

Background: Kinesin-1 motor domain phosphorylation has been linked to impaired transport in axons.
Results: A mechanism by which phosphorylation could affect transport is proposed.
Conclusion: Phosphorylation decreases the stall force of kinesin and stabilizes autoinhibition.
Significance: Kinesin phosphorylation could be used to fine tune the direction of cargo transport and contribute to pathology in neurodegenerative disease.
Keywords: Jun N-terminal Kinase (JNK), Kinesin, Molecular Motors, Phosphorylation, Single-molecule Biophysics
Abstract
Disruptions in microtubule motor transport are associated with a variety of neurodegenerative diseases. Post-translational modification of the cargo-binding domain of the light and heavy chains of kinesin has been shown to regulate transport, but less is known about how modifications of the motor domain affect transport. Here we report on the effects of phosphorylation of a mammalian kinesin motor domain by the kinase JNK3 at a conserved serine residue (Ser-175 in the B isoform and Ser-176 in the A and C isoforms). Phosphorylation of this residue has been implicated in Huntington disease, but the mechanism by which Ser-175 phosphorylation affects transport is unclear. The ATPase, microtubule-binding affinity, and processivity are unchanged between a phosphomimetic S175D and a nonphosphorylatable S175A construct. However, we find that application of force differentiates between the two. Placement of negative charge at Ser-175, through phosphorylation or mutation, leads to a lower stall force and decreased velocity under a load of 1 piconewton or greater. Sedimentation velocity experiments also show that addition of a negative charge at Ser-175 favors the autoinhibited conformation of kinesin. These observations imply that when cargo is transported by both dynein and phosphorylated kinesin, a common occurrence in the cell, there may be a bias that favors motion toward the minus-end of microtubules. Such bias could be used to tune transport in healthy cells when properly regulated but contribute to a disease state when misregulated.
Introduction
Cells use many mechanisms to regulate motor-based transport on microtubules (1). Cargo, tracks, and motors can all be modified to control the flow of traffic within the cell. Cargo attachment is regulated by a variety of scaffolding proteins that can specifically link cargo to a certain motor species (2). Microtubule tracks can be post-translationally modified (3–5) or coated with tau and other microtubule-associated proteins (MAPs) (6–8), which modify transport. In addition, both kinesin and dynein motors may be modified. Dynein, for example, is regulated by a large number of activators, which bind to the dynein heavy chains and modulate transport. Phosphorylation of kinesin-1 light chains is another case that affects the association of cargo with the motor (9–12).
Recent studies imply that the motor domain of kinesin-1 may also play a role in impaired fast axonal transport. In particular, phosphorylation at serine 175/176 via c-Jun N-terminal kinase-3 (referred to as JNK3) (13–17) is associated with Huntington disease and spinal and bulbar muscular atrophy. JNK3 activation is known to be up-regulated in the presence of pathogenic huntingtin or androgen receptor (15, 16). When activated, JNK3 specifically phosphorylates Ser-175 of kinesin-1B, or Ser-176 in kinesin-1A and 1C. Morfini et al. found that perfusion of mutant huntingtin in squid axoplasm results in an increase in JNK3 activation that is accompanied by a decrease in fast axonal transport (15, 16). JNK3 activation can also modify axonal transport by enhancement of the interaction of the JNK-interacting protein Sunday Driver with dynactin following axonal damage (18). For our current studies, we choose to focus on the effect of kinesin-1B phosphorylation.
There are many unanswered questions regarding the mechanism by which Ser-175 modification leads to impaired transport. First, it is not known whether the addition of negative charge due to phosphorylation of kinesin is sufficient to cause impaired cargo transport. Second, it is also unknown what biochemical and biophysical properties of kinesin are modified as a result of Ser-175 phosphorylation. Third, the effect of an altered charge at position 175, which is in the motor domain, cannot be readily inferred by its location on the motor domain (Fig. 1). Specifically, residue 175 does not fall within the microtubule-binding domain or the ATP-binding domain of kinesin (19, 20). Computational and experimental studies of kinesin-microtubule interactions have traditionally focused on mutational studies in which alanine is used to replace positively charged residues in the motor head, but the effect of adding an extra negative charge has not been investigated thoroughly (19, 21). Our investigation answers each of these questions.
FIGURE 1.

Location of serine 175/176. Serine 176 is highlighted in red in the crystal structure of dimeric rat kinesin. The microtubule-binding domain is color-coded yellow, and the ATP-binding site shown in green (Protein Data Bank ID code 3KIN (39)).
To investigate the effect of phosphorylation of serine 175 on kinesin transport, we used optical trapping and single-molecule fluorescence imaging to probe the behavior of individual mouse kinesin-1 molecules. We created a phosphomimetic construct, S175D, in which an aspartic acid residue mimics the negative charge of phosphate. We also created a nonphosphorylatable alanine mutant, S175A. We performed experiments with both full-length protein and a truncated construct lacking residues 889–963 that make up the autoinhibitory C-terminal domain on kinesin. The truncated kinesin (K888)2 allowed us to probe the effect of Ser-175 modification in the absence of autoinhibition (22). In addition to the mutation studies, we also used JNK3 to directly phosphorylate full-length kinesin. We found that both phosphorylation and the S175D mutation attenuated the motor stall force, stabilized the autoinhibited conformation, and biased cargo transport toward the minus-end of microtubules. A force-dependent decrease in velocity coupled with stabilized autoinhibition could play a role in the impaired axonal transport associated with Huntington disease, as well as be a general mechanism for tuning microtubule-motor transport in healthy neurons.
EXPERIMENTAL PROCEDURES
Kinesin Plasmid Construction
Kinesin-888 (K888) heavy chain mutants were created by site-directed mutagenesis with primers CGT TTC GTG TGT GCT CCA GAT GAA GTC A for S175A and CGT TTC GTG TGT GAT CCA GAT GAA GTC A for S175D. The mutations were made to a pAcSG2 baculovirus transfer vector containing the mouse KIF5B kinesin heavy chain, truncated at alanine 888 with a C-terminal biotin tag followed by a FLAG epitope, described previously (22). To create a full-length heavy chain, a N-terminal His6 tag was cloned to mouse KIF5B gene. A C-terminal yellow fluorescent protein was cloned to mouse light chain kinesin. For the K432 construct, a C-terminal His tag and a biotin tag were added to a copy of the human kinesin heavy chain gene that had been truncated to contain the first 432 residues, and the entire gene was inserted into a pET21a vector.
