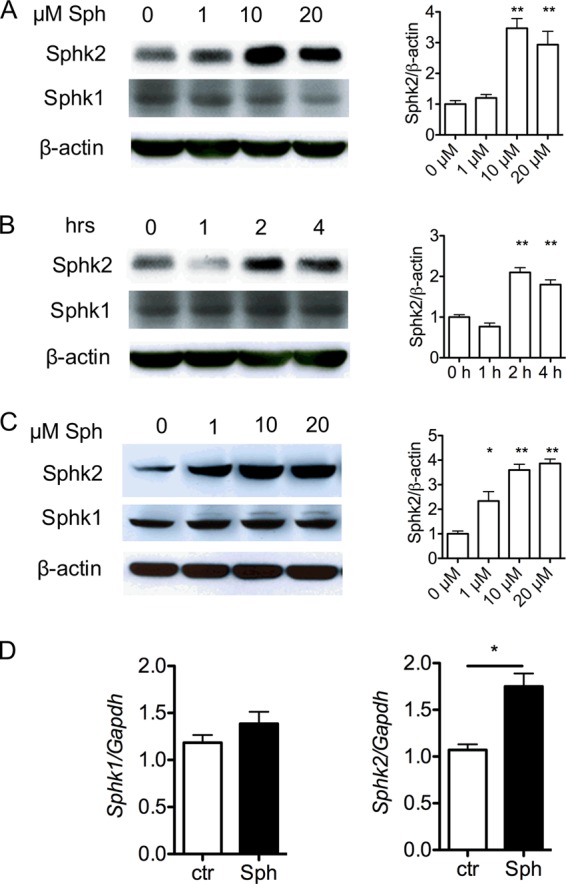
Sphk2 is induced by exogenous Sph treatment. A–C, immunodetection of Sphk1 or Sphk2 protein in C57BL/6 BMDMs (A and B) and 293T cells (C). Cells were exposed to various concentrations of Sph for 4 h or 20 μm Sph for various hours as indicated. On the right is the densitometric quantification of Sphk1/β-actin or Sphk2/β-actin. D, qRT-PCR detection of Sphk1 and Sphk2 mRNA in C57BL/6 BMDMs cultured with 20 μm Sph for 4 h. *, p < 0.05. Data (mean ± S.E.; error bars) are representative of at least three independent experiments (n = 3).
Sphk2 Expression, Cellular Autophagy, and Cell Death
Intracellular accumulation of sphingosine and ceramides could lead to alteration in membrane lipid composition of subcellular organelles. Disturbances in membrane lipid composition cause compensatory autophagy, a process by which organelles are destroyed intracellularly (34). Indeed, autophagic vacuoles, as determined by LC3B staining, were enhanced in Sphk2−/− and MΦ Sphk dKO BMDMs (Fig. 7A). Treatment with exogenous sphingosine further enhanced the accumulation of autophagic vacuoles, especially in MΦ Sphk dKO BMDMs. Similarly, treatment with NH4Cl, which is known to increase the accumulation of autophagic vacuoles, also enhanced the number of LC3+ vesicles in Sphk2−/− and MΦ Sphk dKO BMDMs. Immunoblot analysis using LC3B antibodies demonstrated the presence of active form of LC3B (LC3B-II) in Sphk2−/− and MΦ Sphk dKO BMDMs, which was further increased by exogenous sphingosine and NH4Cl treatment (Fig. 7B). Also, lysosome morphology was determined by the rhodamine dextran uptake assay (Fig. 7C). Under control conditions, normal lysosome morphology was observed in the WT, Sphk2−/−, and MΦ Sphk dKO BMDMs. WT BMDM lysosomes were unaffected following loading the cells with 5 μm sphingosine, whereas a small fraction of cells in the Sphk2−/− and MΦ Sphk dKO BMDMs contained slightly enlarged lysosomes. Upon loading cells with 20 μm sphingosine, BMDMs begin to display enlarged lysosomes. Few macrophages with enlarged lysosomes were observed in the WT BMDMs, whereas in Sphk2−/− and MΦ Sphk dKO BMDMs the majority of the cells exhibited enlarged lysosomes. The most severe phenotype was observed in the MΦ Sphk dKO BMDMs. These data suggest that extremely high intracellular sphingosine levels promoted lysosomal abnormalities that parallel the accumulation of autophagosomes.
