

Background: Point mutations may lead to misfolding of ABC transporters.
Results: Mutations of conserved aspartates in intracellular loops 1 and 3 led to a misfolded P-glycoprotein that could be rescued by short term treatment with substrates/modulators.
Conclusion: Pharmacological chaperones rescue P-glycoprotein in an immunophilin-independent pathway by decreasing its association with Hsp70.
Significance: We reveal a novel mechanism of rescue of misfolded ABC drug transporters.
Keywords: ABC Transporter, Drug Resistance, Molecular Chaperone, Multidrug Transporters, Protein Misfolding, Chemical Chaperones, P-glycoprotein
Abstract
P-glycoprotein (P-gp) is an ATP binding cassette transporter that effluxes a variety of structurally diverse compounds including anticancer drugs. Computational models of human P-gp in the apo- and nucleotide-bound conformation show that the adenine group of ATP forms hydrogen bonds with the conserved Asp-164 and Asp-805 in intracellular loops 1 and 3, respectively, which are located at the interface between the nucleotide binding domains and transmembrane domains. We investigated the role of Asp-164 and Asp-805 residues by substituting them with cysteine in a cysteine-less background. It was observed that the D164C/D805C mutant, when expressed in HeLa cells, led to misprocessing of P-gp, which thus failed to transport the drug substrates. The misfolded protein could be rescued to the cell surface by growing the cells at a lower temperature (27 °C) or by treatment with substrates (cyclosporine A, FK506), modulators (tariquidar), or small corrector molecules. We also show that short term (4–6 h) treatment with 15 μm cyclosporine A or FK506 rescues the pre-formed immature protein trapped in the endoplasmic reticulum in an immunophilin-independent pathway. The intracellularly trapped misprocessed protein associates more with chaperone Hsp70, and the treatment with cyclosporine A reduces the association of mutant P-gp, thus allowing it to be trafficked to the cell surface. The function of rescued cell surface mutant P-gp is similar to that of wild-type protein. These data demonstrate that the Asp-164 and Asp-805 residues are not important for ATP binding, as proposed earlier, but are critical for proper folding and maturation of a functional transporter.
Introduction
Human P-glycoprotein (ABCB1,2 P-gp) is a 170-kDa plasma membrane protein that uses ATP as the energy source to pump various structurally diverse molecules out of cells. P-gp is organized as two homologous halves (43% amino acid identity) that are joined by a linker region. The drug binding pocket is harbored by two transmembrane domains (TMDs), whereas the ATP binding pockets reside in the nucleotide binding domains (NBDs). The TMDs are connected to the NBDs through cytosolic extensions called intracellular loops (ICLs). Interactions between the two halves of P-gp are critical for function, as the binding and hydrolysis of ATP induces conformational changes that are transmitted from the NBDs to the TMDs, resulting in the release of drug to the extracellular medium (1, 2). In the absence of the crystal structure of human P-gp, homology modeling together with mutagenesis studies and biochemical data can serve as an efficient tool to gain insight into the structural and functional details of this transporter.
Based on the crystal structures of MsbA (3) and Sav1866 (4), Becker et al. (5) presented three-dimensional models of P-gp in both nucleotide-bound and nucleotide-free (apo) states. These models help us to map the different residues involved in the catalytic/transport cycle. They proposed two main pathways of transmission that could originate from residues interacting either with adenine or with the γ-phosphate of ATP. In the N- and C-terminal halves of the protein, the adenine ring of ATP makes a hydrogen bond with Asp-164 (ICL1) or Asp-805 (ICL3), respectively. Transmission could also arise through the interactions of adenine with Tyr-444 and Tyr1087, whose side chain hydrogen bonds both to adenine and to Asp-164 of ICL1 and Asp-805 of ICL3 (5). Also, the crystal structure of Caenorhabditis elegans P-gp shows that the TMDs are connected to the NBDs through a “ball-and-socket joint.” ICL1 and -3 were shown to be in close proximity to the NBDs, creating an extensive interaction surface between the TMDs and NBDs (6). Furthermore, the sequence alignment of avian and mammalian P-gps shows these two aspartates to be conserved across all species, which suggested their crucial role in the structure and function of this protein.