Kinesin Expression and Purification
Full-length and K888 constructs were expressed and purified using a baculovirus expression system with Sf9 cells as described previously (22). Briefly, Sf9 cells were co-infected with baculovirus coding for YFP-tagged light chain and either K888 or full-length kinesin heavy chain. Cells were grown in suspension for 72 h, lysed by sonication, and clarified by centrifugation at 200,000 × g for 30 min. Full-length kinesin dimers were applied to a His-Select nickel affinity column (Sigma-Aldrich) and washed with 30 mm imidazole to elute the His-tagged kinesin. K888 constructs were applied to a FLAG affinity resin column (Sigma) and eluted with FLAG peptide (Sigma). Both eluted full-length and K888 fractions were concentrated and dialyzed against 10 mm HEPES, pH 7.3, 200 mm NaCl, 50% glycerol, 1 mm DTT, 10 μm MgATP, and 1 μg/ml leupeptin and stored at −20 °C.
The truncated K432 was purified according to a protocol developed by Qiagen (23). BL-21 (DE3) Escherichia coli cells were transfected with the vector and plated on Luria Broth (LB) agar with ampicillin for selection. A single colony from a plate was used to inoculate 20 ml of LB containing 100 μg/ml ampicillin and incubated at 37 °C overnight in a shaker at 200 rpm. The next day the overnight culture was diluted 1:50 into 1 liter of LB containing ampicillin and grown at 37 °C in a shaker at 300 rpm until an A600 of 0.6 was reached. At A600 of 0.6, expression was induced by adding isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.25 mm. The cells were grown for another 4–5 h and then harvested by centrifugation at 4000 × g for 20 min at 4 °C. The cell pellets were frozen and stored overnight at −80 °C. The next day, the pellets were thawed and purified using a nickel-nitrilotriacetic acid resin (Qiagen, 30210) according to protocol 9 in the QIAexpressionist handbook (23). MgATP was added to all buffers to a final concentration of 40 μm. Eluted fractions enriched in protein were pooled, and 10% sucrose was added as a cryoprotectant. Aliquots were flash frozen in liquid nitrogen and stored at −80 °C.
Dynein Preparation
Purified bovine brain dynein was prepared according to a previously published protocol (24). In accordance with the protocol, bovine brains were homogenized and clarified by centrifugation at 230,000 × g. The supernatant of the centrifugation was flowed over an SP-Sepharose column (GE Healthcare) to separate dynein from other cytoplasmic components. The protein-enriched fractions from the SP-Sepharose column were then added to 10–40% sucrose gradients and centrifuged for 17 h at 140,000 × g. Fractions enriched in dynein were run over a Mono Q column. (GE Healthcare) Dynein was eluted in a buffer consisting of 35 mm Tris, 5 mm MgSO4, pH 7.2, and 1 m KCl. After elution, 0.5 mm ATP and 1.0 mm DTT were added to dynein-enriched fractions. These factions were mixed in a 1:1 ratio with a buffer containing 2.5 m sucrose, 35 mm Tris, 5 mm MgSO4, pH 7.2, and flash frozen for storage at −80 °C.
Microtubule and Axoneme Preparation
Unmodified bovine brain tubulin (Cytoskeleton HTS02), biotinylated porcine brain tubulin (Cytoskeleton T333P), and fluorescent, HiLyte tubulin (Cytoskeleton TL488M) were polymerized in a 25:5:1 ratio in BRB-80 (80 mm PIPES, 1 mm EGTA, 1 mm MgCl2, pH 6.9) containing 1 mm DTT, 1 mm GTP, 25% glycerol, and a final tubulin concentration of 3.1 mg/ml. For microtubule-activated ATPase assay experiments, the biotinylated tubulin and fluorescent tubulin were omitted. The reagents were mixed on ice and then incubated at 37 °C for 30 min to induce polymerization. After polymerization, the sample was diluted with a 4× volume of Taxol solution consisting of room temperature 1 mm GTP and 20 μm Taxol in BRB-80. To remove unpolymerized tubulin monomers, the mixture was centrifuged at 15,000 × g for 30 min at 24 °C. The supernatant was discarded, and the pellet of tubulin polymers was resuspended in Taxol solution.
Axonemes were prepared according to a protocol adapted from Gibbons and Fonk by Pierce and Vale (25). Sea urchins were obtained from the Point Loma Marine Invertebrate Laboratory.
JNK3 Phosphorylation
JNK3 enzyme was purchased from Millipore (JNK3/SAPK1b, 14-501). The concentrated enzyme was diluted to a concentration of 0.1 μg/μl in a buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1 mm EGTA, 270 mm sucrose, and 0.1% 2-mercaptoethanol prior to phosphorylation. Two phosphorylation reaction mixtures were prepared. One large (100 μl) reaction was prepared with no radioactive ATP for use in subsequent experiments. This mixture consisted of 6 μm kinesin, 1 mm ATP, 0.07 μg/μl JNK3 and was mixed on ice in HEM buffer (50 mm HEPES, 1 mm EGTA, 2 mm MgSO4). A smaller radiolabeling quantification reaction with a total volume of 5 μl was also prepared. This reaction was identical in composition to the nonradioactive mixture, but contained an additional 70 nm [γ-32P]ATP (PerkinElmer Life Sciences, NEG502Z250UC). The [γ-32P]ATP had an activity of 6000 Ci/mmol at the time of purchase, and reactions were run within a month of the purchase date. Both mixtures were incubated for 30 min at 37 °C. Sucrose was added to the nonradioactive mixture to a final concentration of 10%, and the resulting aliquots were flash frozen. A 4–20% SDS-polyacrylamide gel of 2-μl fractions the radiolabeled mixture was run at 200 V for 20 min to separate the kinesin from free, excess [γ-32P]ATP and JNK3. After this initial run, a [γ-32P]ATP dilution series was added to the gel and run for an additional 5 min. The gel was exposed on a PhosphorImager plate overnight, and the resulting images were scanned. The intensity of the kinesin and ATP standard bands were measured and compared with determine the efficiency of phosphorylation (see Fig. 3).
FIGURE 3.