FIGURE 7.

Autophagosomes are induced in macrophages that lack Sphk isoenzymes. A, immunostaining of LC3B in BMDMs after 4-h treatment of 5 μm Sph or 20 mm NH4Cl. Scale bar, 10 μm. B, immunodetection of LC3B in BMDMs after treatment with various compounds as indicated for 4 h. Data are representative of at least three independent experiments. C, lysosomes visualized with dextran-rhodamine. Scale bar, 10 μm.
Exogenous sphingosine treatment led to increased cell death in Sphk2−/− and MΦ Sphk dKO BMDMs compared with WT BMDMs (Fig. 8A). In contrast, treatment with exogenous ceramide or S1P did not result in preferential toxicity of Sphk2−/− and MΦ Sphk dKO BMDMs (Fig. 8, C and D). Similarly, treatment of BMDMs with the cytotoxic agent doxorubicin, which induced caspase-dependent cell death, also did not induce preferential death of Sphk2−/− and MΦ Sphk dKO BMDMs (Fig. 8B). These data suggest that accumulation of intracellular sphingosine leads to cell death due to excessive accumulation of sphingoid bases in Sphk2−/− and MΦ Sphk dKO cells.
FIGURE 8.

Sphk-deleted BMDMs are more sensitive to exogenous sphingosine-induced cell death. A–D, BMDMs were exposed to various concentrations of Sph (A), doxorubicin (Dox) (B), ceramide (C), and S1P (D) for 24 h as indicated. The viability of the cells was measured by MTT assay as described under “Experimental Procedures.” **, p < 0.01 (compared with the WT group); n = 3. Data are representative of at least three independent experiments. E, WT BMDMs were exposed to various compounds (20 μm Sph, 20 mm 3-methyladenine (3-MA), 20 mm NH4Cl) as indicated for 24 h, and the viability of the cells was measured by MTT assay. *, p < 0.05; **, p < 0.01 (n = 3). Data are representative of at least three independent experiments. F, cleaved caspase-3 and caspase-8 were immunodetected in BMDMs after cells were treated with various compounds (20 μm Sph, 20 mm 3-methyladenine, 20 mm NH4Cl) as indicated for 4 h (n = 3). Data are representative of at least three independent experiments.
In WT BMDMs, treatment with 3-methyadenine, which inhibits autophagy by attenuation of PI3-kinase (35, 36), as well as NH4Cl, which inhibits lysosomal pH and prevents the fusion of autophagosomes to lysosomes, induced cell death in the presence of exogenous sphingosine (Fig. 8C). Such treatments strongly induced activation of caspase-3 and -8 (Fig. 8D). These data suggest that cellular autophagic pathways are cytoprotective in BMDMs to compensate for intracellular sphingosine accumulation.
DISCUSSION
In this study, we addressed the issue of whether intracellular S1P produced by the Sphk isoenzymes is critical for inflammatory cells from the innate immune system, namely, bone marrow-derived macrophages. During inflammatory reactions, bone marrow-derived monocytes are recruited into tissues in response to chemokine signals and differentiate into inflammatory macrophages. Such cells produce and respond to immune stimuli such as cytokines and are essential for tissue inflammation and resolution (37, 38).
