

Background: The translocation of the Bordetella pertussis CyaA toxin across membrane is still poorly understood.
Results: A membrane-active peptide isolated from the CyaA toxin is characterized by biophysical approaches.
Conclusion: The α-helical peptide is inserted in plane and induces membrane permeabilization.
Significance: The membrane-destabilizing activity of this peptide may assist the initial steps of the CyaA translocation process.
Keywords: Adenylate Cyclase (Adenylyl Cyclase), Bacterial Toxins, Circular Dichroism (CD), Infrared Spectroscopy, Membrane, Molecular Dynamics, Peptide Conformation, Phospholipid Vesicle
Abstract
Bordetella pertussis, the pathogenic bacteria responsible for whooping cough, secretes several virulence factors, among which is the adenylate cyclase toxin (CyaA) that plays a crucial role in the early stages of human respiratory tract colonization. CyaA invades target cells by translocating its catalytic domain directly across the plasma membrane and overproduces cAMP, leading to cell death. The molecular process leading to the translocation of the catalytic domain remains largely unknown. We have previously shown that the catalytic domain per se, AC384, encompassing residues 1–384 of CyaA, did not interact with lipid bilayer, whereas a longer polypeptide, AC489, spanning residues 1–489, binds to membranes and permeabilizes vesicles. Moreover, deletion of residues 375–485 within CyaA abrogated the translocation of the catalytic domain into target cells. Here, we further identified within this region a peptidic segment that exhibits membrane interaction properties. A synthetic peptide, P454, corresponding to this sequence (residues 454–485 of CyaA) was characterized by various biophysical approaches. We found that P454 (i) binds to membranes containing anionic lipids, (ii) adopts an α-helical structure oriented in plane with respect to the lipid bilayer, and (iii) permeabilizes vesicles. We propose that the region encompassing the helix 454–485 of CyaA may insert into target cell membrane and induce a local destabilization of the lipid bilayer, thus favoring the translocation of the catalytic domain across the plasma membrane.
Introduction
The adenylate cyclase (CyaA) produced by Bordetella pertussis, the causative agent of whooping cough, is one of the major virulence factors of this organism (1–3). CyaA plays an important role in the early stages of respiratory tract colonization by B. pertussis. This toxin uses an original intoxication mechanism; secreted by the virulent bacteria, it is able to invade eukaryotic target cells through a unique but poorly understood mechanism that involves a direct translocation of the catalytic domain across the plasma membrane. CyaA is a 1706-residue-long protein organized in a modular fashion. The ATP-cyclizing, calmodulin-activated, catalytic domain is located in the 400 amino-proximal residues (4–6), whereas the C-terminal 1306 residues are endowed with an intrinsic pore-forming (or hemolytic) activity. Several domains can be delineated in the C-terminal “hemolysin” moiety. The hydrophobic region, located from residues 525 to 715, contains several hydrophobic segments that are predicted to adopt α-helical structures and to insert into membranes to form cation-selective pores. The C-terminal part of the molecule (from position 900 to 1706) is involved in toxin binding to a specific cellular receptor (CD11b/CD18). This domain consists of ∼40 copies of a glycine and aspartate-rich nonapeptide repeat (residues 913–1613) (7–14) characteristic of a large family of bacterial cytolysins known as RTX (repeat-in-toxin) toxins (15–17).
The CyaA toxin is synthesized as an inactive precursor, proCyaA, that is converted into the active toxin upon acylation of two lysine residues (Lys-860 and Lys-983) by a specific acyltransferase, CyaC (18). The acylated (i.e. active) CyaA polypeptide is then secreted across the bacterial envelope by a dedicated type I secretion machinery (19, 20). Although CyaA can intoxicate a variety of cell types by a process that is independent of receptor-mediated endocytosis, it is also competent to bind in a calcium-dependent manner and with a high affinity to a cell receptor, the CD11b/CD18 integrin, that is expressed by a subset of leukocytes, including neutrophils, macrophages, and dendritic cells (21, 22).
The molecular mechanism by which CyaA enters into target cells remains, however, largely unknown. It is believed that after binding to the CD11b/CD18 receptor, the hydrophobic region of CyaA, located between amino acids 525 and 715, may insert into the plasma membrane of the target cells (23, 24). Then the catalytic domain is translocated directly across the plasma membrane of the target cells into the cytosol through a process referred to as internalization or intoxication. Upon binding to calmodulin in the cytosol of the infected cell, the catalytic domain produces supraphysiologic levels of cAMP that, in turn, alters cellular physiology (25–28). Numerous studies have highlighted the importance of various amino acid residues and/or the contribution of distinct domains of the CyaA polypeptide in the translocation process (8, 23, 29–32). However, a description at the molecular level of the individual steps that the polypeptide follows to cross the membrane is still missing.
We recently showed that deletion of residues 375–485 within CyaA abrogated the translocation of the catalytic domain into target cells (31). We further showed in vitro that the isolated catalytic domain, AC384 (from residues 1–384 of CyaA), did not bind membranes, whereas a longer construction, AC489, was able to partition from the solution to the membrane and induced permeabilization of lipid bilayers. We suggested that the region encompassing residues 375–485 of CyaA could play an important role in promoting translocation of the catalytic domain across the plasma membrane of target cells.
Here, we further characterized within this region the structural elements that might participate in membrane partitioning and permeabilization. We identified two peptidic segments, located from residue 414 to 440 and from residue 454 to 484, and predicted to adopt α-helical secondary structures as putative membrane interacting sites. Two synthetic peptides modeled from these sequences, hereafter called P414 and P454, respectively, were then analyzed for their structural and functional properties. P454, but not P414, was able to bind to and permeabilize membranes containing negatively charged phospholipids. P454 was mainly unfolded in solution, but upon membrane partitioning, it adopted an α-helical conformation with its axis oriented in the plane of the lipid bilayer. We propose that the helical segment located between residues 454 and 485 of CyaA may insert into the target cell membrane and induce a local destabilization of the lipid bilayer, thus favoring the translocation of the catalytic domain across the plasma membrane.
MATERIALS AND METHODS
Reagents
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC3; 850375C), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE; 850725C), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG; 840475C), and cholesterol (Chol; 700000P) were from Avanti Polar Lipids (Alabaster, AL). ANTS (A-350), DPX (X-1525), and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (N-360) were purchased from Molecular Probes (Eugene, OR). All experiments were performed in 20 mm HEPES, 150 mm NaCl, 2 mm CaCl2, pH 7.4 (buffer A) or 20 mm HEPES, 150 mm NaCl, pH 7.4 (buffer B) unless stated otherwise.
Peptide Synthesis
All synthetic peptides were purchased from Genophere Biotech (France), and their purity (99%) and composition were controlled by reverse-phase HPLC and MALDI-mass spectrometry. All peptides are capped on the N terminus with an acetyl group and on the C terminus with an amide group. We studied two peptides, P414 and P454, corresponding to residues 414–440 and residues 454–484, respectively, numbered from the native sequence of the adenylate cyclase (CyaA) toxin. The P454 amino acid sequence was kept unchanged, with the presence of one single native tryptophan (ASAHWGQRALQGAQAVAAAQRLVHAIALMTQ, positions 454–484 in CyaA), whereas Phe-415 was mutated to tryptophan (underlined) in peptide P414 (SWSLGEVSDMAAVEAAELEMTRQVLHA, positions 414–440 in CyaA). The P414 peptide has 27 amino acid residues, with a molecular mass of 2972 g/mol, a pK = 4.2, and a valence z = −4e at pH 7. The P454 peptide has 31 amino acids, with a molecular mass of 3268 g/mol, a pK = 11, and a valence z = +2.5e at pH 7.
Preparation of Lipid Vesicles
Multilamellar vesicles were prepared at a lipid concentration of 10–20 mm from DOPC, DOPE, DOPG, and cholesterol at a molar ratio of 6:3:0:1 or 4:3:2:1 in buffer B (20 mm HEPES, 150 mm NaCl, pH 7.4) by reverse phase evaporation and then submitted to extrusion through 0.8-, 0.4-, and 0.2-μm polycarbonate filters to produce large unilamellar vesicles (LUVs), as described previously (31). The suspensions of LUVs were monodisperse with mean hydrodynamic diameters of 180 nm, as measured by dynamic light scattering on a NanoZS instrument (Malvern) (11, 33). LUVs or multilamellar vesicles were then sonicated using a sonicator tip to form monodisperse small unilamellar vesicles (SUVs) with mean hydrodynamic diameters of 40 nm.
Tryptophan Fluorescence Titrations
Fluorescence titrations were performed with a FP-6200 Jasco spectrofluorimeter, equipped with a Peltier-thermostatted cell holder (25 °C). A bandwidth of 5 nm was used for both excitation and emission beams. Fluorescence emission spectra of 1–10 μm peptide solutions in buffer A, in the absence or in the presence of lipids (LUVs or SUVs), were recorded between 300 and 400 nm at a scan rate of 125 nm·min−1, with an excitation wavelength of 290 nm. The maximum emission wavelength (λmax) and fluorescence intensity ratio at 380 nm over 340 nm represent the average of three values obtained from emission spectra that were corrected for blank measurements. Fluorescence spectra were also corrected for light scattering, by subtracting the corresponding spectra of vesicles at similar final concentrations. Changes in tryptophan intrinsic fluorescence were used to evaluate peptide membrane partition.
Partition Coefficient (KX) of the Peptides from the Solution to the Membrane
The value for KX, which is defined as the ratio of peptide concentrations in the membrane and buffer phases, respectively, is given (see Ref. 34 for a review) by the following equation.
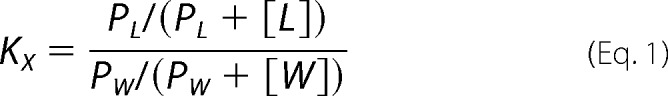 |
Taking into consideration that in our experiments, L ≫ PL and W ≫ PW, KX is expressed as follows,
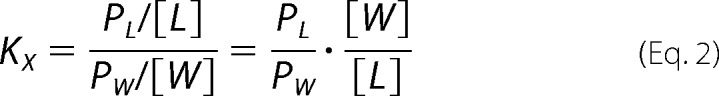 |
with PL and PW in mol or in mol/liter, where PW and PL denote the soluble peptide (peptide in water phase, PW) and the membrane-bound peptide (peptide in lipid phase, PL), respectively; W is the concentration of water (55.5 m); L is the concentration of lipid.