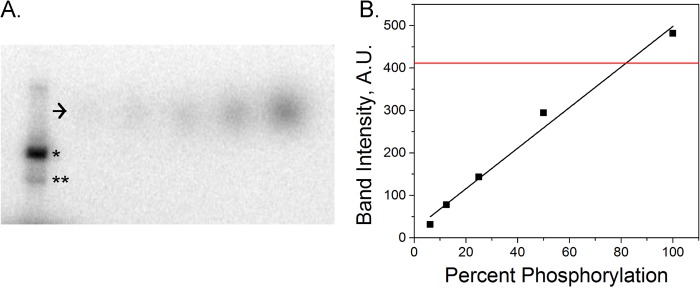
Estimation of phosphorylation efficiency using radiolabeling and SDS-PAGE. Phosphorylation was measured using radiolabeling. Kinesin was phosphorylated by JNK3 in the presence of [γ-32P]ATP. The result of the reaction was run through an SDS-polyacrylamide gel to separate kinesin from JNK3, which autophosphorylates. The resulting intensity of the kinesin band of known concentration was compared with bands of [γ-32P]ATP standards of known concentration to calculate the percentage of kinesin that had been phosphorylated. A, SDS-PAGE gel of radiolabeled kinesin-1. *, [γ-32P]ATP standards corresponding to labeling efficiencies varying from 6 to 100% (arrow). JNK3 also autophosphorylates (**). B, ATP standard band intensity as a function of P32 labeling of kinesin (black line) and kinesin band intensity (red line).
Analytical Ultracentrifugation
The sedimentation coefficient of expressed full-length mouse kinesin (WT and S175D) was determined using an Optima XL-I analytical ultracentrifuge (Beckman Coulter). Sedimentation velocity runs were performed in the An60Ti rotor at 35,000 rpm and 20 °C, in 10 mm imidazole, pH 7.5, 2 mm MgCl2, 1 mm DTT, and varying potassium chloride concentrations. Sedimentation coefficients were determined by curve fitting to one species, using the dc/dt program (26). Sedimentation values were corrected for density and viscosity of the solvent.
Single-molecule Motility Assay
Flow chambers were prepared by attaching coverglasses to glass slides with double-sided tape. Axonemes were flowed over a glass flow chamber and incubated at 4 °C for 20 min. The chamber was washed with dynein motility buffer (DMB) which consisted of 30 mm HEPES, 50 mm potassium acetate, 2 mm magnesium acetate, and 1 mm EGTA at pH 7.2 to which 8 mg/ml BSA (DMB-BSA) and 1 mg/ml casein had been added for blocking. Kinesin protein was diluted to 0.8 μm in BRB-80 containing 10 mg/ml BSA and 10 mm DTT. 0.5 μl was mixed with 0.5 μl of a 2 μm solution of 655 streptavidin-coated quantum dots (Invitrogen, Q10021MP) and incubated at 4 °C for 30 min. After 30 min, the kinesin and quantum dot mixture was diluted 1:5 into 100 μm biotin in BRB-80 and incubated for 10 min on ice. Meanwhile, the imaging buffer was prepared. This consisted of DMB-BSA containing 1 mg/ml 3,4-dihydroxybenzoic acid (Fluka 37580), 100 nm protocatechuate 3,4-dioxygenase (Sigma P8279), 2 mm creatine phosphate, 5 units/ml creatine kinase, 10 mm DTT, and varying ATP concentrations from 10 μm to 1 mm. The quantum dot-kinesin mixture was diluted 1:100 into the imaging buffer and flowed into the chamber. Motor-coated quantum dots were tracked using custom-written Matlab software and kinetic parameters from ATPase activity data by fitting the average quantum dot velocity as a function of ATP concentration to a Michaelis-Menten curve in Matlab.
Microtubule-activated ATPase Assay
ATPase activity was assayed using an ATPase Kinetic ELIPA Assay Kit from Cytoskeleton, Inc (BK051). A starting ELIPA mix was made consisting of 100 μl of the ELIPA reaction buffer with 20 μm Taxol, 24 μl of 1 nm 2-amino-6-mercapto-7-methylpurine ribonucleoside, and 1.2 μl of 100 units/ml purine nucleotide phosphorylase. This starting ELIPA mix was combined with 50 nm kinesin, 0.5 mm ATP and 0.1–8 mm microtubules. The absorbance at 360 nm was recorded at 3-s intervals for 5 min. A linear fit was applied to the first 30 s to obtain the rate of ATP turnover/head/s at each microtubule concentration. The KmMT was extracted by performing a nonlinear fit in Matlab of a Michaelis-Menten curve to the ATP turnover/head/s as a function of microtubule concentration.
Run Length Measurement
K888 motors were attached to streptavidin-coated quantum dots as in the single-molecule ATPase assay. Full-length kinesin was diluted to 1 μm and mixed with 4 μm carboxyl quantum dots in BRB-80 containing 10 mg/ml BSA and 10 mm DTT and allowed to incubate for 30 min.
Sample chambers were prepared by flowing 1 mg/ml BSA-biotin into a flow chamber and incubating for 5 min. The chamber was washed to remove excess BSA-biotin and then functionalized with 0.5 mg/ml neutravidin. Biotinylated, fluorescent microtubules were diluted to a final tubulin concentration of 100 nm in BRB-80 containing 20 μm Taxol and flowed into the chamber. This mixture was incubated for 10 min and then flushed with BRB-80 plus Taxol. Motor-coated quantum dots were diluted into 1:100 in imaging buffer with 2 mm ATP and flowed over prepared microtubule sample chambers. Images were recorded at a frame rate of 67 Hz. Run lengths were measured using custom-written Matlab tracking software. The processivity of kinesin carried quantum dots was obtained by fitting the run lengths to an exponential distribution using a maximum likelihood fit in Matlab.
Two-motor Fluorescence Assay
Immediately prior to all two-motor experiments, kinesin was affinity-purified to remove dead motors. 6–20 μm kinesin, 10 μl of axonemes, 17 mm AMP-PNP (10102547001 Roche Applied Science), 1 mm DTT, and 30 μl of BRB-80 were mixed on ice for 10 min. The mixtures was centrifuged at 14,000 rpm at 4 °C for 30 min. The supernatant was discarded, and the pellet was resuspended in 40 μl of BRB-80 containing 20 mm DTT and 8 mm ATP. The mixture was incubated on ice for 10 min and then spun in the centrifuge a second time at 14,000 rpm at 4 °C for 30 min. The supernatant was saved, and the concentration of kinesin was measured using a Bradford assay.