The function of intracellular S1P is controversial. Prior to the cloning and characterization of the G protein-coupled receptors for S1P (39, 40), S1P was considered to be a “second messenger” (41–43), in a fashion analogous to the well characterized intracellular signaling molecules such as diacylglycerol and cAMP. However, it is now clear that many of the biological effects of S1P, for example, regulation of lymphocyte egress, endothelial cell barrier function, angiogenesis, fibroblast proliferation and survival require the action of the G protein-coupled S1P receptors (1, 2, 44). Indeed, specialized transporters for S1P, such as Spns2, mediate the export of S1P and maintain the vascular S1P gradient in vertebrates (45, 46). In addition to the extracellular signaling mode, S1P and dihydro-S1P are utilized as intermediates in critical intracellular lipid metabolic pathways. For example, during de novo sphingolipid synthesis or metabolic breakdown of exogenously derived sphingolipids, metabolism of phosphorylated sphingoid bases by the S1P lyase enzyme is important in the downstream utilization of fatty acyl-CoA and phosphoethanolamine into complex phospholipid synthesis (47, 48). In contrast to well characterized role of S1P, i.e. extracellular ligand for G protein-coupled receptors and a metabolic intermediate, the physiological relevance of S1P as a classical second messenger that activates intracellular transducer systems to modulate cellular responses is not as well established. Recent studies proposed specific intracellular targets of S1P, for example, TRAF2, HDAC1, and HDAC2 (8, 9).
In this study, we developed a novel system to examine the intracellular role of S1P in macrophages. Because Sphk1 and 2 carry out redundant functions of producing S1P and can compensate for each other in the absence of one isoform, we developed a model in which both isoenzymes are lacking in macrophages. In MΦ Sphk dKO BMDMs, S1P levels are markedly attenuated whereas sphingosine and ceramide levels were up-regulated, suggesting metabolic pileup. Indeed, low intracellular levels of S1P and higher levels of sphingosine and ceramide, which are at least 1 and 2 orders of magnitude higher, respectively, suggest the high catalytic turnover of Sphk enzymes under basal conditions. Interestingly, macrophage proliferation, differentiation, and survival were not affected by lack of Sphk enzymes. Thus, lack of S1P or enhanced sphingosine or ceramide in and of itself is not sufficient to alter cell proliferation and/or death.
Importantly, stimulation of BMDMs or elicited peritoneal macrophages from MΦ Sphk dKO mice did not exhibit any defect in TNFα- and LPS-induced inflammatory responses. In particular, the NFκB pathway was activated to a similar extent, suggesting that intracellular S1P is not necessary for the activation of this critical inflammatory signaling pathway. Moreover, Sphk1−/− and MΦ Sphk dKO did not protect from LPS-induced systemic inflammation and death. This is particularly important because several reports have concluded that Sphk1 is an important proinflammatory enzyme necessary for TNFα- and LPS-induced NFκB activation (9) and LPS-induced inflammation through Sphk1/S1P3 signaling (26). Indeed, Sphk inhibitors were proposed as important novel therapeutics in the treatment of chronic inflammatory disease and sepsis (26, 50, 51). Our work using genetic models (27) questions the validity of this hypothesis. It is possible that the use of sphingosine kinase inhibitors with poor specificity as well as siRNAs, which can have off-target effects, contributed to these hypotheses. It is also worth noting that several reports dealing with sphingosine kinases and inflammation have been retracted from the literature. These factors, together with many reports (25, 27, 28), including the data shown in this report, suggest that intracellular S1P activation of NFκB and inflammatory responses is not a major, physiologically relevant pathway in inflammation.