The fraction of peptide partitioned into the membrane phase is equal to the following,
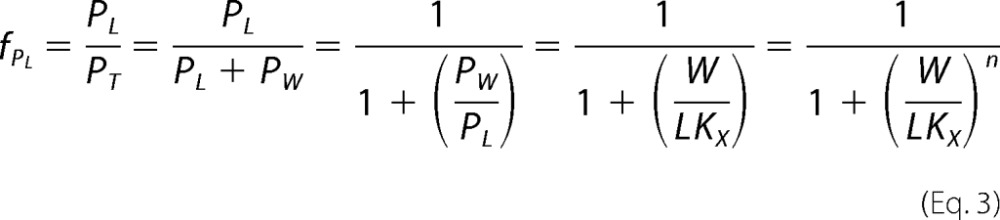 |
where n is the Hill coefficient in the case of cooperativity and PT is the total peptide, PT = PW + PL. This equation was fitted to the experimental data using Kaleidagraph software.
Once the partition coefficient is calculated, the free energy (ΔG) of transfer of the peptide from water to membrane was calculated according to (31), ΔG = −RTln(KX), where ΔG is the free energy (kcal/mol), R is the gas constant (R = 1.98 10−3 kcal·mol−1·K−1), T is the temperature in Kelvin, and KX is the partition coefficient.
The partition coefficient is related to the apparent dissociation constant as follows.
with
 |
Surface Plasmon Resonance
Binding kinetics were determined by surface plasmon resonance (SPR) using a BIAcore 2000 instrument (GE Healthcare), as described previously (35, 36). Briefly, lipid monolayer reconstitution on a hydrophobic acyl surface (chip HPA; GE Healthcare) was achieved at 25 °C by the injection of a total volume of 80 μl at a flow rate of 2 μl/min for 40 min of a lipid vesicle suspension (1 g/liter) composed of POPC/DOPE/DOPG/Chol (molar ratio 4:3:2:1). The lipid layer was washed and stabilized with four pulses of NaOH (50 mm, 15 μl, flow rate 100 μl/min). Samples of 1 and 10 μm P454 were injected onto the chip for 300 s (25 μl, flow rate 5 μl/min).
Isothermal Titration Calorimetry (ITC)
ITC experiments were carried out using a Microcal VP-ITC high sensitivity titration calorimeter (Microcal, Northampton, MA). All solutions were degassed under vacuum to eliminate air bubbles. The temperature was fixed at 25 °C, and the VP-ITC device was electrically calibrated. The reference power was set to 15 μcal/s using a syringe stirring speed of 307 rpm. The syringe was loaded with 20 mm SUVs (DOPC/DOPE/DOPG/Chol, 6:3:0:1 or 4:3:2:1), and the sample cell was loaded with 1.41 ml of P454 or P414 peptide solutions at a concentration of 42 μm. Twenty-nine injections of 5 μl of SUVs were performed into the sample cell. To determine ΔH, six injections of 5 μl of the P454 peptide at 85 μm were performed into the sample cell containing 1.41 ml of anionic SUVs at 5 mm. For both experiments, the duration of each injection was 10 s with a spacing of 600 s. The heats of dilution were determined by titrating (i) SUVs into buffer, (ii) buffer into P454 or P414 peptide solutions, (iii) peptides into buffer, and (iv) buffer into SUVs. Data were analyzed using Microcal Origin software by fitting a one-site binding model to the experimental data.
Circular Dichroism Spectroscopy
CD spectra were recorded on an Aviv circular dichroism spectropolarimeter model 215. CD measurements were carried out at 25 °C at a scan rate of 0.5 nm·s−1 (step, 0.5 nm; integration time, 1 s) with a time constant of 100 ms and a bandwidth of 1 nm. Rectangular quartz Suprasil cells of 1-mm path lengths (110.QS, Hellma) were used for measuring CD spectra in the far-UV region (200–250 nm). Each far-UV CD spectrum represents the average of five scans (11). Working peptide concentration was 40 μm, and SUVs were used at final concentrations up to 2 mm (DOPC/DOPE/DOPG/Chol 6:3:0:1 or 4:3:2:1).
Synchrotron Radiation Circular Dichroism Spectroscopy
Synchrotron radiation CD experiments were carried out at the synchrotron facility SOLEIL (DISCO beamline, Gif-sur-Yvette, France). Synchrotron radiation CD spectra were recorded at 25 °C, with an integration time of 1.2 s and a bandwidth of 1 nm with a 1 nm resolution step. Each far-UV spectrum represents the average of at least three individual scans. QS cells (Hellma) with a path length of 50 μm were used to record CD signals in the far-UV region (from 180 to 260 nm) (14). Buffer B in the absence or in the presence of SUVs (DOPC/DOPE/DOPG/Chol, 4:3:2:1) were used as blank and subtracted from peptide/SUV spectra.
The CD units used are the mean residue ellipticity, expressed in kilodegrees square centimeters per decimole and per amino acid ((kdeg·cm2)/(dmol·aa)). P454 peptide concentration was 200 μm, and lipids were at 10 mm.
The mean residue ellipticity is calculated from the relation,
 |
where θm is the measured ellipticity in degrees, C is the concentration in mol/liter, l is the path length of the cell in cm, and N is the number of residues. The value 100 arises from the conversion of the concentration in mol/liter to dmol/cm3.
Fourier Transform Infrared Spectroscopy
FTIR was performed as described elsewhere (11). Spectra were acquired on an FP-6100 Jasco spectrometer, equipped with a ceramic source, a Ge/KBr beam splitter, and a deuterated, l-alanine-doped triglycine sulfate detector. A water-cooled Peltier unit was used to control the temperature of the cell compartment, which was constantly purged with dried air. The peptide samples were twice lyophilized and solubilized (first in buffer prepared in D2O and then in D2O only) and finally dialyzed against the experimental buffer (20 mm HEPES, 150 mm NaCl). Peptides were used at a concentration of 150 μm in the absence or in the presence of 5 mm SUVs. A drop of 60 μl of sample was deposited between two calcium fluoride windows, separated by a cell path length spacer of 100 μm. Each spectrum corresponds to the accumulation of 256 scans, recorded from 1000 to 6000 cm−1 at a resolution of 4 cm−1. Background measurement was done against air. The buffer spectrum was subtracted from the sample spectrum prior to subtraction of a scaled spectrum of CO2 and water vapor. The major bands of the protein FTIR spectra were then identified by standard deconvolution and second derivative procedures as described previously (11). The maximum wave number values were used to deconvolute absorption bands from the experimental spectra. A deconvoluted spectrum consists of the sum of bands, each band made of fractions of a Gaussian and of a Lorentzian curve. The area under each absorption band was used to quantify the secondary structure content, taking into account the molar absorption of each band and according to the following assignments: intermolecular β-sheets (EM = 1000/m/cm) from 1612 to 1627 cm−1, intramolecular β-sheets (EM = 700/m/cm) from 1625 to 1640 cm−1, disordered regions (EM = 330/m/cm) from 1640 to 1650 cm−1, α-helices (EM = 540/m/cm) from 1650 to 1660 cm−1, turns (EM = 300/m/cm) between 1660 and 1680 cm−1, and finally, antiparallel β-sheet (EM = 300/m/cm) from 1680 to 1697 cm−1. We also consider the amino acids, such as glutamines at 1635 cm−1 (EM = 550/m/cm), asparagines at 1648 cm−1 (EM = 570/m/cm), and arginines at 1609 cm−1 (EM = 480/m/cm) (11, 37–43). All acquisitions and data processing were done using Spectra Analysis (Jasco). The deconvoluted absorption bands were redrawn using Kaleidagraph (Synergy Software, Reading, PA).
Orientation of the Secondary Structures by ATR-FTIR
Attenuated total reflection infrared (ATR-FTIR) spectra were recorded on a FP-6100 Jasco spectrometer, equipped with a liquid nitrogen-cooled mercury cadmium telluride detector. The internal reflection element was a germanium ATR plate (50 × 20 × 2 mm; Pike Technologies) with an incident angle of 45°. The penetration depth of the evanescent wave is ∼0.4 μm in the wave number region of investigation. A total of 256 scans were accumulated at a nominal resolution of 4 cm−1. The spectrophotometer was continuously purged with dried air at a flow rate of 10 liters/min.
For ATR-FTIR experiments, 100 μg of peptides were dissolved in TFE/water at a final concentration of 10 g/liter and then mixed with 500 μg of a lipid mixture, DOPC/DOPE/DOPG/Chol (4:3:2:1), resuspended in chloroform. Oriented thin multilayer films were obtained by applying the solution onto the surface of a germanium internal reflection element and drying it under a stream of nitrogen. Spectra were recorded with the incident light polarized parallel and perpendicular with respect to the incidence plane using a germanium polarizer (Pike Technologies).
We first determined the angle θ of the secondary structure axis with respect to the membrane plane from the difference between the polarized ATR absorbance values, ΔATR, in both amide I′ and II′ bands (44). Dichroism spectra were computed by subtracting the perpendicular polarized spectrum from the parallel polarized spectrum, ΔATR = Abs‖ − RATRiso × AbsT, with the isodichroic ratio RATRiso from the lipid ester v(C=O) stretching band at 1740 cm−1. RATRiso was chosen so that the area of the lipid ester at 1740 cm−1 equals zero on the dichroism spectrum to take into account the difference in the relative power of the evanescent field for each polarization. The dichroic ratio RATR is defined as the ratio of the band area recorded for the parallel polarization (A‖; i.e. at 90°) and perpendicular polarization (A⊥; i.e. at 0°), RATR = A‖/A⊥.
A positive ΔATR profile in the amide I′ region and a negative ΔATR profile in the amide II′ band indicate that the helix axis is oriented parallel to the normal of the ATR plate and hence adopts a transmembrane orientation; on the contrary, a negative ΔATR amide I′ band and a positive ΔATR amide II′ band indicate that the helix axis adopts an orientation close to the plane of the ATR plate and hence adopts an in-plane orientation on the membrane surface (43–46).