Motors were nonspecifically attached to carboxylated beads. 500-nm red carboxylated microspheres (beads) were diluted 2:3 into DMB containing 8 mg/ml BSA (DMB-BSA) and briefly sonicated in an ice bath. Kinesin and dynein were diluted to the desired concentrations and were mixed with the beads in a ratio of 1:1:1. For experiments involving the K888 mutants, 500 nm dynein and 10 nm kinesin were incubated with beads. For experiments involving full-length kinesin, which included the autoinhibition domain, different concentrations of kinesin and dynein were required to produce beads with bidirectional motion. For these experiments, 165 nm dynein and 30 nm kinesin were used. The bead and motor mixtures were incubated at 4 °C for 30 min prior to imaging.
To measure the polarity of microtubules, a reference kinesin, K432, was used. This was coupled to a fluorescent antibody as cargo. 1 μl of 4.4 μm K432 with a C-terminal His tag was mixed with 1 μl of Alexa Fluor 647-labeled anti-His (Qiagen 126244141). Prior to imaging, the motor-coated beads and reference K432 were mixed in imaging buffer containing 2 mm ATP. The motor-bead mixture was added to the buffer at a 1:50 dilution, and the reference K432 was added to a final concentration of 3 nm. Sample chambers containing biotinylated, fluorescent microtubules were prepared as in the run length measurements.
TIRF Imaging of Quantum Dots and Fluorescent Beads
Images were acquired on an inverted Olympus Ixon 70 microscope with a 100× 1.45 numerical aperture objective (PlanApo 100×1.45 NA ∞/0.17) plus an extra 1.5× sliding lens coupled to an Andor EM-CCD camera (DV-897E-CS0). A 488-nm argon ion laser (Melles Griot, 60 milliwatts, 543-AP-01) was used to excite the HiLyte488 microtubules, a 532-nm diode laser (World Star Tech, 30 milliwatts, model TECGL-30) was used to excite the fluorescent beads, and a 633 nm HeNe laser (Coherent HeNe laser, 4 milliwatts, model 31-2041-000) was used for imaging the Alexa Fluor 647 dye attached to the reference K432. In the microscope, optical filters were used to separate the laser lines from the sample fluorescence. A z488/532/633rpc (Chroma) triple bandpass dichroic mirror was used in combination with a z488/532/635m (Chroma) triple bandpass emission filter. For viewing the microtubules, an additional sliding filter (HQ525/50 M Chroma) was used to block the emission of the fluorescent beads, which was very broad. Motor-coated beads were tracked using Matlab software. For the two-motor bead assay, a reversal was defined as a change in direction of the bead with at least 500 nm traveled on either side of reversal point. Significance tests were performed using Matlab.
Optical Trapping Assay
The optical trap setup was used as described previously with the addition of a force-feedback loop to maintain a constant load for force-velocity curve measurements (27). Motors were nonspecifically attached to carboxylated beads in a manner similar to that used in fluorescence imaging experiments. 500-nm red carboxylated microspheres (beads) were diluted 2:3 into DMB containing 8 mg/ml BSA (DMB-BSA) and briefly sonicated in an ice bath to dissociate any large aggregates of beads and then mixed in a 1:1 ratio with diluted kinesin. Kinesin was diluted such that ∼30% of the beads in a given sample were motile to ensure that no more than one motor was attached to a bead. After incubating the beads and kinesin for 30 min at 4 °C, an imaging buffer was prepared containing DMB-BSA, 20 mm DTT, 4 mm ATP, 1 mg/ml 3,4-dihydroxybenzoic acid, and 100 nm protocatechuate 3,4-dioxygenase. The bead and kinesin mixture was diluted 1000× in the imaging buffer and flowed over an axoneme slide. Trap data were acquired at room temperature with a data acquisition rate of 4000 Hz. The power spectrum method was used to measure trap stiffness (28, 29). To analyze the stall force data, the force exerted by a single kinesin molecule was plotted as a function of time. A stall was defined as a period at least 0.5 s long with less than 1-pN variation in force followed by a sharp drop back to the center of the trap. For force feedback data, velocities were measured for runs of longer than 0.5 s in duration.
RESULTS
Addition of a Negative Charge at Residue 175 Does Not Change the ATPase Activity of Kinesin or the Affinity of Kinesin for Microtubules
To test the relationship between the charge of Ser-175 and the ATPase activity of kinesin, we performed a single-molecule motility assay in which we measured the mean velocity of individual quantum dot-labeled kinesin motors (S175D and S175A) as a function of ATP concentrations between 10 μm and 1 mm. The velocity of a single kinesin molecule as a function of ATP concentration followed Michaelis-Menten kinetics (30, 31). At each ATP concentration, for each mutant, the velocities of at least 40 quantum dots were averaged. For each mutant, the extracted kinetic parameters, namely, the maximum velocity and KmATP (ATP concentration at half-maximal velocity) were measured. Neither the S175A nor S175D mutation significantly changed either parameter compared with wild-type, truncated kinesin, using an analysis of covariance test of significance (p > 0.95, Fig. 2A and Table 1). This result indicates that an additional negative charge at this position does not alter the affinity of kinesin for ATP or the speed at which it walks processively on microtubules.
FIGURE 2.
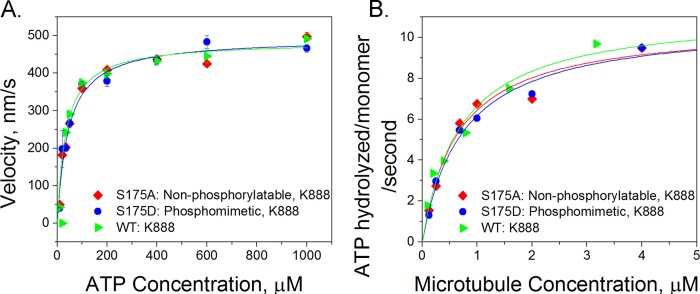
Ser-175 modification does not alter the chemical kinetics of kinesin. Measurements of the affinity of the wild-type K888 (green triangles), S175A (red diamonds), and S175D (blue circles) mutants for ATP and microtubules are shown. A, velocity versus ATP concentration. We measured the velocity of quantum dot cargos moved by single kinesin motors and found no significant difference in the velocity of the three different kinesin mutants as a function of ATP concentration. The error bars shown at each point represent the S.E. velocity at each ATP concentration. The velocities of at least 40 quantum dots were measured and averaged for each data point. B, microtubule-stimulated ATPase activity in which the concentrations of kinesin and ATP were constant but the amount of microtubules in bulk solution was titrated in an enzyme-linked phosphatase assay. For each curve, two or three concentration points were picked, and the assay was repeated to ensure the consistency of the results.