Based on the homology modeling studies, we explored the role of these negatively charged residues by mutating the conserved Asp-164 and Asp-805 individually or together to cysteine in cysteine-less P-gp. Our insect cell studies show that the double mutant D164C/D805C displays no change in Km for ATP binding, thus contradicting the suggested direct interaction of these residues with ATP (5). We used BacMam baculovirus-transduced HeLa cells to study the expression and function of these mutant proteins. The conserved aspartates, when mutated to cysteine singly (D164C, D805C) or together (D164C/D805C), affected the processing and trafficking of P-gp to the cell membrane. We discovered that the maturation defect associated with the D164C/D805C mutant was sensitive to growth temperature. When cells expressing the D164C/D805C mutant were incubated at 27 °C (similar to growth conditions for High-five insect cells), normal maturation of P-gp was observed. These cells exhibited substrate transport similar to cells expressing the cysless-WT P-gp. Subsequently, we observed that the incubation of cells expressing the D164C/D805C mutant in the presence of pharmacological chaperones or substrates such as cyclosporine A (CsA) completely rescued the misfolded protein as a functional protein to the cell surface. We also report that the presence of chemical chaperones (e.g. CsA) is not required for the entire 18-h growth period. Rather, a treatment with CsA or FK506 for 4–6 h is enough to rescue the trapped protein (in the ER) to the cell surface. Our results provide evidence for an immunophilin-independent mechanism of rescue of misfolded P-gp unlike in the case of CFTR, where FKBP38 is shown to play a major role in the regulation of post-translational folding of CFTR through its peptidyl prolyl cis-trans isomerase activity (7). The treatment with CsA results in decreased association of misfolded mutant protein with chaperone Hsp70. A similar mechanism may be involved in the rescue of ABCG2 mutants by corrector molecules. This study is the first experimental report that establishes the role of residues Asp-164 (ICL1) and Asp-805 (ICL3) in proper folding and maturation of P-gp. In contrast to previous reports, we did not find these residues to play a role in ATP binding. Our results reveal their importance in interdomain interactions and assembly of a functional transporter.
EXPERMENTAL PROCEDURES
Chemicals
Cyclosporine A was purchased from Alexis Corp. (Switzerland). Calcein-AM was purchased from Invitrogen. ATP, rhodamine 123, daunorubicin, and all other chemicals were obtained from Sigma. Endo H and PNGase F were purchased from New England Biolabs Inc. (Ipswich, MA). The P-gp-specific monoclonal antibody C219 was obtained from Fujirebio Diagnostic Inc. (Malvern, PA). MRK16 antibody was purchased from Kyowa Medex Company, Tokyo, Japan, and UIC2 antibody was from eBioscience (San Diego, CA). FITC-labeled IgG2a anti-mouse secondary antibody was purchased from BD Biosciences. Polyclonal and monoclonal Hsp70, monoclonal Hsc70, and polyclonal calnexin antibodies were purchased from Abcam (Cambridge, MA). Tariquidar (XR9576) was kindly provided by Dr. Susan Bates (NCI, National Institutes of Health). NBD-CsA was a generous gift from Drs. Anika Hartz and Bjorn Bauer (University of Minnesota, Duluth, MN).
Preparation of Crude Membranes from Baculovirus-infected High-Five Insect Cells
High-Five insect cells (Invitrogen) were infected at multiplicity of infection 10 with recombinant baculovirus carrying the human cysless-MDR1 cDNA (all seven cysteines replaced with alanine) and D164C/D805C (in a cysless background) with a His6 tag at the C terminus as described previously. Cells were grown at 27 °C for 56–64 h and collected at 60% cell viability (8). Crude membranes were prepared as described previously (9, 10). The cells were disrupted using a Dounce homogenizer, and the undisrupted cells and nuclear debris were removed by centrifugation at 500 × g for 10 min. The crude membranes were collected by centrifugation for 60 min at 100,000 × g and resuspended in resuspension buffer containing 10% glycerol. These membranes were stored in aliquots at −80 °C.