As a complementary procedure, we quantitatively interpreted the dichroic ratios to determine the angle θ of the secondary structure axis with respect to the normal of the membrane plane from the orientational order parameter, S, and the dichroic ratio, RATR, at the wave numbers of the helix (43, 44, 47–49). The measured order parameter S is expressed as a set of uniaxial S distributions,
where Smb describes the distribution function of the lipid membrane with respect to the internal reflection element; because the ordering of the membrane on the ATR crystal is usually quite good, previous measurements indicated that Smb is close to 1; the order parameter S of the helix is made of two contributions, an order function Shorder, assumed to scale at 1, and Shθ, the helix orientation with respect to the lipid bilayer normal; Sdipol is the dipole orientation of the amide I band with respect to the helix long axis, with a transition dipole of a unique distribution of angle α, which ranges between 35 and 39° for α-helices. Taken together, the S values are expressed as follows,
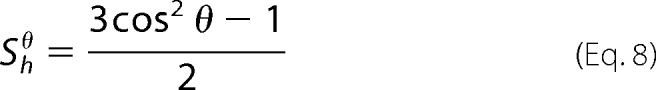 |
and
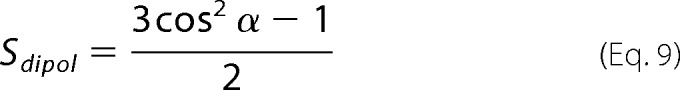 |
and
leading to the simplification of the orientational order parameter S.
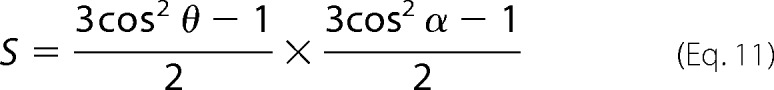 |
The angle θ of the secondary structure axis with respect to the membrane normal can be expressed as follows,
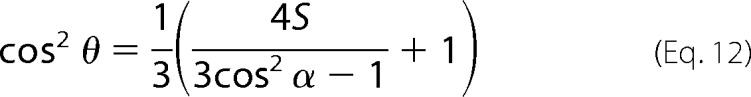 |
using the double-angle formula,
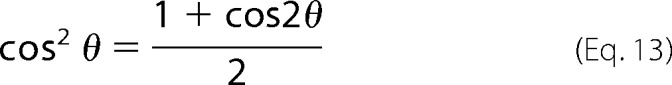 |
given the following,
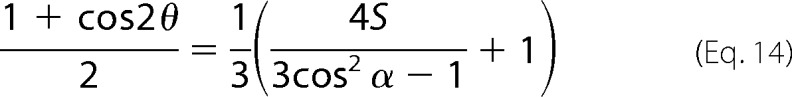 |
from which the angle θ is extracted.
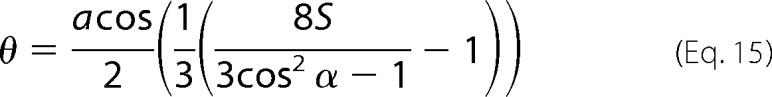 |
The orientational order parameter S is related to the dichroic ratio RATR,
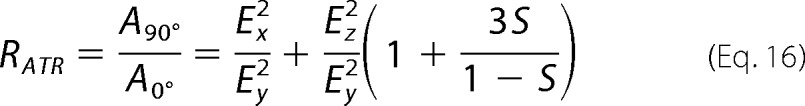 |
from which the orientational order parameter S for any helix with a polarization is extracted,
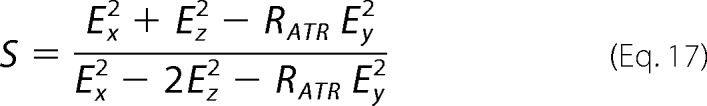 |
where Ex2, Ey2, and Ez2 are the time-averaged square electric field amplitudes of the evanescent wave at the interface between the sample and the internal reflection element (44). Ex and Ey are easily tabulated, depending on if the thin (RATRiso < 1.4) or thick (RATRiso > 1.4) model applies. Ez is estimated, taking into consideration that for RATRiso, S = 0 and the following,
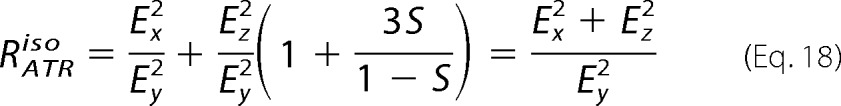 |
given the following.
Again, the isodichroic ratio RATRiso is taken from the lipid ester v(C=O) stretching band at 1740 cm−1.
The values Ex, Ey, Ez, and RATR are input into Equation 17, and then S is inserted into Equation 15 to give θ in radians. The angle θ of the helix axis is normal to the bilayer plane for θ = 0° and is parallel to the lipid bilayer plan for θ = 90°.
LUV Permeabilization Assay
Permeabilization followed by dye leakage was assayed with ANTS (fluorescent probe) and DPX (quencher) entrapped into LUVs. Lipid vesicles (10 mm lipids) were prepared from a mixture of DOPC, DOPE, DOPG, and cholesterol (6:3:0:1 or 4:3:2:1) containing 20 mm ANTS and 60 mm DPX (31). Non-entrapped probes were removed by buffer exchange of the LUV preparation through a G-25 Sephadex column eluted with buffer B. For permeabilization assays, the LUVs were incubated at 25 °C under constant stirring. ANTS fluorescence was recorded continuously (λex = 360 nm, λem = 510 nm, bandwidths 5 nm) to set the base line. Then peptides or CyaA proteins were added to the sample, and fluorescence was recorded.
Molecular Dynamics
The coarse grained simulations were carried out with Gromacs version 4.5.5 (50) using the Martini force field (51–53). The coarse grained models of P414 and P454 were directly derived from the all-atom NMR models. First, 1000 lipids were randomly mixed with water and charged beads mimicking counterions. Two ratios of DOPC/DOPG/cholesterol were used, 800:100:100 and 900:0:100, in order to study the influence of negatively charged lipids. The lipid/water mixture was equilibrated over 20 ns using a 20-fs integration time step. The temperature was maintained at 323.15 K using velocity rescaling (54) and a coupling constant of 1 ps. The pressure was maintained at 1 bar using a semi-isotropic Berendsen barostat (55), a coupling constant of 2 ps, and a compressibility of 3e−4 bar−1. Van der Waals interaction was cut off at 12 Å with a shift function applied beyond 9 Å. Electrostatics was cut off at 14 Å with long range electrostatics treated by the particle mesh Ewald method (56, 57) using a grid spacing of ∼1 Å. The dielectric constant was set to 15, in agreement with previous studies (52). The two systems were simulated over 0.5 μs (i.e. until a lipid bilayer at equilibrium was obtained). It should be noted that bilayers were self-assembled in ∼0.1 μs. Second, the resulting bilayers were extracted, and the P414 and P454 peptides were placed at ∼4.8 nm from the bilayer center (i.e. 0.8 nm above the bilayer surface). The systems were fully hydrated and neutralized with charged beads. The final size of the lipid/peptide systems was ∼16 × 20 × 23.5 nm. Each system was equilibrated over 20 ns in conditions identical to the lipid/water mixture and then simulated over 1 μs. Visualization of the trajectories was done with VMD version 1.9.1 (58). Their analysis was carried out with Gromacs version 4.5.5 (50), R version 2.14.1 (59), and the ggplot2 package (60). The peptide structures were generated by MODELLER 9v8 (61, 62).
RESULTS
Membrane Partition of the P454 Peptide
We have investigated two peptides, P414 and P454, corresponding to residues 414–440 and residues 454–484, respectively, numbered from the known sequence of the adenylate cylase (CyaA) toxin. Their amino acid sequences and characteristics are listed under “Materials and Methods.”
We first analyzed the partition from the solution to the membrane of the P414 (z = −4e) and P454 (z = +2.5e) peptides by measuring tryptophan intrinsic fluorescence at 25 °C. We used LUVs and SUVs, respectively, in the absence and in the presence of anionic lipids. Neutral vesicles were made of DOPC/DOPE/cholesterol at a ratio 6:3:1, whereas negatively charged lipid bilayers were made of DOPC/DOPE/DOPG/cholesterol at a ratio of 4:3:2:1. The tryptophan fluorescence of P414 peptide was not affected by the lipid composition or the vesicle size (supplemental Fig. S1). Similarly, the fluorescence of P454 did not change in the presence of neutral SUVs and LUVs (supplemental Fig. S1, A and B). However, the ratio of tryptophan fluorescence changes upon the addition of negatively charged SUVs and LUVs (Fig. 1 and supplemental Fig. S2), indicating partition of P454 from the solution to the membrane in these conditions. Binding of P454 to negatively charged membrane was confirmed by SPR spectroscopy on HPA chips (supplemental Fig. S3). The SPR traces clearly indicate that membrane interaction of P454 is a reversible process, as illustrated by association and dissociation phases at both 1 and 10 μm P454, the range of peptide concentrations studied hereafter. The reversibility of the process indicates that fluorescence data can be used to perform a thermodynamic analysis of the interaction between P454 and membranes.
FIGURE 1.
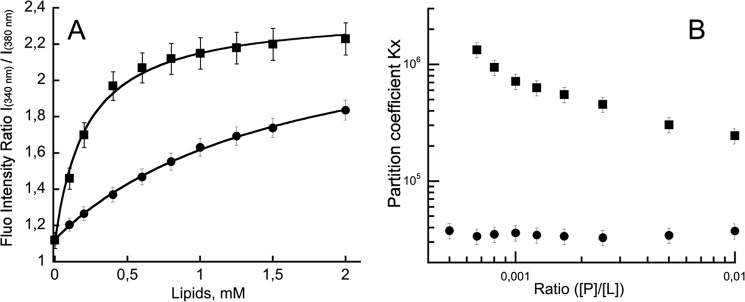
P454 membrane partition followed by fluorescence. A, ratio of fluorescence intensity at 340 and 380 nm as a function of lipid concentration. The negatively charged small unilamellar vesicles (squares) or large unilamellar vesicles (circles) were made of DOPC/DOPE/DOPG/Chol (4:3:2:1). The P454 fluorescence intensity ratio values were fitted as described under “Materials and Methods,” providing partition coefficient KX and the Hill number (KX = 2.9 × 105, nH = 1.35 for SUVs; KX = 3.9 × 104, nH = 1.04 for LUVs). B, partition coefficient KX as a function of peptide/lipid ratio. Thermodynamic parameters are reported in supplemental Table S1. Error bars correspond to S.D.