TABLE 1.
Summary of the kinetics of K888 mutants
Kinetics are from single-molecule motility studies and microtubule-activated ATPase assays of K888 mutants. The results from the motility studies are given in the left two columns. The microtubule-activated ATPase kinetics are in the right two columns. Errors are given as S.E.
| Kinesin | KmATP | Vmax | KmMT | VmaxMT |
|---|---|---|---|---|
| μm | nm/s | μm | ATP molecules/kinesin monomer/s | |
| WT K888 | 35 ± 8 | 484 ± 23 | 0.70 ± 0.26 | 10.6 ± 1.4 |
| S175A K888 | 44 ± 9 | 493 ± 24 | 0.65 ± 0.25 | 10.8 ± 1.1 |
| S175D K888 | 44 ± 10 | 494 ± 28 | 0.74 ± 0.22 | 11.3 ± 1.6 |
Mutating Ser-175 also did not change the ability of kinesin to bind to microtubules. At a fixed ATP concentration, the microtubule dependence of ATPase activity follows Michaelis-Menten kinetics (32, 33). The affinity of kinesin for microtubules can be characterized by an apparent Michaelis-Menten constant, KmMT. Mutations in kinesin that affect microtubule binding, but do not affect the overall rate of ATP hydrolysis, are expected to result in altered KmMT values (19). An enzyme-linked phosphate assay was used to measure the rate of free phosphate released from ATP at different microtubule concentrations. Fig. 2B shows the resulting microtubule-activated ATPase curves, and Table 1 summarizes the kinetic parameters. Our results show that microtubule affinity is not affected by the charge at residue 175 (p = 0.21, using an analysis of covariance test).
Altering Residue 175 Does Not Alter Processive Run Length
We performed single-molecule motility assays in which we measured the run length of quantum dot cargo attached to individual motors. The S175A and S175D mutants in the K888 backbone had a C-terminal biotin tag for attachment to streptavidin-coated quantum dots. Full-length unphosphorylated and JNK3-phosphorylated kinesin were bound nonspecifically to carboxylated quantum dots. Radiolabeling experiments indicate that we achieved ∼80% phosphorylation of kinesin monomers (Fig. 3). All experiments were performed at saturating levels of ATP. We found that modifying Ser-175 had no significant impact on the kinesin processive run length using a Komolgorov-Smirnov test of significance (p = 0.48, Fig. 4).
FIGURE 4.
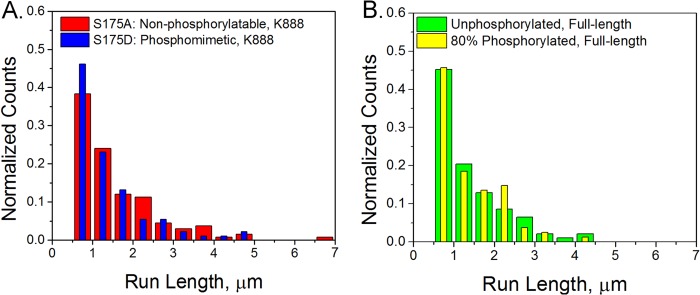
Ser-175 modification does not alter processivity. Run lengths of quantum dot cargos carried by a single kinesin were measured. A, for K888 mutants, we measured a run length of 1.06 ± 0.08 μm for S175A (red, n = 133) and 0.89 ± 0.08 μm for S175D (blue, n = 91). B, for full-length kinesin, we measured a run length of 0.97 ± 0.12 μm for unphosphorylated kinesin (green, n = 93) and 0.86 ± 0.11 μm for 80% phosphorylated kinesin (yellow, n = 81). Error bars are S.E.
Modifying Residue 175 Decreases the Kinesin Stall Force and Its Velocity While Working against a Load
An optical trap was used to measure the stall force of kinesin attached to 500-nm-diameter carboxylated polystyrene beads. The phosphomimetic S175D stalled at 5.3 ± 0.1 pN, whereas the nonphosphorylatable S175A mutant stalled at ∼6.6 ± 0.2 pN, and the wild-type protein stalled at 7.2 ± 0.1 pN (Fig. 5A, where uncertainties are given as S.E.). Experiments using JNK3 phosphorylated full-length kinesin showed a decreased stall force of 5.5 ± 0.1 pN compared with 6.4 ± 0.1 pN for untreated full-length kinesin (Fig. 5B). The shift due to an additional negative charge for 80% phosphorylated kinesin is not as dramatic as the shift seen in the S175D and wild-type K888 stall forces. However, in these data there will be broadening of the JNK3-phosphorylated kinesin stall force distribution due to the presence of three subpopulations of kinesin, i.e. motors where both heads are phosphorylated, only one head is phosphorylated, and neither head is phosphorylated. For all comparisons, the shift toward lower stall forces due to Ser-175 modification is statistically significant with p < 0.001 (Kolmogorov-Smirnov test), indicating that addition of a negative charge at residue 175 decreases the stall force of kinesin.
FIGURE 5.

Modification of Ser-175 reduces the stall force of kinesin. Optical trap measurements of stall forces and velocities are shown at constant force. A and B, stall force histograms of K888 S175A (red, n = 147), S175D (blue, n = 249) mutants, and WT (green, n = 84) (A) and full-length unphosphorylated kinesin (green, n = 271) and 80% phosphorylated kinesin (yellow, n = 440) (B). C, example trace of a stall of full-length kinesin. D, velocity of K888 mutants under application of a constant load of WT K888 (green triangles), S175A (red diamonds), and S175D (blue circles). Error bars are S.E. Open data points at 0 pN are velocities measured from fluorescence video.
We also measured the velocity of kinesin under various loads by using optical tweezers to exert a constant force on polystyrene beads attached to individual kinesin molecules. Force-velocity curves were measured for S175D, S175A, and wild-type K888 (Fig. 5C). When pulling against a load of 1 pN or greater, the S175D mutant displayed lower velocity than K888 or S175A. The S175A mutant also had a decreased velocity, compared with wild-type, at resisting forces greater than 1 pN, and a lower stall force, suggesting that any mutation of Ser-175 may affect the ability of kinesin to move under an external force.