Although we found no role for sphingosine kinases in inflammatory responses, our results suggest a novel homeostatic role for sphingosine kinase in macrophages. We found that BMDMs that lack Sphk2 or both Sphk isoenzymes show exaggerated autophagic vesicles. Addition of exogenous sphingosine further enhanced the autophagic response. Accumulation of autophagosomes may result from abnormal levels of sphingosine-inducing abnormal lysosomes (55) and/or increased ceramide levels inducing enhanced autophagy (56). Indeed, enlarged lysosomes were more common in MΦ Sphk dKO BMDMs treated with exogenous sphingosine. Interestingly, exogenous sphingosine treatment also induced apoptosis of BMDMs. This suggests that accumulation of sphingolipid metabolites such as ceramides that modulate membrane domains trigger a compensatory autophagy response, which promotes cell survival. Indeed, inhibition of autophagy leads to caspase-dependent apoptosis. Previous work pointed out the importance of sphingolipids in the regulation of autophagy (56–58). Indeed, manipulation of sphingolipid metabolic enzymes modulates autophagy in various systems (22, 34, 49, 52–54). We speculate that pharmacologic inhibition of Sphk isoenzymes could lead to intracellular sphingolipid accumulation and compensatory autophagic responses in cells with high flux in the sphingolipid metabolic pathways.
We also found that enhanced sphingolipid accumulation due to Sphk dKO led to the induction of Sphk2 expression. Indeed, exogenous sphingosine treatment induced Sphk2 protein and mRNA. These data suggest that cells up-regulate Sphk2 in response to exogenous sphingosine so that the sphingoid base may be metabolized to the phosphorylated metabolite, which can be secreted out of the cell or further metabolized by the S1P lyase. Specific mechanisms involved, i.e. how sphingolipid metabolites induce expression of Sphk2 transcription and/or protein expression, are not known and need to be investigated further.
In conclusion, our studies indicate that intracellular S1P generated by Sphk isoenzymes in macrophages is not needed for inflammatory responses. In contrast, Sphk isoenzymes maybe involved in a metabolic pathway that maintains membrane homeostasis. If Sphk levels are attenuated, enhanced sphingolipid levels could lead to enhanced Sphk2 expression and compensatory autophagy. These data are consistent with an important metabolic role for Sphk2 in cellular membrane homeostasis.
Acknowledgments
We are grateful to Coralie Guerin and Paula Michea for technical assistance and the PARCC ERI for animal care. We also thank Dr. Shaun Coughlin for then gift of conditional gene targeted mice for Sphk1 and 2.
This work was supported, in whole or in part, by National Institutes of Health Grants HL67330, HL70694, and HL89934 (to T. H). This work was also supported by INSERM Avenir, Marie Curie Actions, the French National Research Agency, Fondation de France, and the Ile-de-France Region (to E. C.). Sphingolipid measurements were supported by the Lipidomics Shared Resource, Hollings Cancer Center, Medical University of South Carolina Grant P30 CA138313 and the Lipidomics Core in the South Carolina Lipidomics and Pathobiology COBRE Grant P20 RR017677.
- S1P
- sphingosine 1-phosphate
- BMDM
- bone marrow-derived macrophage
- dKO
- double knock-out
- MΦ
- macrophage
- MS/MS
- tandem MS
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- qRT-PCR
- quantitative RT-PCR
- Sph
- sphingosine
- Sphk
- spingosine kinase.