The partition coefficient KX of P454 for anionic membranes was estimated from the tryptophan intrinsic fluorescence titration curves (Fig. 1A) and at each peptide/lipid ratio (Fig. 1B, [P]/[L]). The partition coefficient of P454 for anionic LUVs is 3.4 104 (ΔG = −6.2 kcal/mol) from the titration curve (Fig. 1A). Similar partition coefficients of P454 for anionic LUVs were calculated at each peptide/lipid ratio (Fig. 1B), indicating that the partition process of the peptide from the solution to the negatively charged LUVs is similar for all tested concentrations.
However, the KX values for P454 from the solution to the anionic SUVs show an anticooperativity trend (34), suggesting that repulsive interactions may occur between P454 peptides at the membrane surface as the peptide concentration increases (Fig. 1B, squares). The KX of P454 for anionic SUVs at infinite dilution (peptide/lipid ratio below 0.001) is around 106, corresponding to ΔG of approximately −8 kcal/mol (supplemental Table S1). At higher peptide/lipid ratios, KX decreases to 2.5 × 105 (ΔG = −7.3 kcal/mol). Similar KX and ΔG values are obtained by a global fitting to the data set (KX = 2.9 105 and ΔG = −7.4 kcal/mol; Fig. 1A).
The differential binding of P454 to anionic SUVs or LUVs might be explained by the weak lateral pressure experienced by the lipid headgroups in SUVs and/or by the occurrence of the first acyl chain methyls in the vicinity of the cis lipid headgroup region in SUVs (63–65). Both effects (i.e. packing default and hydrophobicity) may favor the membrane partition of P454 into SUVs rather than into LUVs due to enhanced hydrophobic effects with SUVs.
We performed the same experiments in the presence of DOPS instead of DOPG (supplemental Fig. S4). Similar results were obtained, indicating that the presence of anionic lipids and not the chemical nature of the lipid headgroup is the critical parameter. Taken together, these data suggest that both hydrophobic forces and attractive electrostatic interactions are required for P454 to partition from the solution to the membrane. In contrast, the P414 peptide, which was also predicted to exhibit membrane interacting features, did not bind to the artificial membranes.
Thermodynamics of P454 Membrane Interactions Followed by ITC
Isothermal titration calorimetry was performed on both P414 and P454 peptides with negatively charged and neutral SUVs at 25 °C to measure the thermodynamic parameters of the peptide membrane interactions (Fig. 2). The binding isotherm titration of P454 peptide by negatively charged SUVs is exothermic, as shown by the negative heat flow pattern (Fig. 2A). On the contrary, P454 titrated by neutral SUVs (off-set black trace in Fig. 2B) and P414 titrated by anionic SUVs (gray trace in Fig. 2B) exhibit almost no heat exchange. Integration of the heat flow for negatively charged SUVs injected into the P454 solution provides an ITC association constant KITC of ∼5500 m−1, giving a KD of ∼180 μm and a ΔGITC of approximately −5.1 kcal/mol. It is noteworthy that conversion of the association constant KITC to the partition coefficient (see “Materials and Methods”) yields a KX of ∼3 × 105 and a ΔG of about −7.4 kcal/mol, values very similar to those obtained by fluorescence titrations (supplemental Figs. S1 and S2).
FIGURE 2.

Isothermal titration calorimetry of peptide membrane partition. The heat flow traces show the calorimetric titrations of P454 (A) and P414 (B, gray traces) by consecutive injections of negatively charged SUVs (DOPC/DOPE/DOPG/Chol, 4:3:2:1). B also shows the heat flow produced by injections of neutral SUVs (DOPC/DOPE/Chol, 6:3:1) into P454 (black traces; offset −0.5 μcal/s in the figure for a better visualization). Each peak corresponds to the injection of 5 μl of 20 mm SUVs into the ITC reaction cell containing 1.41 ml of 42 μm peptide in buffer B at 25 °C. C, integrated binding isotherm of P454 with negative SUVs (filled circles) or with neutral SUVs (empty circles) and P414 with negative SUVs (filled squares). D, heat flow trace of anionic SUVs titrated by a solution of P454. Each peak corresponds to the injection of 5 μl of 85 μm P454 into the ITC reaction cell containing 1.41 ml of 5 mm anionic SUVs in buffer B at 25 °C. Thermodynamic parameters are reported in supplemental Table S2.
We then measured the enthalpy change of P454 upon membrane partitioning in an independent experiment by performing successive injections of P454 into a solution of negatively charged SUVs (66, 67). Integration of the heat flow indicates that the exothermic reaction has an enthalpy change (ΔH) of approximately −3.3 kcal/mol (Fig. 2D and supplemental Table S2). Taken together, ΔGITC and ΔH values from ITC experiments allow us to calculate the entropy change, ΔS, of 5.9 cal/mol/K, corresponding to an entropic contribution to the free energy, −TΔS, of −1.8 kcal/mol (see “Materials and Methods”).
These data indicate that the membrane insertion process of P454 is a spontaneous reaction (i.e. driven by both enthalpic and entropic forces). The main contribution to the ΔG is the exothermic enthalpy change, which accounts for about two-thirds of the free energy (ΔH/ΔG = −3.3/−5.1). This enthalpic contribution may be assigned to the peptide-membrane complex formation followed by acquisition of P454 secondary structure (34) (see below).
The entropic contribution to the free energy, −TΔS, represents about one-third of the free energy (−TΔS/ΔG = −1.8/−5.1) of the membrane insertion process. Such a favorable entropic change suggests that P454 undergoes a partial dehydration upon partition from the solution to the membrane, the released water molecules recovering their free motion in solution. Moreover, both cationic P454 and anionic lipid membrane have on their respective surfaces condensed counterions of opposite signs, which can also be released upon P454 membrane interaction, leading to an entropic gain (68, 69). However, both effects on the entropy (i.e. peptide dehydration upon membrane insertion and electrostatic compensation) are rather low, probably due to the dynamic state of the lipid headgroups in the outer leaflet of SUVs. Taken together, fluorescence and ITC data indicate that the membrane insertion process of P454 is a spontaneous reaction, which involves both hydrophobic effects and electrostatic interactions between the peptide and the membrane.
Secondary Structure Changes of the Peptides Followed by Circular Dichroism
Conformational changes of the peptides induced by the addition of membrane were then investigated by circular dichroism. The far-UV CD spectrum of P414 is typical of a mainly unfolded peptide in solution (supplemental Fig. S5). Its far-UV CD spectrum did not change upon the addition of SUVs up to 2 mm lipid, whatever the lipid composition. P454 is also mainly disordered in solution (Fig. 3A) and exhibited weak changes upon the addition of neutral SUVs up to 2 mm lipids (data not shown). However, the addition of negatively charged SUVs induced significant changes in the far-UV CD spectra of P454, as shown by the strong intensity increase of the n-π* band around 222 nm (Fig. 3A), indicating that P454 acquired secondary structures upon membrane interaction. Unfortunately, high lipid concentrations preclude the use of conventional CD due to vesicle light scattering in the far-UV region, thus preventing the extraction of structural information from the π-π* band (∼190–210 nm).
FIGURE 3.

Circular dichroism and FTIR spectra of P454. A, far-UV CD spectra of P454 (40 μm) in the absence (solid line) or in the presence (dashed lines) of increasing concentrations (0.5, 1, and 2 mm from top to bottom) of anionic SUVs made of DOPC/DOPE/DOPG/cholesterol (4:3:2:1) in buffer B at 25 °C. B, synchrotron radiation CD spectra of P454 (200 μm) in the absence (solid line) or in the presence (dashed line) of anionic SUVs (10 mm) in buffer B at 25 °C. C and D, transmission FTIR spectra and band deconvolutions of P454 (150 μm) in D2O buffer B (C) or in the presence (D) of anionic SUVs (5 mm lipids) in D2O buffer B at 25 °C. The S.D. value is within 5%. Secondary structure content analysis is reported in supplemental Table S3.
The use of synchrotron radiation circular dichroism (SRCD) improves data quality and allows recording of polypeptide CD signal down to 180 nm in the presence of lipid vesicles. The SRCD spectra clearly indicate that P454 adopts an α-helical conformation upon partitioning from solution to anionic membranes (Fig. 3B). Indeed, the far-UV SRCD spectrum of P454 in the presence of negatively charged SUVs exhibits a split π-π* band characteristic of an α-helix conformation, and the maximum wavelength of the n-π* band (222 nm) is typical of α-helix polarity. In solution, P454 is characterized by high structural disorder, as revealed by a poorly resolved π-π* band centered at 200 nm and a weak n-π* band around 220 nm. Estimations of secondary structure contents using the K2D3 prediction server (70) indicate a strong increase of the α-helical content upon binding to anionic SUVs (supplemental Table S3). The far-UV SRCD difference spectrum between the P454 peptide in the presence and in the absence of SUVs, corresponding to the membrane-induced secondary structure content, is typical of an α-helical spectrum (Fig. 3B, inset).
Secondary Structure Changes of P454 and Its Orientation with Respect to the Membrane Plane Followed by FTIR Spectroscopy
We further investigated the secondary structure changes of P454 upon membrane insertion by FTIR spectroscopy. P454 FTIR spectra were obtained by transmission using CaF2 windows in the absence and in the presence of negatively charged SUVs in D2O buffer (Fig. 3, C and D). Analysis of the amide I′ bands (11) (see “Materials and Methods”) of the P454 peptide in both conditions shows that the partition of P454 from the aqueous phase to the membrane environment is characterized by an increase of the helical content at the expense of disordered conformations (Table S4), in agreement with SRCD data.