A Negative Charge at Ser-175 Stabilizes the Autoinhibited Conformation
When autoinhibited, kinesin folds into a compact conformation in which the ATPase activity and microtubule-binding ability of the motor are inhibited (34–36). The tail contains a conserved positively charged motif that binds and docks to the motor domain near residue 175 (31, 37, 38). Addition of a negative charge at Ser-175 could stabilize the ionic interactions between the tail and motor domains. Analytical ultracentrifugation was used to determine the effect of negative charge at serine 175 on autoinhibition of full-length kinesin. The sedimentation coefficient of a phosphomimetic S175D construct was compared with wild-type kinesin. At low (<200 mm KCl) or high salt levels (>350 mm KCl), there was no difference between wild-type and the S175D mutant, indicating that both constructs adopted a compact conformation at low salt and an extended conformation in high salt (Fig. 6). We found that at “intermediate salt” concentrations of 200–350 mm KCl, the S175D mutant had a higher sedimentation coefficient than wild-type kinesin. This indicates that placement of a negative charge at residue 175 enhanced the fraction of kinesin that adopted a compact, autoinhibited conformation. Furthermore, the intermediate salt concentrations approximate the salt concentration of mammalian cytosol. We conclude that the S175D modification stabilizes autoinhibition at physiologically relevant salt concentrations.
FIGURE 6.
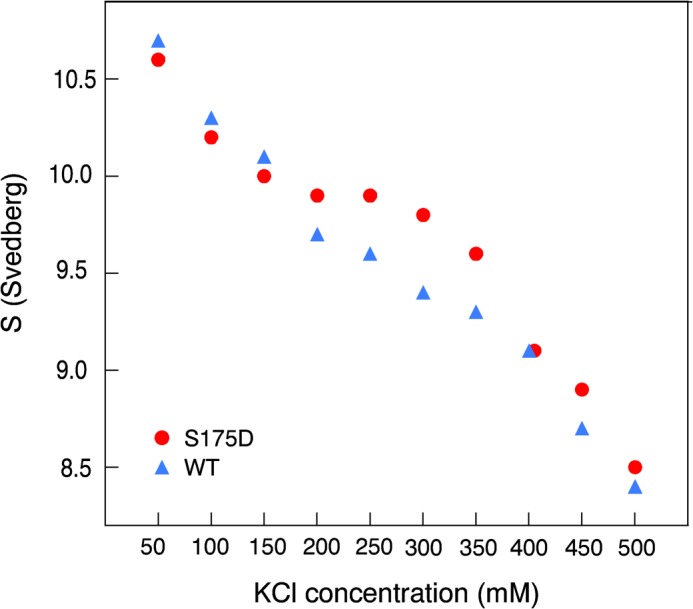
Autoinhibition is stabilized by Ser-175 modification. Analytical ultracentrifugation was used to measure the sedimentation coefficient of full-length wild-type kinesin (blue) and a full-length S175D mutant (red) as a function of the ionic strength of the buffer. Higher sedimentation coefficients were observed for the Ser-175 mutant, indicating an increase in the fraction of autoinhibited S175D relative to wild-type kinesin. The S.E. of the fit produced error bars smaller than the size of the symbols used in the plot.
Ser-175 Modification Biases Transport toward Microtubule Minus-ends When Both Kinesin and Dynein Are Present
We attached kinesin and cytoplasmic dynein to 500-nm carboxylated fluorescent polystyrene beads and observed the resulting motion. Beads coated with the phosphomimetic S175D kinesin mutant and dynein were more likely to undergo minus-end-directed motion than beads coated with dynein and either wild-type kinesin or the alanine mutant: 40% of the S175A mixture moved toward the plus-end compared with 23% in the S175D sample. Phosphorylated, full-length kinesin was also more likely to move in the minus direction than unphosphorylated kinesin (Fig. 7, A and B). Also, on average, phosphomimetic or phosphorylated kinesin and dynein-coated cargo traveled farther toward the minus-end of microtubules than cargo coated with the S175A mutant and dynein (Fig. 7, C and E, and Table 2). The distributions of net displacements of phosphorylated kinesin and unphosphorylated kinesin-containing samples differ statistically (p < 0.001, Kolmogorov-Smirnov test). The mean velocities of cargo carried by phosphomimetic or phosphorylated kinesin and dynein were also shifted: more minus-end motion was observed in cargo carried by modified kinesin and dynein (Fig. 7, D and F, and Table 2). These results are consistent with previous measurements in axoplasm which suggest that phosphorylation of residue 175 inhibits kinesin-driven transport (15, 16). Tests performed with kinesin truncated to contain the first 560 amino acids did not result in any motors binding to carboxylated beads. From this, we conclude that the motor domain of kinesin does not interact with beads, but rather a region on the stalk binds to beads. Hence, observed differences in motility of kinesin mutants, which differ only in the motor domain at Ser-175, are due to motor-microtubule interactions, not motor-bead interactions (data not shown).
FIGURE 7.

A negative charge at Ser-175 can tune the direction of two-motor transport. Beads were coated with both dynein and kinesin with different Ser-175 modifications. A, in the case of K888 mutants, the S175D and dynein (blue, n = 195) beads moved toward the plus-end less often than beads with either S175A and dynein (red, n = 110) or WT kinesin and dynein (green, n = 72). B, phosphorylation of full-length kinesin also led to less plus-end motion in an 80% phosphorylated sample (green, n = 113) compared with an unphosphorylated sample (yellow, n = 127). C and D, comparison of WT K888 and dynein, S175A K888 and dynein (red) and S175D K888 and dynein (blue) in terms of the total distance traveled (C) and mean velocities (D) is shown. E and F, comparison of full-length unmodified protein (green) and 80% phosphorylated protein (yellow) in term of the total distance traveled (E) and mean velocities (F) is shown. Positive values represent motion toward the microtubule plus-end, and negative values represent motion toward the minus-end.
TABLE 2.