Cell Lines
HeLa cells were cultured in DMEM media supplemented with 10% FBS, 1% glutamine, and 1% penicillin.
Cloning, Amplification of BacMam-P-gp Baculovirus, and Transduction of HeLa Cells
The expression clones for P-gp were generated in pDest-625, as previously described (11). These were then transformed into Escherichia coli DH10Bac cells (Invitrogen) and plated on gentamycin-, kanamycin-, tetracycline-, isopropyl 1-thio-β-d-galactopyranoside-, and X-gal-selective media as per the manufacturer's protocols. The bacmid DNA was prepared by the alkaline lysis method, which was further verified by PCR amplification. HeLa cells were transduced with BacMam P-gp mutant viruses as described previously (11). Briefly, either cysless-WT, single (D164C, D805C), or double mutant (D164C/D805C) baculovirus was added to HeLa cells at a ratio of 50–60 viral particles per cell in 3 ml of media. DMEM medium was added after 1 h, and these infected cells were further incubated. After 3–4 h, 10 mm butyric acid was added, and the cells were grown overnight at 37 °C. For rescue at lower temperatures, the baculovirus-transduced cells were incubated at 27 °C for 39 h. In both cases, the cells were trypsinized, washed, counted, and analyzed by flow cytometry for cell surface expression and transport function of cysless-WT and mutant P-gps.
Detection of Cell Surface Expression of P-gp by MRK-16 Antibody
Cell surface expression of cysless-WT and mutant P-gp was examined using MRK16 antibody, as described earlier (12, 13). P-gp-expressing cells (250,000 cells) were incubated with MRK16 antibody (1 μg/100,000 cells) for 60 min. Cells were subsequently washed and incubated with FITC-labeled IgG2a anti-mouse secondary antibody (1 μg/100,000 cells) for 30 min at 37 °C. The cells were washed with cold PBS and analyzed by green fluorescence detector.
UIC2 Shift Assay
The UIC2 shift assay was performed as described earlier (14). Briefly, 250,000 cells were resuspended in 0.5 ml of Iscove's modified Dulbecco's medium in sample and control tubes and allowed to equilibrate at 37 °C for 5 min with CsA (20 μm) or DMSO, respectively. UIC2 antibody (2 μg/100,000 cells) was then added to this suspension, and the tubes were incubated at 37 °C for another 30 min. Cells were then washed with excess Iscove's modified Dulbecco's medium, resuspended in 250 μl of Iscove's modified Dulbecco's medium, and incubated with FITC-labeled anti-mouse secondary antibody (1 μg/100,000 cells) for 30 min at 37 °C. Cells were washed, and fluorescence of FITC was measured on a FACSort flow cytometer equipped with a 488-nm argon laser and 530-nm bandpass filter. The UIC2 shift was defined as the difference between UIC2 binding in the presence and in the absence of CsA.
Rescue of Mutant P-gp Using Cyclosporine A
HeLa cells were transduced with D164C, D805C, or D164C/D805C mutant P-gps as described above. 6.25 μm CsA was added 6 h after the transduction, and the cells were grown for 16–18 h. The medium containing CsA and virus was removed, and cells were incubated in fresh medium for 2 h just before trypsinization. For time course experiments, the cells were transduced with D164C/D805C mutant P-gp and grown at 37 °C for 24 h as described above. The virus was then removed, and fresh medium with 15 μm CsA was added. The cell surface expression of the mutant P-gp was then monitored at different time points (0.5, 1, 2, 4, 6, and 8 h). Also, to check if preformed protein was being rescued by CsA, HeLa cells were transduced with the D164C/D805C P-gp. After 24 h, the virus was removed, and fresh medium with 5 μm cycloheximide was added to ensure the inhibition of new protein synthesis. After 2 h, 15 μm CsA was added to initiate the rescue of the mutant P-gp. The cell surface expression of the cysless-WT and double mutant P-gp was measured 4 h after the addition of CsA.