REFERENCES
- 1. Blaho V. A., Hla T. (2011) Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem. Rev. 111, 6299–6320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cyster J. G., Schwab S. R. (2012) Sphingosine 1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 30, 69–94 [DOI] [PubMed] [Google Scholar]
- 3. Kohama T., Olivera A., Edsall L., Nagiec M. M., Dickson R., Spiegel S. (1998) Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 273, 23722–23728 [DOI] [PubMed] [Google Scholar]
- 4. Liu H., Sugiura M., Nava V. E., Edsall L. C., Kono K., Poulton S., Milstien S., Kohama T., Spiegel S. (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275, 19513–19520 [DOI] [PubMed] [Google Scholar]
- 5. Zhou J., Saba J. D. (1998) Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem. Biophys. Res. Commun. 242, 502–507 [DOI] [PubMed] [Google Scholar]
- 6. Le Stunff H., Galve-Roperh I., Peterson C., Milstien S., Spiegel S. (2002) Sphingosine-1-phosphate phosphohydrolase in regulation of sphingolipid metabolism and apoptosis. J. Cell Biol. 158, 1039–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brindley D. N., English D., Pilquil C., Buri K., Ling Z. C. (2002) Lipid phosphate phosphatases regulate signal transduction through glycerolipids and sphingolipids. Biochim. Biophys. Acta 1582, 33–44 [DOI] [PubMed] [Google Scholar]
- 8. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine 1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alvarez S. E., Harikumar K. B., Hait N. C., Allegood J., Strub G. M., Kim E. Y., Maceyka M., Jiang H., Luo C., Kordula T., Milstien S., Spiegel S. (2010) Sphingosine 1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strub G. M., Paillard M., Liang J., Gomez L., Allegood J. C., Hait N. C., Maceyka M., Price M. M., Chen Q., Simpson D. C., Kordula T., Milstien S., Lesnefsky E. J., Spiegel S. (2011) Sphingosine 1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mizugishi K., Yamashita T., Olivera A., Miller G. F., Spiegel S., Proia R. L. (2005) Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 25, 11113–11121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allende M. L., Sasaki T., Kawai H., Olivera A., Mi Y., van Echten-Deckert G., Hajdu R., Rosenbach M., Keohane C. A., Mandala S., Spiegel S., Proia R. L. (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 279, 52487–52492 [DOI] [PubMed] [Google Scholar]
- 13. Zemann B., Kinzel B., Müller M., Reuschel R., Mechtcheriakova D., Urtz N., Bornancin F., Baumruker T., Billich A. (2006) Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 107, 1454–1458 [DOI] [PubMed] [Google Scholar]
- 14. Olivera A., Eisner C., Kitamura Y., Dillahunt S., Allende L., Tuymetova G., Watford W., Meylan F., Diesner S. C., Li L., Schnermann J., Proia R. L., Rivera J. (2010) Sphingosine kinase 1 and sphingosine 1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. J. Clin. Invest. 120, 1429–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liang J., Nagahashi M., Kim E. Y., Harikumar K. B., Yamada A., Huang W. C., Hait N. C., Allegood J. C., Price M. M., Avni D., Takabe K., Kordula T., Milstien S., Spiegel S. (2013) Sphingosine 1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kharel Y., Raje M., Gao M., Gellett A. M., Tomsig J. L., Lynch K. R., Santos W. L. (2012) Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem. J. 447, 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P. H., Keats J. J., Wang H., Vignali D. A., Bergsagel P. L., Karin M. (2008) Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 9, 1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Devin A., Lin Y., Yamaoka S., Li Z., Karin M., Liu Z. (2001) The α and β subunits of IκB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol. Cell. Biol. 21, 3986–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu S. F., Malik A. B. (2006) NF-κB activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L622–645 [DOI] [PubMed] [Google Scholar]