Distances and velocities of beads coated with both kinesin and dynein
Velocities and total distances traveled of dynein and kinesin coated beads are shown. Top three rows correspond to datasets with K888 and dynein. Bottom two rows correspond to datasets with full-length kinesin and dynein. Errors are S.D.
| Mixture of motors | Number of observations | Net displacement | Velocity |
|---|---|---|---|
| μm | nm/s | ||
| WT (K888) + dynein | 93 | 0.0 ± 3.6 | −22.2 ± 192 |
| S175A (K888) + dynein | 74 | 0.4 ± 3.8 | 23.5 ± 146 |
| S175D (K888) + dynein | 151 | −1.7 ± 4.3 | −55.6 ± 127 |
| Unphosphorylated kinesin (full-length) + dynein | 105 | 3.4 ± 4.0 | 169.8 ± 251 |
| 80% phosphorylated kinesin (full-length) + dynein | 117 | 0.6 ± 4.4 | 51.7 ± 210 |
DISCUSSION
Modifying Ser-175 Tunes Kinesin by Stabilizing Autoinhibition and Attenuating Kinesin under a Load
We propose that phosphorylation of Ser-175 modulates kinesin by stabilizing autoinhibition and decreasing its stall force. We found that phosphomimetic full-length kinesin had a higher sedimentation coefficient than wild-type protein, indicating that Ser-175 phosphorylation stabilizes the ionic interaction that occurs when the tail domain binds to the motor domains. We also found that modification of residue 175 leads to a decreased stall force and slower velocities at loads >1 pN in optical trap experiments. However, in the absence of an external force, there was no difference in the processivity or microtubule-activated ATPase activity of phosphorylated or phosphomimetic kinesin compared with kinesin without a negative charge at residue 175.
Both the reduction in stall force and stabilized autoinhibition may contribute to our observed bias toward increased minus-end motility of beads coated with both dynein and either S175D or phosphorylated kinesin. In the study with full-length kinesin and dynein bound to a common cargo, stabilized autoinhibition would may decrease the amount of kinesin in an active state and available to bind to cargo. Force-induced effects are also present as demonstrated from the study of truncated kinesin (K888) and dynein. Even in the absence of an autoinhibitory tail domain, the S175D kinesin- and dynein-coated beads demonstrated a bias toward minus-end transport. A recent study indicated that dynein remains bound to the microtubule during plus-end transport, dragging behind kinesin (27). Dynein dragging acts as an opposing force on the order of a few pN that kinesin must work against. Our data show that the ability of kinesin to pull against a ∼1 pN or greater force is reduced by phosphorylation of Ser-175. A phosphorylated kinesin with a decreased stall force likely cannot pull against a few dynein as effectively as unphosphorylated kinesin. It would therefore lose the “tug-of-war,” thus shifting the balance toward more minus-end directed transport as seen in our data.
Addition of a Negative Charge at Serine 175 Can Modulate Microtubule-based Transport
Our experiments were performed in an in vitro system. The lack of external signaling molecules that would be present in a cell or cytoplasm allowed us to isolate the effect of Ser-175 phosphorylation on the motor domain of kinesin in the absence of any cofactors. Thus, we conclude that phosphorylation alone is sufficient to affect kinesin motility. We propose a model in which Ser-175 phosphorylation directs traffic in the axon by altering both the autoinhibition and the ability of kinesin to transport cargo under a load (Fig. 8). If constitutively up-regulated in a disease state, as may occur in Huntington disease or spinal and bulbar muscular atrophy, Ser-175 phosphorylation of kinesin could result in long term inhibition of kinesin-mediated axonal transport, resulting in signaling defects and contributing to eventual neuronal death. However, even in healthy cells, microtubule transport must be tightly regulated to ensure that cargo is delivered to an appropriate destination. By decreasing the velocity of kinesin under a load and stabilizing autoinhibition, Ser-175 phosphorylation can fine tune kinesin-driven transport.
FIGURE 8.
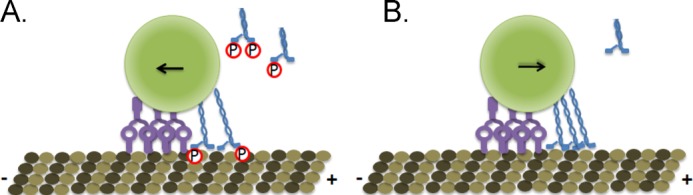
Ser-175 phosphorylation tunes kinesin transport. A, phosphorylation (red circle) stabilizes the autoinhibition of kinesin (blue) and decreases its stall force. Phosphorylated kinesin loses a tug-of-war against dynein (purple), leading to an increase in minus-end directed transport. B, unphosphorylated kinesin can exert higher forces, leading to more plus-end-directed transport, and is less strongly autoinhibited than Ser-175-phosphorylated kinesin.
Acknowledgments
We thank Yong Wang for creation of Fig. 1 and Tobias Rosenkranz and Melinda Tonks Hoffman for discussions and advice.
This work was supported, in whole or in part, by National Institutes of Health Grants GM068625 (to P. R. S.), GM078097 (to K. M. T.), GM101066 (to C. L. B.), ES016865 (to Y. L.), and GM044589 (to T. A. S.). This work was also supported by National Science Foundation Grants 1063188 and 0822613 (to P. R. S.).
- K888
- kinesin-888
- AMP-PNP
- adenylyl-imidodiphosphate
- DMB
- dynein motility buffer
- ELIPA
- enzyme-linked phosphatase assay
- pN
- piconewton(s).