Rescue of Mutant P-gp Using Various Substrates/Modulators or CFTR Correctors
HeLa cells were transduced with D164C/D805C as described earlier. After 24 h, the virus was removed, and fresh medium was added to the cells. The cells were treated with 15 μm CsA, 15 μm FK506, 15 μm tariquidar, 15 μm vinblastine, 15 μm paclitaxel, 25 μm curcumin, 25 μm valinomycin, 15 μm thapsigargin, 15 μm verapamil, 10 μm nilotinib, 20 μm cisplatin, or 15 μm camptothecin for 6 h. 20 μm corrector VRT-325 or corr-4a was added 6 h after infection, and the cells were grown overnight. In all cases cell surface expression of mutant P-gp was analyzed by flow cytometry using MRK16 antibody followed by incubation with FITC-conjugated anti-mouse secondary antibody. The rescue of trapped mutant protein to the cell surface was detected by flow cytometry.
Determination of Transport Function of Cysless WT and Mutant P-gps Using Fluorescent Substrates
The transport function of P-gp in transduced HeLa cells was determined by flow cytometry as previously described (11, 15). Briefly, cells were trypsinized and incubated with calcein-AM (0.5 μm) for 10 min or rhodamine 123 (Rh123, 0.5 μg/ml), NBD-cyclosporine A (NBD-CsA, 0.5 μm), and daunorubicin (0.5 μm) for 45 min. Cells were washed with cold PBS before analysis. Fluorescence of substrates was measured on a FACSort flow cytometer equipped with a 488-nm argon laser and 530-nm bandpass filter.
Efflux Rate of Rhodamine 123 and NBD-cyclosporine A
300,000 transfected cells were incubated with 20 mm 2-deoxyglucose and 5 mm sodium azide in 2 ml of PBS without glucose for 10 min at 37 °C to deplete all the cellular energy. Rh123 (0.5 μg/ml) or NBD-CsA (0.5 μm) was added to each tube and incubated at 37 °C for another 20 min. The cells were then washed with PBS without glucose and chilled on ice. The efflux of the substrates was initiated at 37 °C by the addition of 2 ml of PBS with 50 mm glucose, 0.1 mm Cacl2, and 1 mm MgCl2. The efflux was allowed to continue for 0, 1, 2, 3, 5, 7, 10, 20, 30, or 45 min. The cells were then washed with cold PBS and analyzed by flow cytometry.
Immunofluorescence and Confocal Microscopy
250,000 transduced cells were cultured per well in a 24-well plate on a glass coverslip. The cells were grown in the absence or presence of 6.25 μm CsA at 37 °C for 24 h or at 27 °C for 39 h. These coverslips were rinsed once with PBS, pH 7.5, fixed with 2% formaldehyde-PBS for 30 min, treated with 100 mm glycine-PBS for 30 min, and then washed 5 times with PBS. After the washes the samples were blocked with 0.5% BSA for 20 min, washed 5 times, then incubated with the primary antibody MRK16 at a 1:50 dilution for 1 h and then washed 3 times. The samples were then incubated with FITC-conjugated anti-mouse IgG2a at a 1:50 dilution for 30 min, washed 3 times with PBS, and mounted on glass slides using Vectashield®. Slides were examined on a Leica TCS 4D CLSM confocal microscope equipped with an argon-krypton laser; 488-nm light was used for excitation of the FITC-labeled antibody, and images were collected using a 100× oil immersion objective (Leica Lasertechnik, Heidelberg, Germany).