In order to gain insight into the orientation of the P454 helix in the lipid bilayer, we analyzed infrared spectra of P454 in oriented membrane multilayers. Oriented multilayers of DOPC/DOPE/DOPG/cholesterol (4:3:2:1) membranes were prepared on the surface of a germanium internal reflection element in the presence of P454. Perpendicular and parallel polarized ATR-FTIR infrared spectra were then recorded (Fig. 4, A and B). The amide I′ band is centered at 1655 cm−1, which confirms that P454 is predominantly folded in α-helix in the presence of membranes. The weaker amide II′ band is located at 1547 cm−1, in agreement with an α-helical conformation (44). The two other major bands in this region are assigned to the lipid ester stretch vibration v(C=O) at 1737 cm−1 and to the lipid bending motion δ(CH2) at 1464 cm−1. A qualitative analysis of the secondary structure orientation can be performed by calculating the dichroic spectrum (Fig. 4C) obtained by subtracting the parallel from the perpendicular polarized spectrum corrected by the isodichroic ATR ratio (RATRiso) to take into account the differences in the relative power of the evanescent field (45) (see “Materials and Methods”).
FIGURE 4.

ATR-FTIR spectra of P454. The ATR-FTIR spectra of P454 mixed with lipids (DOPC/DOPE/DOPG/cholesterol, 4:3:2:1) were recorded with perpendicular (top) or parallel (middle) polarization of the infrared light. The resulting dichroic spectrum of P454 in membranes (bottom) is multiplied by a scaling factor of 8 for convenience (see “Materials and Methods”). The S.D. value is within 5%. Secondary structure content analysis is reported in supplemental Table S4.
The negative dichroism of the P454 amide I′ band at 1654 cm−1 as well as the positive amide II′ band at 1547 cm−1 indicates that the α-helical P454 peptide is oriented in parallel to the membrane plane (for details, see “Materials and Methods”). It is noteworthy that the lipid v(C=O) dichroism is resolved into two components at 1745 and 1732 cm−1, indicating that the lipid bilayers are stacked in parallel multilayers, in agreement with previous studies (45). We also calculated the θ angle of the helix axis with respect to the normal to the bilayer plane (see “Materials and Methods”) and found a value of 89 ± 5° (mean of three independent experiments), which confirms that the helix axis of P454 is parallel to the membrane plan.
Coarse Grained Molecular Dynamics of the P454 Peptide
Coarse grained simulations were performed with the two peptides P414 and P454 in the presence of neutral and negatively charged lipid bilayers. In agreement with the previous experiments, most of the P414 peptides remained at the surface of the bilayer, where they were initially deposited (data not shown). Noticeably, the helical axis of most P454 peptides remains almost parallel to the membrane plane after 1 μs of molecular dynamics simulation (Fig. 5), in agreement with the experimental data obtained by polarized ATR-FTIR spectroscopy. Moreover, Fig. 5 suggests that the P454 peptides are located in the lipid headgroup region.
FIGURE 5.
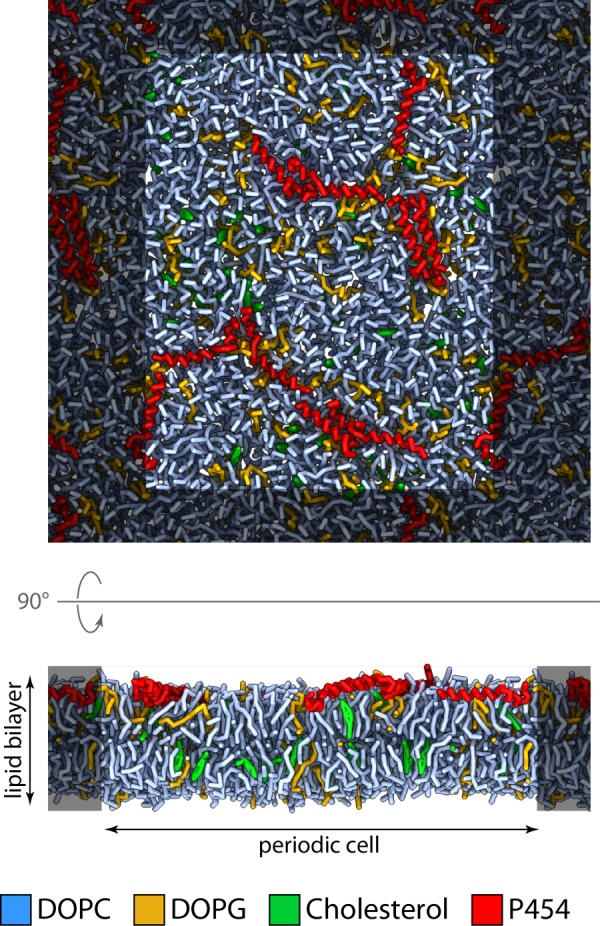
Molecular dynamics of P454 inserted into an anionic lipid bilayer. Snapshot of the system after 1 μs of molecular dynamics simulation at constant temperature (323.15 K) and pressure (1 bar). Red, P454 peptides; light blue, DOPC; green, cholesterol; yellow-orange, DOPG. For clarity, water is not depicted.
The radial distribution function of the lipids around the peptides was calculated to better characterize the direct environment around P454 (Fig. 6). In the presence of DOPG, the first and the highest density peak occurs at ∼0.52 nm from the center of mass of P454 for the three types of membrane compounds (DOPC, DOPG, and cholesterol). This peak corresponds to the direct lipid environment of P454. The peaks of cholesterol and DOPC have approximately the same intensity, whereas the peak of DOPG is about 2.2 times higher. This indicates that DOPG is more densely packed around P454 than DOPC or cholesterol. This in silico observation may be related to the in vitro result showing that anionic lipids are required to induce the partition and the helical folding of P454.
FIGURE 6.
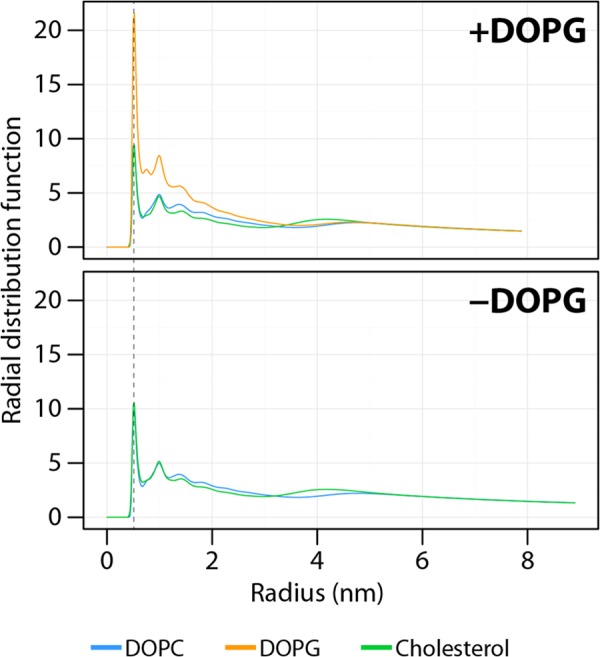
Radial distribution function of the lipids around the center of mass of P454 in the presence or absence of DOPG in the lipid bilayer. The radial distribution function was calculated using the phosphate beads for DOPC and DOPG or the hydroxyl beads for cholesterol. The dashed line indicates the first shell of lipids around the P454 mass center. The small and wide peak at ∼4 nm corresponds to lipids on the opposite side of the bilayer.
LUV Permeabilization
Finally, the ability of the peptides to alter the membrane permeability of vesicles was investigated. The permeabilization of LUVs, containing the co-entrapped probe/quencher ANTS/DPX, was assessed using the fluorescence recovery of ANTS after release of the probe in the extravesicular milieu. As expected, P414, which did not interact with vesicles, did not show any permeabilization activity at concentrations up to 10 μm (data not shown), whereas LUV permeabilization was quickly induced at low concentrations of P454 (Fig. 7A). The presence of anionic lipids greatly enhanced the permeabilization activity of P454 as compared with neutral lipid membranes (Fig. 7A, dashed line). Noteworthy, membrane permeabilization by the P454 peptide showed a concentration-dependent activity (Fig. 7A). For each peptide concentration, a single exponential was fitted to the kinetic traces of permeabilization to determine the maximal amplitude of fluorescence and the observed rate constants (kobs). Supplemental Fig. S6 shows that the maximal amplitude of fluorescence linearly increased with P454 concentration, whereas the kobs values remained roughly similar (0.03 s−1). These results suggest that the vesicle permeabilization process by P454 is similar at each peptide concentration.
FIGURE 7.

Membrane permeabilization by P454. A, time course of ANTS and DPX efflux from negatively charged LUVs (0.45 mm lipids) made of DOPC/DOPE/DOPG/Chol (4:3:2:1 lipid ratio) in the presence of the indicated concentrations of P454 (solid lines). Dashed line, permeabilization of neutral LUVs (DOPC/DOPE/Chol at a 6:3:1 ratio) in the presence of 1 μm P454. B, time course of ANTS and DPX efflux from negatively charged LUVs (0.45 mm lipids) made of DOPC/DOPE/DOPG/Chol (4:3:2:1) in the presence of 0.3 μm P454, AC384, or AC489.
It is noteworthy that the average observed rate constant of dye release by P454 corresponds to a half-permeabilization time of about 20 s, a kinetics much faster than that observed for membrane permeabilization by AC489 (see Fig. 7B). This difference in permeabilization kinetics may be related to the fact that, whereas P454 may immediately permeabilize the membrane as it binds to the vesicles, the AC489 protein may have to undergo conformational changes after membrane partitioning before inducing LUV permeabilization, and this would thus delay the observed dye release.