REFERENCES
- 1. Chevalier-Larsen E., Holzbaur E. L. (2006) Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108 [DOI] [PubMed] [Google Scholar]
- 2. Hirokawa N., Noda Y., Tanaka Y., Niwa S. (2009) Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696 [DOI] [PubMed] [Google Scholar]
- 3. Hammond J. W., Cai D., Verhey K. J. (2008) Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 20, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hammond J. W., Huang C. F., Kaech S., Jacobson C., Banker G., Verhey K. J. (2010) Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Konishi Y., Setou M. (2009) Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 12, 559–567 [DOI] [PubMed] [Google Scholar]
- 6. McVicker D. P., Chrin L. R., Berger C. L. (2011) The nucleotide-binding state of microtubules modulates kinesin processivity and the ability of tau to inhibit kinesin-mediated transport. J. Biol. Chem. 286, 42873–42880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dixit R., Ross J. L., Goldman Y. E., Holzbaur E. L. (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vershinin M., Xu J., Razafsky D. S., King S. J., Gross S. P. (2008) Tuning microtubule-based transport through filamentous MAPs: the problem of dynein. Traffic 9, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kardon J. R., Vale R. D. (2009) Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol. Cell Biol. 10, 854–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sato-Yoshitake R., Yorifuji H., Inagaki M., Hirokawa N. (1992) The phosphorylation of kinesin regulates its binding to synaptic vesicles. J. Biol. Chem. 267, 23930–23936 [PubMed] [Google Scholar]
- 11. Hollenbeck P. J. (1993) Phosphorylation of neuronal kinesin heavy and light chains in vivo. J. Neurochem. 60, 2265–2275 [DOI] [PubMed] [Google Scholar]
- 12. Morfini G., Szebenyi G., Elluru R., Ratner N., Brady S. T. (2002) Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szebenyi G., Morfini G. A., Babcock A., Gould M., Selkoe K., Stenoien D. L., Young M., Faber P. W., MacDonald M. E., McPhaul M. J., Brady S. T. (2003) Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40, 41–52 [DOI] [PubMed] [Google Scholar]
- 14. Piccioni F., Simeoni S., Andriola I., Armatura E., Bassanini S., Pozzi P., Poletti A. (2001) Polyglutamine tract expansion of the androgen receptor in a motoneuronal model of spinal and bulbar muscular atrophy. Brain Res. Bull. 56, 215–220 [DOI] [PubMed] [Google Scholar]
- 15. Morfini G., Pigino G., Szebenyi G., You Y., Pollema S., Brady S. T. (2006) JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci. 9, 907–916 [DOI] [PubMed] [Google Scholar]
- 16. Morfini G. A., You Y. M., Pollema S. L., Kaminska A., Liu K., Yoshioka K., Björkblom B., Coffey E. T., Bagnato C., Han D., Huang C. F., Banker G., Pigino G., Brady S. T. (2009) Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 12, 864–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gunawardena S., Her L. S., Brusch R. G., Laymon R. A., Niesman I. R., Gordesky-Gold B., Sintasath L., Bonini N. M., Goldstein L. S. (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40 [DOI] [PubMed] [Google Scholar]
- 18. Cavalli V., Kujala P., Klumperman J., Goldstein L. S. (2005) Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Woehlke G., Ruby A. K., Hart C. L., Ly B., Hom-Booher N., Vale R. D. (1997) Microtubule interaction site of the kinesin motor. Cell 90, 207–216 [DOI] [PubMed] [Google Scholar]
- 20. Sosa H., Dias D. P., Hoenger A., Whittaker M., Wilson-Kubalek E., Sablin E., Fletterick R. J., Vale R. D., Milligan R. A. (1997) A model for the microtubule-Ncd motor protein complex obtained by cryo-electron microscopy and image analysis. Cell 90, 217–224 [DOI] [PubMed] [Google Scholar]
- 21. Li M., Zheng W. (2011) Probing the structural and energetic basis of kinesin-microtubule binding using computational alanine-scanning mutagenesis. Biochemistry 50, 8645–8655 [DOI] [PubMed] [Google Scholar]
- 22. Lu H., Ali M. Y., Bookwalter C. S., Warshaw D. M., Trybus K. M. (2009) Diffusive movement of processive kinesin-1 on microtubules. Traffic 10, 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crowe J., Henco K. (1992) The QIAexpressionist, pp. 79, 81,, and 82, Qiagen Inc., Chatsworth, CA [Google Scholar]
- 24. Bingham J. B., King S. J., Schroer T. A. (1998) Purification of dynactin and dynein from brain tissue. Methods Enzymol. 298, 171–184 [DOI] [PubMed] [Google Scholar]
- 25. Pierce D. W., Vale R. D. (1998) Assaying processive movement of kinesin by fluorescence microscopy. Methods Enzymol. 298, 154–171 [DOI] [PubMed] [Google Scholar]
- 26. Philo J. S. (2000) A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal. Biochem. 279, 151–163 [DOI] [PubMed] [Google Scholar]
- 27. Blehm B. H., Schroer T. A., Trybus K. M., Chemla Y. R., Selvin P. R. (2013) In vivo optical trapping indicates kinesin's stall force is reduced by dynein during intracellular transport. Proc. Natl. Acad. Sci. U.S.A. 110, 3381–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berg-Sørensen K., Flyvbjerg H. (2004) Power spectrum analysis for optical tweezers. Rev. Sci. Instrum. 75, 594–612 [Google Scholar]
- 29. Tolic-Nørrelykke S. F., Schaffer E., Howard J., Pavone F. S., Julicher F., Flyvbjerg H. (2006) Calibration of optical tweezers with positional detection in the back focal plane. Rev. Sci. Instrum. 77, 103101–103111 [Google Scholar]
- 30. Coy D. L., Wagenbach M., Howard J. (1999) Kinesin takes one 8-nm step for each ATP that it hydrolyzes. J. Biol. Chem. 274, 3667–3671 [DOI] [PubMed] [Google Scholar]
- 31. Hua W., Young E. C., Fleming M. L., Gelles J. (1997) Coupling of kinesin steps to ATP hydrolysis. Nature 388, 390–393 [DOI] [PubMed] [Google Scholar]
- 32. Huang T. G., Hackney D. D. (1994) Drosophila kinesin minimal motor domain expressed in Escherichia coli: purification and kinetic characterization. J. Biol. Chem. 269, 16493–16501 [PubMed] [Google Scholar]
- 33. Gilbert S. P., Johnson K. A. (1993) Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. Biochemistry 32, 4677–4684 [DOI] [PubMed] [Google Scholar]
- 34. Hackney D. D., Levitt J. D., Suhan J. (1992) Kinesin undergoes a 9 S to 6 S conformational transition. J. Biol. Chem. 267, 8696–8701 [PubMed] [Google Scholar]
- 35. Stock M. F., Guerrero J., Cobb B., Eggers C. T., Huang T. G., Li X., Hackney D. D. (1999) Formation of the compact confomer of kinesin requires a COOH-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J. Biol. Chem. 274, 14617–14623 [DOI] [PubMed] [Google Scholar]
- 36. Coy D. L., Hancock W. O., Wagenbach M., Howard J. (1999) Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1, 288–292 [DOI] [PubMed] [Google Scholar]
- 37. Hackney D. D., Stock M. F. (2000) Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat. Cell Biol. 2, 257–260 [DOI] [PubMed] [Google Scholar]
- 38. Dietrich K. A., Sindelar C. V., Brewer P. D., Downing K. H., Cremo C. R., Rice S. E. (2008) The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. Proc. Natl. Acad. Sci. U.S.A. 105, 8938–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kozielski F., Sack S., Marx A., Thormählen M., Schönbrunn E., Biou V., Thompson A., Mandelkow E. M., Mandelkow E. (1997) The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell 91, 985–994 [DOI] [PubMed] [Google Scholar]