ATPase Assay
ATPase activity of cysless-WT and mutant P-gp was measured by end-point inorganic phosphate assay as described previously (16). Briefly, crude membranes (100 μg of protein/ml) from High-Five insect cells expressing cysless-WT, and double mutant P-gp were prepared and incubated in the absence or presence of 0.3 mm orthovanadate at 37 °C in ATPase buffer (50 mm MES-Tris, pH 6.8, 50 mm KCl, 5 mm NaN3, 1 mm EGTA, 1 mm ouabain, 2 mm DTT, and 10 mm MgCl2). Both basal and drug-stimulated ATPase activity were measured in the absence of any drug or in the presence of CsA (10 μm), valinomycin (10 μm), nilotinib (5 μm), verapamil (30 μm), or tariquidar (5 μm) in the reaction mixture. The reaction was started by adding 5 mm or the indicated concentration of ATP and stopped at 20 min by 5% SDS. The amount of inorganic phosphate released was quantified by the colorimetric method as described previously (16). Km and Vmax values were determined by fitting the data to a Michaelis-Menten equation using GraphPad Prism 5.0.
Immunoprecipitation, Deglycosylation, and Western Blotting
Cell lysates from 2 × 106 cells were immunoprecipitated in radio-immunoprecipitation assay buffer with protease inhibitors. For immunoprecipitation, either P-gp-specific antibody C219 (17) or 4007 (18) or Hsp70-specific antibody was added, and the samples were incubated at room temperature for 30 min with rocking. Protein A-Sepharose beads were washed with radio-immunoprecipitation assay buffer and then added to each sample with further incubation at 4 °C overnight with rocking. The samples were then centrifuged to remove any supernatant, and the beads were washed twice with radio-immunoprecipitation assay buffer. The beads were further washed twice with 50 mm Tris-HCl, pH 7.5. The bound protein was eluted by adding 2× SDS/PAGE buffer to the beads and incubating at 37 °C for 30 min. The beads were incubated at 37 °C for an additional 30 min after diluting with running buffer. Cell lysates were subjected to Endo H and PNGase F digestion (19) for 60 min at 37 °C according to the manufacturer's protocol. Samples were denatured in sample buffer for 30 min at 37 °C. Cell lysates, immunoprecipitated, or deglycosylated samples were analyzed by SDS-PAGE and Western blot by resolving on 7% Tris-acetate gels. These immunoblots were used for detection of P-gp with the monoclonal antibody C219 or the polyclonal antibody 4007 at 1:2000 dilutions. Hsp70, Hsc70, and calnexin antibodies were used at 1:2500, 1:500, and 1:1000 dilutions, respectively. The immunoblots were developed using horseradish peroxidase-conjugated secondary antibodies and an ECL detection kit (GE Healthcare).
RESULTS
The Asp-164 and Asp-805 Residues in Intracellular Loops 1 and 3, respectively, Assist in Creating an Extensive Interaction Surface between the TMDs and NBDs
The homology model of human P-gp based on bacterial Sav1866 showed the close proximity of intracellular loops of the TMDs to the ATP-binding pockets of the NBDs, suggesting that the TMDs could directly influence the structure and movement of NBD motifs involved in ATP binding and hydrolysis and vice versa (20). Also, on the basis of MsbA and Sav1866 structures, Becker et al. (5) proposed that the adenine ring of ATP interacts either directly or through nearby aromatic residues of the NBDs with the coupling helix in ICL1 and ICL3, which itself contacts residues in ICL4 and ICL2. This interaction strongly implies Asp-164 is an important residue. Previously, ICL1 and ICL3 were shown to contact both NBDs, spanning the ATP binding pockets, whereas ICL2 and ICL4 contacted only NBD2 and NBD1, respectively (20). In a homology model of human P-gp based on Sav1866 structure, it was seen that residues Asp-164 and Asp-805 interact with the adenine group of ATP bound in NBD1 and NBD2, respectively (Fig. 1, A and D). Additionally, a homology model of human P-gp based on the recent crystal structure of C. elegans P-gp suggests that residues in ICL1 and ICL3 also interact with the NBDs, thus creating a more extensive network surface between the TMDs and NBDs (6). In this model Asp-164 and Asp-805 in ICL1 and ICL3, respectively, appear to form an interface between the TMDs and NBDs (Fig. 1, B and E). Furthermore, this interface might play a role in causing conformational changes upon ATP hydrolysis to facilitate substrate translocation. Based on the crystal structure of mouse P-gp, we developed a homology model of human P-gp, which gives us a better idea of its molecular structure and function. Along the lines of the two above-mentioned homology models of human P-gp, this model also shows the interactions of TMDs with NBDs via Asp-164 and Asp-805 in the respective homologous halves (Fig. 1, C and F) but fails to show the interactions between the two halves of the protein (21). This can be attributed to the poor resolution of the mouse P-gp structure. To explore the role of these charged residues in ATP-binding to, and hydrolysis by P-gp, we used a cysteine-less P-gp (referred to as cysless-WT) for the introduction of single as well as paired cysteines at these proposed conserved positions (Asp-164 and Asp-D805).