DISCUSSION
Transport of proteins across cellular membranes is a central process in cell physiology. Many protein toxins from poisonous plants or from pathogenic bacteria are also able to penetrate into the cytosol of their target cells, where they exert their toxic effects (71, 72). Although most of these toxins exploit the cellular machinery of endocytosis and intracellular sorting to gain access to the cell cytosol, a few others, like diphtheria, botulinum, anthrax, and adenylate cyclase (CyaA) toxins, carry their own translocation apparatus (73). These latter toxins constitute excellent model systems to analyze the molecular mechanisms and the physico-chemical principles underlying the membrane insertion process of proteins (74, 75) and transport across biological membranes (76). To date, CyaA is unique among other bacterial toxins in its ability to translocate its catalytic domain directly across the plasma membrane of the target cells from the extracellular milieu to the cytosol. The other toxins are at least partially dependent on endocytosis (like diphtheria and botulinum toxins) or intracellular sorting (ricin) to reach the cytosol (73, 77, 78). How the CyaA catalytic domain crosses the membrane is a fascinating but still unsolved question.
We have previously shown that the region of CyaA encompassing residues 384–489 (see supplemental Figs. S7 and S8 for details on the primary sequence of CyaA) is crucial for the translocation of the catalytic domain into the cytosol (31). Indeed, a recombinant CyaA protein deleted of this region, CyaAΔ375–489, was still able to induce cell hemolysis, but it failed to translocate its catalytic domain across the plasma membrane, as revealed by the loss of cAMP production. We further showed that AC489, a polypeptide encompassing residues 1–489 of CyaA, could partition from the solution to the membrane in vitro and permeabilize lipid bilayers, whereas the isolated catalytic domain, AC384 (residues 1–384 of CyaA) could not. Finally, we showed that AC489 could not translocate its catalytic domain in vivo, indicating that the C-terminal part of CyaA (i.e. residues 490–1706) is required to deliver the catalytic domain into the cytosol. We proposed that although the enzymatic activity is confined to residues 1–384, the translocation module per se extends from residue 1 to 489 of CyaA. Unfortunately, our attempts to produce the CyaA region 384–489 in order to characterize more precisely its membrane binding and permeabilization capabilities were unsuccessful (31).
As an alternative strategy, described in the present work, we attempted to identify amino acid segments within this CyaA region that might exhibit membrane-interacting properties with the aim of characterizing the biophysical properties of the corresponding synthetic peptides. We hypothesized that regions able to partition from solution to membrane should exhibit some canonical features like secondary structure propensity and amphiphilicity (79, 80). Two regions with potential membrane-interacting properties were thus identified, located from residue 414 to 440 and from residue 454 to 484. Two synthetic peptides, corresponding to these two amino acid sequences, P414 and P454, respectively, were obtained by chemical synthesis and characterized by a combination of biophysical approaches.
Fluorescence, SPR, and ITC data indicate that P414 does not interact with membranes, whereas P454 exhibits a strong propensity to interact with membranes containing negatively charged lipids. The presence of anionic lipids was crucial for the partitioning of P454 from the solution to the membrane, as reflected by the partition coefficient and the ΔG values obtained by fluorescence and ITC experiments, although the precise chemical nature of the anionic lipid head group was not important. Coarse grained molecular dynamics provide further evidence for the role of anionic lipids in membrane partitioning of P454. Indeed, the radial distribution functions of lipids revealed that the anionic DOPG lipids are sequestered around P454. P454 contains two positively charged arginine residues that most likely contribute to the recruitment of anionic lipids by attractive electrostatic effects, thus generating the asymmetric lateral distribution of anionic lipids observed by molecular dynamics.
Synchrotron radiation CD and FTIR spectroscopy studies revealed that P454 is mainly unfolded in solution but adopts an α-helical conformation in the presence of anionic but not in the presence of neutral membranes, indicating that anionic lipids are also mandatory for the secondary structure folding of P454. The orientation of P454 in negatively charged membranes was then investigated by ATR-FTIR and molecular dynamics. Both approaches clearly indicate that the helix axis of P454 is oriented in the plane of the lipid bilayer. ITC experiments further indicate that the P454 membrane partitioning in negatively charged membrane is driven by both entropy and enthalpy contributions. These thermodynamic data suggest that P454 is partially dehydrated and acquires secondary structures upon membrane insertion. Finally, we also showed that P454 permeabilizes lipid vesicles in a concentration-dependent manner in the presence of negatively charged lipids. The strict dependence on anionic lipids for the membrane binding and insertion process of P454 might be explained as follows. Both membrane partitioning and folding steps of P454 should overcome the cost of transferring two positively charged arginine side chains in the vicinity of the membrane. This thermodynamic penalty may be compensated by the presence of negatively charged molecules on the membrane surface, such as anionic lipids. The guanidinium group from the arginine side chains and the negatively charged lipids may act as reciprocal counterions upon P454 membrane insertion. Altogether, our results indicate that P454 (i) binds to negatively charged membranes, (ii) adopts an α-helical structure oriented in plane with respect to the lipid bilayer, and (iii) permeabilizes negatively charged vesicles.
Thus, the segment encompassing residues 454–485 displays properties of membrane-active peptides. We propose that this segment may directly contribute to the membrane insertion/destabilization activity of the AC489 translocating domain. Interestingly, the isolated P454 peptide exhibits a much stronger membrane permeabilization activity than the full AC489 translocating domain but only in the presence of negatively charged lipids. The fact that anionic lipids are required for P454 peptide partitioning, folding, and membrane permeabilization suggests that the membrane-destabilizing activity of this CyaA region might be favored by the occurrence of negatively charged lipids in the outer leaflet of the plasma membrane. Although anionic lipids are usually kept in the inner leaflet of the plasma membrane of healthy cells, it has been reported previously that the full-length CyaA toxin is endowed with a lipid flip-flop activity that probably results from the insertion of the hydrophobic region (residues 525–715) of CyaA in the membrane bilayer (81). This lipid mixing effect may contribute to expose anionic lipids to the outer leaflet of the plasma membrane. Subsequently, the presence of anionic lipids exposed to CyaA in the extracellular medium might trigger the membrane insertion and permeabilization activity of the region encompassing the helix, 454–485. These events may lead to a local disorder of the lipid bilayer in the vicinity of the catalytic domain, favoring the initiation of its translocation across the plasma membrane.
Alternatively, in the context of the full-length CyaA toxin, the membrane binding of the 454–485 helical segment may not be as critically dependent on the presence of anionic lipids as is the membrane binding of the isolated P454 peptide observed here. Indeed, after CyaA binding to its receptor and insertion of the hydrophobic region within the cell membrane, the AC489 N-terminal domain is retained in close proximity to the plasma membrane. This may be thermodynamically sufficient to promote interaction of the 454–485 region with the nearby lipid bilayer and initiate the local destabilization of the membrane required for the translocation of the catalytic domain.
It is noteworthy that P454 exhibits common features with antimicrobial peptides (AMPs) and the so-called cell-penetrating peptides (CPPs). AMPs are components of the innate immune response and constitute potent peptidic agents against bacteria, some fungi, and viruses. They mostly act as membrane pore-forming peptides or by targeting intracellular macromolecules (82). CPPs are short peptides that facilitate cellular uptake of various molecular cargo across the plasma membrane (83–86). They are believed to bind to the cell membrane through electrostatic interactions and hydrophobic forces and to destabilize the lipid bilayer so as to allow the cargo protein to cross the membrane. However, their exact mechanisms of action still remain controversial despite the wide utilization of CPPs in cell biology (87). Most CPPs are derived from protein transduction domains that exhibit the ability to interact with membranes and to traverse various types of mammalian cell membranes. Clearly, P454 does not possess the translocation properties of CPPs. Indeed, we previously showed that AC489 (adenylate cyclase acting as a native cargo for P454) is not able to translocate the enzymatic moiety into eukaryotic cells (31), indicating that P454 is not a protein transduction domain. However, like CPPs and AMPs, P454 exhibits membrane-interacting properties. P454 can be classified in the arginine-rich peptide group, like the TAT and Penetratin CPP (88) or the Magainin and Melittin AMP (89). P454 also shares with membrane-active peptides the property to fold upon membrane interaction (90). Moreover, P454 induces anionic lipid clustering upon membrane interaction and lipid bilayer permeabilization, as observed for many CPPs and AMPs (91–94). Our present results therefore suggest that the CyaA toxin harbors an intrinsic membrane-active segment appended close to the catalytic “cargo” domain that has to be delivered across the plasma membrane into the cytosol of the target cells to exert its biological function.
Acknowledgments
We acknowledge SOLEIL for provision of synchrotron radiation facilities (Proposal ID 20110586 and 20120444), and we thank Frank Wien for assistance in using the Disco beamline. We thank Sylviane Hoos and Patrick England for help and expertise on ITC. We thank Agnes Ullmann for critical reading of the manuscript.
This work was supported by the Institut Pasteur (Projet Transversal de Recherche, PTR374), CNRS (UMR 3528, Biologie Structurale et Agents Infectieux), and the Agence Nationale de la Recherche, program Jeunes Chercheurs (Grant ANR-09-JCJC-0012).

This article contains supplemental Tables S1–S4 and Figs. S1–S8.
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOPG
- 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- Chol
- cholesterol
- ANTS
- 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt
- DPX
- p-xylene-bis-pyridinium bromide
- LUV
- large unilamellar vesicle
- SUV
- small unilamellar vesicle
- ITC
- isothermal titration calorimetry
- ATR
- attenuated total reflection
- SPR
- surface plasmon resonance
- SRCD
- synchrotron radiation circular dichroism
- AMP
- antimicrobial peptide
- CPP
- cell-penetrating peptide.