- 20. Nayak D., Huo Y., Kwang W. X., Pushparaj P. N., Kumar S. D., Ling E. A., Dheen S. T. (2010) Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 166, 132–144 [DOI] [PubMed] [Google Scholar]
- 21. Limaye V., Xia P., Hahn C., Smith M., Vadas M. A., Pitson S. M., Gamble J. R. (2009) Chronic increases in sphingosine kinase-1 activity induce a pro-inflammatory, pro-angiogenic phenotype in endothelial cells. Cell. Mol. Biol. Lett. 14, 424–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oskouian B., Saba J. D. (2010) Cancer treatment strategies targeting sphingolipid metabolism. Adv. Exp. Med. Biol. 688, 185–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prakash H., Lüth A., Grinkina N., Holzer D., Wadgaonkar R., Gonzalez A. P., Anes E., Kleuser B. (2010) Sphingosine kinase-1 (SphK-1) regulates Mycobacterium smegmatis infection in macrophages. PLoS One 5, e10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grin'kina N. M., Karnabi E. E., Damania D., Wadgaonkar S., Muslimov I. A., Wadgaonkar R. (2012) Sphingosine kinase 1 deficiency exacerbates LPS-induced neuroinflammation. PLoS One 7, e36475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wadgaonkar R., Patel V., Grinkina N., Romano C., Liu J., Zhao Y., Sammani S., Garcia J. G., Natarajan V. (2009) Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niessen F., Schaffner F., Furlan-Freguia C., Pawlinski R., Bhattacharjee G., Chun J., Derian C. K., Andrade-Gordon P., Rosen H., Ruf W. (2008) Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 452, 654–658 [DOI] [PubMed] [Google Scholar]
- 27. Michaud J., Kohno M., Proia R. L., Hla T. (2006) Normal acute and chronic inflammatory responses in sphingosine kinase 1 knockout mice. FEBS Lett. 580, 4607–4612 [DOI] [PubMed] [Google Scholar]
- 28. Zemann B., Urtz N., Reuschel R., Mechtcheriakova D., Bornancin F., Badegruber R., Baumruker T., Billich A. (2007) Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol. Lett. 109, 56–63 [DOI] [PubMed] [Google Scholar]
- 29. Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 [DOI] [PubMed] [Google Scholar]
- 30. Pappu R., Schwab S. R., Cornelissen I., Pereira J. P., Regard J. B., Xu Y., Camerer E., Zheng Y. W., Huang Y., Cyster J. G., Coughlin S. R. (2007) Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine 1-phosphate. Science 316, 295–298 [DOI] [PubMed] [Google Scholar]
- 31. Michaud J., Im D. S., Hla T. (2010) Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J. Immunol. 184, 1475–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mendelson K., Zygmunt T., Torres-Vázquez J., Evans T., Hla T. (2013) Sphingosine 1-phosphate receptor signaling regulates proper embryonic vascular patterning. J. Biol. Chem. 288, 2143–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Majumdar A., Cruz D., Asamoah N., Buxbaum A., Sohar I., Lobel P., Maxfield F. R. (2007) Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol. Biol. Cell 18, 1490–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bedia C., Levade T., Codogno P. (2011) Regulation of autophagy by sphingolipids. Anticancer Agents Med. Chem. 11, 844–853 [DOI] [PubMed] [Google Scholar]
- 35. Seglen P. O., Gordon P. B. (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 79, 1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blommaart E. F., Krause U., Schellens J. P., Vreeling-Sindelárová H., Meijer A. J. (1997) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243, 240–246 [DOI] [PubMed] [Google Scholar]
- 37. Bellingan G. J., Caldwell H., Howie S. E., Dransfield I., Haslett C. (1996) In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J. Immunol. 157, 2577–2585 [PubMed] [Google Scholar]
- 38. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hla T., Maciag T. (1990) An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G protein-coupled receptors. J. Biol. Chem. 265, 9308–9313 [PubMed] [Google Scholar]
- 40. Lee M. J., Van Brocklyn J. R., Thangada S., Liu C. H., Hand A. R., Menzeleev R., Spiegel S., Hla T. (1998) Sphingosine 1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 279, 1552–1555 [DOI] [PubMed] [Google Scholar]