FIGURE 1.

In silico location and interactions of conserved aspartates Asp-164 in ICL1 and Asp-805 in ICL3. Shown is the interaction between NBD-TMD via Asp-164 or Asp-805 as seen in the homology model of human P-gp based on the crystal structure of Sav1866 (2HYD.pdb) (4) A, and D, C. elegans P-gp (4F4C.pdb) (6) B and E, and mouse mdr1a (3G60.pdb) C and F, (21). The top panels of A–C show the interactions of Asp-164, whereas the bottom panels of D–F show those of Asp-805. The ADP molecule is shown in black sticks in the homology model based on Sav1866. ICL1 (green), ICL4 (cyan) and NBD1 (orange) are shown as schematic models in panels A–C, whereas ICL2 (green), ICL3 (cyan), and NBD2 (magenta) are shown as schematic models in panels D–F. Nitrogen and oxygen atoms are colored as blue and red, respectively. The figure was prepared with PyMOL v1.4.
Mutant P-gps Display Lower Cell Surface Expression and Defective Transport Function When Expressed in HeLa Cells
The Baculovirus BacMam system was used to express single (D164C or D805C) and double (D164C/D805C) mutant P-gps in HeLa cells. The cell surface expression and function of mutant P-gp was checked using flow cytometry as described under “Experimental Procedures.” Based on UIC2 antibody staining, cell surface expression of 35–40, 0–10, and 10–20% with respect to the level of cysless-WT was observed for D164C, D805C, and D164C/D805C, respectively (Fig. 2, A–C). An increase in UIC2 detection upon treatment with P-gp substrate (CsA) confirms that the cell surface-expressed P-gp retains its normal conformation (Fig. 2, A–C).
FIGURE 2.

BacMam baculovirus-transduced HeLa cells exhibit altered cell surface expression and transport function of single (D164C and D805C) and double (D164C/D805C) mutant P-gp at 37 °C. P-gp-virus-transduced HeLa cells were incubated with UIC2 antibody (2 μg/100,000 cells) for 30 min at 37 °C followed by incubation with FITC-conjugated anti-mouse IgG2a secondary antibody. The detection with UIC2 was carried out both in the absence and presence of 20 μm CsA, as described in the UIC2 shift assay under “Experimental Procedures.” The cell surface localization of cysless-WT along with the three mutant P-gps (D164C, D805C, and D164C/D805C), as detected by UIC2 antibody, is shown in panels A–C, respectively. The blue and green histograms show the UIC2 detection of cysless-WT and mutant-Pgp, respectively, in the absence of CsA, whereas the orange and red histograms mark the UIC2 shift upon CsA treatment for cysless-WT and mutant P-gps. Panels D–F show typical histograms for accumulation of daunorubicin in HeLa cells expressing D164C, D805C, or D164C/D805C. The transport function of the three mutant P-gps was compared with cysless-WT (blue, positive control) and E556Q/E1201Q (referred to as EQ)-mutant P-gp (red, negative control), which is known to be completely functionless (22). The quantification of data and number of experiments are given in Table 1.