REFERENCES
- 1. Ladant D., Brezin C., Alonso J. M., Crenon I., Guiso N. (1986) Bordetella pertussis adenylate cyclase. Purification, characterization, and radioimmunoassay. J. Biol. Chem. 261, 16264–16269 [PubMed] [Google Scholar]
- 2. Glaser P., Ladant D., Sezer O., Pichot F., Ullmann A., Danchin A. (1988) The calmodulin-sensitive adenylate cyclase of Bordetella pertussis. Cloning and expression in Escherichia coli. Mol. Microbiol. 2, 19–30 [PubMed] [Google Scholar]
- 3. Ladant D., Ullmann A. (1999) Bordatella pertussis adenylate cyclase. A toxin with multiple talents. Trends Microbiol. 7, 172–176 [DOI] [PubMed] [Google Scholar]
- 4. Guo Q., Shen Y., Lee Y. S., Gibbs C. S., Mrksich M., Tang W. J. (2005) Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 24, 3190–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo Q., Jureller J. E., Warren J. T., Solomaha E., Florián J., Tang W. J. (2008) Protein-protein docking and analysis reveal that two homologous bacterial adenylyl cyclase toxins interact with calmodulin differently. J. Biol. Chem. 283, 23836–23845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karst J. C., Sotomayor Pérez A. C., Guijarro J. I., Raynal B., Chenal A., Ladant D. (2010) Calmodulin-induced conformational and hydrodynamic changes in the catalytic domain of Bordetella pertussis adenylate cyclase toxin. Biochemistry 49, 318–328 [DOI] [PubMed] [Google Scholar]
- 7. Rose T., Sebo P., Bellalou J., Ladant D. (1995) Interaction of calcium with Bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 270, 26370–26376 [DOI] [PubMed] [Google Scholar]
- 8. Bauche C., Chenal A., Knapp O., Bodenreider C., Benz R., Chaffotte A., Ladant D. (2006) Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 281, 16914–16926 [DOI] [PubMed] [Google Scholar]
- 9. Chenal A., Guijarro J. I., Raynal B., Delepierre M., Ladant D. (2009) RTX calcium binding motifs are intrinsically disordered in the absence of calcium. Implication for protein secretion. J. Biol. Chem. 284, 1781–1789 [DOI] [PubMed] [Google Scholar]
- 10. Szilvay G. R., Blenner M. A., Shur O., Cropek D. M., Banta S. (2009) A FRET-based method for probing the conformational behavior of an intrinsically disordered repeat domain from Bordetella pertussis adenylate cyclase. Biochemistry 48, 11273–11282 [DOI] [PubMed] [Google Scholar]
- 11. Chenal A., Karst J. C., Sotomayor Pérez A. C., Wozniak A. K., Baron B., England P., Ladant D. (2010) Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys. J. 99, 3744–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sotomayor Pérez A. C., Karst J. C., Davi M., Guijarro J. I., Ladant D., Chenal A. (2010) Characterization of the regions involved in the calcium-induced folding of the intrinsically disordered RTX motifs from the Bordetella pertussis adenylate cyclase toxin. J. Mol. Biol. 397, 534–549 [DOI] [PubMed] [Google Scholar]
- 13. Sotomayor-Pérez A. C., Ladant D., Chenal A. (2011) Calcium-induced folding of intrinsically disordered repeat-in-toxin (RTX) motifs via changes of protein charges and oligomerization states. J. Biol. Chem. 286, 16997–17004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sotomayor-Pérez A. C., Subrini O., Hessel A., Ladant D., Chenal A. (2013) Molecular crowding stabilizes both the intrinsically disordered calcium-free state and the folded calcium-bound state of a repeat in toxin (RTX) protein. J. Am. Chem. Soc. 135, 11929–11934 [DOI] [PubMed] [Google Scholar]
- 15. Welch R. A. (1991) Pore-forming cytolysins of Gram-negative bacteria. Mol. Microbiol. 5, 521–528 [DOI] [PubMed] [Google Scholar]
- 16. Coote J. G. (1992) Structural and functional relationships among the RTX toxin determinants of Gram-negative bacteria. FEMS Microbiol. Rev. 8, 137–161 [DOI] [PubMed] [Google Scholar]
- 17. Linhartová I., Bumba L., Mašín J., Basler M., Osička R., Kamanová J., Procházková K., Adkins I., Hejnová-Holubová J., Sadílková L., Morová J., Sebo P. (2010) RTX proteins. A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 34, 1076–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westrop G. D., Hormozi E. K., Da Costa N. A., Parton R., Coote J. G. (1996) Bordetella pertussis adenylate cyclase toxin. ProCyaA and CyaC proteins synthesised separately in Escherichia coli produce active toxin in vitro. Gene 180, 91–99 [DOI] [PubMed] [Google Scholar]
- 19. Glaser P., Sakamoto H., Bellalou J., Ullmann A., Danchin A. (1988) Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J. 7, 3997–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masure H. R., Au D. C., Gross M. K., Donovan M. G., Storm D. R. (1990) Secretion of the Bordetella pertussis adenylate cyclase from Escherichia coli containing the hemolysin operon. Biochemistry 29, 140–145 [DOI] [PubMed] [Google Scholar]
- 21. Guermonprez P., Khelef N., Blouin E., Rieu P., Ricciardi-Castagnoli P., Guiso N., Ladant D., Leclerc C. (2001) The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the α(M)β(2) integrin (CD11b/CD18). J. Exp. Med. 193, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guermonprez P., Fayolle C., Rojas M. J., Rescigno M., Ladant D., Leclerc C. (2002) In vivo receptor-mediated delivery of a recombinant invasive bacterial toxoid to CD11c + CD8 α-CD11b high dendritic cells. Eur. J. Immunol. 32, 3071–3081 [DOI] [PubMed] [Google Scholar]
- 23. Basler M., Knapp O., Masin J., Fiser R., Maier E., Benz R., Sebo P., Osicka R. (2007) Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 282, 12419–12429 [DOI] [PubMed] [Google Scholar]
- 24. Fiser R., Masín J., Basler M., Krusek J., Spuláková V., Konopásek I., Sebo P. (2007) Third activity of Bordetella adenylate cyclase (AC) toxin-hemolysin. Membrane translocation of AC domain polypeptide promotes calcium influx into CD11b+ monocytes independently of the catalytic and hemolytic activities. J. Biol. Chem. 282, 2808–2820 [DOI] [PubMed] [Google Scholar]
- 25. Ladant D., Michelson S., Sarfati R., Gilles A. M., Predeleanu R., Bârzu O. (1989) Characterization of the calmodulin-binding and of the catalytic domains of Bordetella pertussis adenylate cyclase. J. Biol. Chem. 264, 4015–4020 [PubMed] [Google Scholar]
- 26. Sakamoto H., Bellalou J., Sebo P., Ladant D. (1992) Bordetella pertussis adenylate cyclase toxin. Structural and functional independence of the catalytic and hemolytic activities. J. Biol. Chem. 267, 13598–13602 [PubMed] [Google Scholar]
- 27. Vojtova J., Kamanova J., Sebo P. (2006) Bordetella adenylate cyclase toxin. A swift saboteur of host defense. Curr. Opin. Microbiol. 9, 69–75 [DOI] [PubMed] [Google Scholar]
- 28. Uribe K. B., Etxebarria A., Martín C., Ostolaza H. (2013) Calpain-mediated processing of adenylate cyclase toxin generates a cytosolic soluble catalytically active N-terminal domain. PLoS One 8, e67648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karimova G., Fayolle C., Gmira S., Ullmann A., Leclerc C., Ladant D. (1998) Charge-dependent translocation of Bordetella pertussis adenylate cyclase toxin into eukaryotic cells. Implication for the in vivo delivery of CD8(+) T cell epitopes into antigen-presenting cells. Proc. Natl. Acad. Sci. U.S.A. 95, 12532–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Osicková A., Osicka R., Maier E., Benz R., Sebo P. (1999) An amphipathic α-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J. Biol. Chem. 274, 37644–37650 [PubMed] [Google Scholar]
- 31. Karst J. C., Barker R., Devi U., Swann M. J., Davi M., Roser S. J., Ladant D., Chenal A. (2012) Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 287, 9200–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holubova J., Kamanova J., Jelinek J., Tomala J., Masin J., Kosova M., Stanek O., Bumba L., Michalek J., Kovar M., Sebo P. (2012) Delivery of large heterologous polypeptides across the cytoplasmic membrane of antigen-presenting cells by the Bordetella RTX hemolysin moiety lacking the adenylyl cyclase domain. Infect. Immun. 80, 1181–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sotomayor-Pérez A. C., Karst J. C., Ladant D., Chenal A. (2012) Mean net charge of intrinsically disordered proteins. Experimental determination of protein valence by electrophoretic mobility measurements. Methods Mol. Biol. 896, 331–349 [DOI] [PubMed] [Google Scholar]
- 34. White S. H., Wimley W. C., Ladokhin A. S., Hristova K. (1998) Protein folding in membranes. Determining energetics of peptide-bilayer interactions. Methods Enzymol. 295, 62–87 [DOI] [PubMed] [Google Scholar]
- 35. Béven L., Adenier H., Kichenama R., Homand J., Redeker V., Le Caer J. P., Ladant D., Chopineau J. (2001) Ca2+-myristoyl switch and membrane binding of chemically acylated neurocalcins. Biochemistry 40, 8152–8160 [DOI] [PubMed] [Google Scholar]
- 36. Roy M. O., Pugniere M., Jullien M., Chopineau J., Mani J. C. (2001) Study of hydrophobic interactions between acylated proteins and phospholipid bilayers using BIACORE. J. Mol. Recognit. 14, 72–78 [DOI] [PubMed] [Google Scholar]
- 37. Goormaghtigh E., Cabiaux V., Ruysschaert J. M. (1994) Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Subcell. Biochem. 23, 405–450 [DOI] [PubMed] [Google Scholar]
- 38. Goormaghtigh E., Cabiaux V., Ruysschaert J. M. (1994) Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. I. Assignments and model compounds. Subcell. Biochem. 23, 329–362 [DOI] [PubMed] [Google Scholar]
- 39. Marsh D. (1999) Quantitation of secondary structure in ATR infrared spectroscopy. Biophys. J. 77, 2630–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barth A. (2000) The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 74, 141–173 [DOI] [PubMed] [Google Scholar]
- 41. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 42. Barth A., Zscherp C. (2002) What vibrations tell us about proteins. Q. Rev. Biophys. 35, 369–430 [DOI] [PubMed] [Google Scholar]