- 41. Spiegel S., Milstien S. (1995) Sphingolipid metabolites: members of a new class of lipid second messengers. J. Membr. Biol. 146, 225–237 [DOI] [PubMed] [Google Scholar]
- 42. Mattie M., Brooker G., Spiegel S. (1994) Sphingosine 1-phosphate, a putative second messenger, mobilizes calcium from internal stores via an inositol trisphosphate-independent pathway. J. Biol. Chem. 269, 3181–3188 [PubMed] [Google Scholar]
- 43. Olivera A., Spiegel S. (1993) Sphingosine 1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 365, 557–560 [DOI] [PubMed] [Google Scholar]
- 44. Olivera A., Allende M. L., Proia R. L. (2013) Shaping the landscape: metabolic regulation of S1P gradients. Biochim. Biophys. Acta 1831, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fukuhara S., Simmons S., Kawamura S., Inoue A., Orba Y., Tokudome T., Sunden Y., Arai Y., Moriwaki K., Ishida J., Uemura A., Kiyonari H., Abe T., Fukamizu A., Hirashima M., Sawa H., Aoki J., Ishii M., Mochizuki N. (2012) The sphingosine 1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest. 122, 1416–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawahara A., Nishi T., Hisano Y., Fukui H., Yamaguchi A., Mochizuki N. (2009) The sphingolipid transporter Spns2 functions in migration of zebrafish myocardial precursors. Science 323, 524–527 [DOI] [PubMed] [Google Scholar]
- 47. Worgall T. S., Juliano R. A., Seo T., Deckelbaum R. J. (2004) Ceramide synthesis correlates with the posttranscriptional regulation of the sterol-regulatory element-binding protein. Arterioscler. Thromb. Vasc. Biol. 24, 943–948 [DOI] [PubMed] [Google Scholar]
- 48. Dobrosotskaya I. Y., Seegmiller A. C., Brown M. S., Goldstein J. L., Rawson R. B. (2002) Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 296, 879–883 [DOI] [PubMed] [Google Scholar]
- 49. Fyrst H., Oskouian B., Bandhuvula P., Gong Y., Byun H. S., Bittman R., Lee A. R., Saba J. D. (2009) Natural sphingadienes inhibit Akt-dependent signaling and prevent intestinal tumorigenesis. Cancer Res. 69, 9457–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Edmonds Y., Milstien S., Spiegel S. (2011) Development of small-molecule inhibitors of sphingosine 1-phosphate signaling. Pharmacol. Ther. 132, 352–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Neill L. A. (2010) Stopping sepsis by targeting sphingosine kinase 1. Sci. Transl. Med. 2, 36ps29. [DOI] [PubMed] [Google Scholar]
- 52. Colié S., Van Veldhoven P. P., Kedjouar B., Bedia C., Albinet V., Sorli S. C., Garcia V., Djavaheri-Mergny M., Bauvy C., Codogno P., Levade T., Andrieu-Abadie N. (2009) Disruption of sphingosine-1-phosphate lyase confers resistance to chemotherapy and promotes oncogenesis through Bcl-2/Bcl-xL up-regulation. Cancer Res. 69, 9346–9353 [DOI] [PubMed] [Google Scholar]
- 53. Pattingre S., Bauvy C., Levade T., Levine B., Codogno P. (2009) Ceramide-induced autophagy: to junk or to protect cells? Autophagy 5, 558–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lavieu G., Scarlatti F., Sala G., Carpentier S., Levade T., Ghidoni R., Botti J., Codogno P. (2008) Sphingolipids in macroautophagy. Methods Mol. Biol. 445, 159–173 [DOI] [PubMed] [Google Scholar]
- 55. Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 56. Sims K., Haynes C. A., Kelly S., Allegood J. C., Wang E., Momin A., Leipelt M., Reichart D., Glass C. K., Sullards M. C., Merrill A. H., Jr. (2010) Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J. Biol. Chem. 285, 38568–38579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coward J., Ambrosini G., Musi E., Truman J. P., Haimovitz-Friedman A., Allegood J. C., Wang E., Merrill A. H., Jr., Schwartz G. K. (2009) Safingol (l-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy 5, 184–193 [DOI] [PubMed] [Google Scholar]
- 58. Zheng W., Kollmeyer J., Symolon H., Momin A., Munter E., Wang E., Kelly S., Allegood J. C., Liu Y., Peng Q., Ramaraju H., Sullards M. C., Cabot M., Merrill A. H., Jr. (2006) Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 1758, 1864–1884 [DOI] [PubMed] [Google Scholar]