- 43. Tatulian S. A. (2013) Structural characterization of membrane proteins and peptides by FTIR and ATR-FTIR spectroscopy. Methods Mol. Biol. 974, 177–218 [DOI] [PubMed] [Google Scholar]
- 44. Goormaghtigh E., Raussens V., Ruysschaert J. M. (1999) Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1422, 105–185 [DOI] [PubMed] [Google Scholar]
- 45. Bechinger B., Ruysschaert J. M., Goormaghtigh E. (1999) Membrane helix orientation from linear dichroism of infrared attenuated total reflection spectra. Biophys. J. 76, 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Güldenhaupt J., Rudack T., Bachler P., Mann D., Triola G., Waldmann H., Kötting C., Gerwert K. (2012) N-Ras forms dimers at POPC membranes. Biophys. J. 103, 1585–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tamm L. K., Tatulian S. A. (1997) Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 30, 365–429 [DOI] [PubMed] [Google Scholar]
- 48. le Coutre J., Narasimhan L. R., Patel C. K., Kaback H. R. (1997) The lipid bilayer determines helical tilt angle and function in lactose permease of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 94, 10167–10171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vinchurkar M. S., Chen K. H., Yu S. S., Kuo S. J., Chiu H. C., Chien S. H., Chan S. I. (2004) Polarized ATR-FTIR spectroscopy of the membrane-embedded domains of the particulate methane monooxygenase. Biochemistry 43, 13283–13292 [DOI] [PubMed] [Google Scholar]
- 50. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4. Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 51. Marrink S. J., de Vries A. H., Mark A. E. (2004) Coarse grained model for semi-quantitative lipid simulations. J. Phys. Chem. B 108, 750–760 [Google Scholar]
- 52. Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H. (2007) The MARTINI force field. Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 [DOI] [PubMed] [Google Scholar]
- 53. Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., Marrink S.-J. (2008) The MARTINI coarse-grained force field. Extension to proteins. J. Chem. Theory Comput. 4, 819–834 [DOI] [PubMed] [Google Scholar]
- 54. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101–014107 [DOI] [PubMed] [Google Scholar]
- 55. Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 56. Darden T., York D., Pedersen L. (1993) Particle Mesh Ewald. An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 57. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method,. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 58. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 59. R Development Core Team (2005) R: A Language and Environment for Statistical Computing, version 2.14.1, R Foundation for Statistical Computing, Vienna [Google Scholar]
- 60. Wickham H. (2009) ggplot2: Elegant Graphics for Data Analysis, Springer, New York [Google Scholar]
- 61. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 62. Eswar N., Marti-Renom M. A., Webb B., Madhusudhan M. S., Eramian D., Shen M., Pieper U., Sali A. (2006) Current Protocols in Bioinformatics, pp. 5.6.1–5.6.30, John Wiley & Sons, Inc., New York: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chenal A., Vernier G., Savarin P., Bushmarina N. A., Gèze A., Guillain F., Gillet D., Forge V. (2005) Conformational states and thermodynamics of α-lactalbumin bound to membranes. A case study of the effects of pH, calcium, lipid membrane curvature and charge. J. Mol. Biol. 349, 890–905 [DOI] [PubMed] [Google Scholar]
- 64. Chiantia S., London E. (2012) Acyl chain length and saturation modulate interleaflet coupling in asymmetric bilayers. Effects on dynamics and structural order. Biophys. J. 103, 2311–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chiantia S., Klymchenko A. S., London E. (2012) A novel leaflet-selective fluorescence labeling technique reveals differences between inner and outer leaflets at high bilayer curvature. Biochim. Biophys. Acta 1818, 1284–1290 [DOI] [PubMed] [Google Scholar]
- 66. Wenk M. R., Seelig J. (1998) Magainin 2 amide interaction with lipid membranes. Calorimetric detection of peptide binding and pore formation. Biochemistry 37, 3909–3916 [DOI] [PubMed] [Google Scholar]
- 67. Wieprecht T., Apostolov O., Seelig J. (2000) Binding of the antibacterial peptide magainin 2 amide to small and large unilamellar vesicles Biophys. Chem. 85, 187–198 [DOI] [PubMed] [Google Scholar]
- 68. Jeppesen B., Smith C., Gibson D. F., Tait J. F. (2008) Entropic and enthalpic contributions to annexin V-membrane binding. A comprehensive quantitative model. J. Biol. Chem. 283, 6126–6135 [DOI] [PubMed] [Google Scholar]
- 69. Tolokh I. S., Vivcharuk V., Tomberli B., Gray C. G. (2009) Binding free energy and counterion release for adsorption of the antimicrobial peptide lactoferricin B on a POPG membrane. Phys. Rev. E 80, 031911 [DOI] [PubMed] [Google Scholar]
- 70. Louis-Jeune C., Andrade-Navarro M. A., Perez-Iratxeta C. (2011) Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 80, 374–381 [DOI] [PubMed] [Google Scholar]
- 71. Sandvig K., van Deurs B. (2005) Delivery into cells. Lessons learned from plant and bacterial toxins. Gene. Ther. 12, 865–872 [DOI] [PubMed] [Google Scholar]
- 72. Watson P., Spooner R. A. (2006) Toxin entry and trafficking in mammalian cells. Adv. Drug Deliv. Rev. 58, 1581–1596 [DOI] [PubMed] [Google Scholar]
- 73. Falnes P. O., Sandvig K. (2000) Penetration of protein toxins into cells. Curr. Opin. Cell Biol. 12, 407–413 [DOI] [PubMed] [Google Scholar]
- 74. Chenal A., Prongidi-Fix L., Perier A., Aisenbrey C., Vernier G., Lambotte S., Haertlein M., Dauvergne M. T., Fragneto G., Bechinger B., Gillet D., Forge V., Ferrand M. (2009) Deciphering membrane insertion of the diphtheria toxin T domain by specular neutron reflectometry and solid-state NMR spectroscopy. J. Mol. Biol. 391, 872–883 [DOI] [PubMed] [Google Scholar]
- 75. Galloux M., Vitrac H., Montagner C., Raffestin S., Popoff M. R., Chenal A., Forge V., Gillet D. (2008) Membrane Interaction of botulinum neurotoxin A translocation (T) domain. The belt region is a regulatory loop for membrane interaction. J. Biol. Chem. 283, 27668–27676 [DOI] [PubMed] [Google Scholar]
- 76. Sandvig K., Skotland T., van Deurs B., Klokk T. I. (2013) Retrograde transport of protein toxins through the Golgi apparatus. Histochem. Cell Biol. 140, 317–326 [DOI] [PubMed] [Google Scholar]
- 77. Chenal A., Nizard P., Gillet D. (2002) Structure and function of diphtheria toxin. From pathology to engineering. J. Toxicol. Tox. Rev. 21, 321–359 [Google Scholar]
- 78. Reig N., van der Goot F. G. (2006) About lipids and toxins. FEBS Lett. 580, 5572–5579 [DOI] [PubMed] [Google Scholar]
- 79. White S. H., Wimley W. C. (1999) Membrane protein folding and stability. Physical principles. Annu. Rev. Biophys. Biomol. Struct. 28, 319–365 [DOI] [PubMed] [Google Scholar]
- 80. Montagner C., Perier A., Pichard S., Vernier G., Ménez A., Gillet D., Forge V., Chenal A. (2007) Behavior of the N-terminal helices of the diphtheria toxin T domain during the successive steps of membrane interaction. Biochemistry 46, 1878–1887 [DOI] [PubMed] [Google Scholar]
- 81. Martín C., Requero M. A., Masin J., Konopasek I., Goñi F. M., Sebo P., Ostolaza H. (2004) Membrane restructuring by Bordetella pertussis adenylate cyclase toxin, a member of the RTX toxin family. J. Bacteriol. 186, 3760–3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Diamond G., Beckloff N., Weinberg A., Kisich K. O. (2009) The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 15, 2377–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nakase I., Tanaka G., Futaki S. (2013) Cell-penetrating peptides (CPPs) as a vector for the delivery of siRNAs into cells. Mol. Biosyst. 9, 855–861 [DOI] [PubMed] [Google Scholar]
- 84. Trabulo S., Cardoso A. L., Cardoso A. M., Morais C. M., Jurado A. S., Pedroso de Lima M. C. (2013) Cell-penetrating peptides as nucleic acid delivery systems. From biophysics to biological applications. Curr. Pharm. Des. 19, 2895–2923 [DOI] [PubMed] [Google Scholar]
- 85. Lindgren M., Langel U. (2011) Classes and prediction of cell-penetrating peptides. Methods Mol. Biol. 683, 3–19 [DOI] [PubMed] [Google Scholar]
- 86. Johnson R. M., Harrison S. D., Maclean D. (2011) Therapeutic applications of cell-penetrating peptides. Methods Mol. Biol. 683, 535–551 [DOI] [PubMed] [Google Scholar]
- 87. Bechara C., Sagan S. (2013) Cell-penetrating peptides. 20 years later, where do we stand? FEBS Lett. 587, 1693–1702 [DOI] [PubMed] [Google Scholar]
- 88. Terrone D., Sang S. L., Roudaia L., Silvius J. R. (2003) Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry 42, 13787–13799 [DOI] [PubMed] [Google Scholar]
- 89. Bechinger B., Lohner K. (2006) Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta 1758, 1529–1539 [DOI] [PubMed] [Google Scholar]
- 90. Caesar C. E., Esbjörner E. K., Lincoln P., Nordén B. (2006) Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques. Correlations of structure to cellular uptake. Biochemistry 45, 7682–7692 [DOI] [PubMed] [Google Scholar]
- 91. Tiriveedhi V., Butko P. (2007) A fluorescence spectroscopy study on the interactions of the TAT-PTD peptide with model lipid membranes. Biochemistry 46, 3888–3895 [DOI] [PubMed] [Google Scholar]
- 92. Ziegler A., Seelig J. (2008) Binding and clustering of glycosaminoglycans. A common property of mono- and multivalent cell-penetrating compounds. Biophys. J. 94, 2142–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bobone S., Piazzon A., Orioni B., Pedersen J. Z., Nan Y. H., Hahm K. S., Shin S. Y., Stella L. (2011) The thin line between cell-penetrating and antimicrobial peptides. The case of Pep-1 and Pep-1-K. J. Pept. Sci. 17, 335–341 [DOI] [PubMed] [Google Scholar]
- 94. Wadhwani P., Epand R. F., Heidenreich N., Bürck J., Ulrich A. S., Epand R. M. (2012) Membrane-active peptides and the clustering of anionic lipids. Biophys. J. 103, 265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]