The transport function of these single and double mutants was checked by accumulation assays using various fluorescent substrates of P-gp, as described under “Experimental Procedures.” Fig. 2, D–F, shows a typical histogram for accumulation of daunorubicin in single (D164C, D805C) and double (D164C/D805C) mutants. Cysless-WT and the non-functional mutant E556Q/E1201Q (22) were used as positive and negative controls, respectively. The ability of D164C, D805C, and D164C/D805C mutant P-gp to transport other substrates such as Rh123, NBD-CsA, and calcein-AM was also analyzed. D164C, which shows only 35–40% cell surface expression, displayed fair to normal transport at steady state for all the drugs tested. On the contrary, D805C completely fails to transport any of the substrates (Table 1). The double mutant D164C/D805C was defective in transport for almost all these substrates to varying degrees except Rh123, which was near normal. D164C/D805C was severely defective in the transport of NBD-CsA and daunorubicin (∼30%), whereas calcein-AM was partially transported (∼60%) (Table 1). These results were corroborated by MRK16 antibody detection, which also displays a lower surface expression for both single and double mutant P-gp. Either a total loss or significantly lower cell surface expression of D164C, D805C, and D164C/D805C strongly suggests that the substitution of these negatively charged residues with cysteine has an effect on synthesis, translocation, proper folding, or trafficking of these mutant proteins.
TABLE 1.
Cell surface expression and transport function of mutant P-gps when transduced HeLa cells were grown at either 37, 27, or at 37 °C in the presence of cyclosporine A
The Bacmam baculovirus-transduced HeLa cells were evaluated for cell surface P-gp expression using MRK-16 antibody as represented by histograms in Fig. 2, A–C. The expression of cysless-WT was taken as 100%, and the cell surface expression of other variants was calculated accordingly. For transport function, the accumulation of various substrates including Rh123 (0.5 μg/ml), NBD-CsA (0.5 μm), dauno (0.5 μm), and cal-AM (0.5 μm) was followed as depicted in Fig. 2, D–F. The transport function of cysless-WT was taken as 100%, and the transport by other variants was calculated with respect to it. For both assays, the cells were washed and analyzed by flow cytometry. The values indicate the range of cell surface expression and steady-state accumulation of substrates compared to cysless-WT P-gp from the number of independent experiments given in the table. HeLa cells were transduced with baculovirus at a ratio of 50:1 to achieve same level of expression of mutants as well as the cysless-WT. dauno, daunorubicin; cal-AM, calcein-AM; ND, not detectable.
| Mutation(s) | Growth condition | No. of Repeats | Cell surface expression (%) | Transport function (%) |
|||
|---|---|---|---|---|---|---|---|
| Rh123 | NBD-CsA | Dauno | Cal-AM | ||||
| D164C | 37 °C | n = 3 | 35–40 | 90–100 | 75–85 | 80–90 | 90–100 |
| D805C | n = 3 | ND | ND | ND | ND | ND | |
| D164C/D805C | n = 10 | 10–20 | 80–90 | 20–30 | 10–20 | 30–40 | |
| D164C | 27 °C | n = 3 | 40–50 | 90–100 | 70–80 | 80–90 | 95–100 |
| D805C | n = 3 | ND | ND | ND | ND | ND | |
| D164C/D805C | n = 5 | 70–80 | 90–100 | 70–80 | 80–90 | 95–100 | |
| D164C | 37 °C + 6.25 μm CsA | n = 3 | 70–80 | 90–100 | 80–90 | 90–100 | 95–100 |
| D805C | n = 3 | 10–15 | 5–10 | ND | ND | ND | |
| D164C/D805C | n = 10 | 90–100 | 90–100 | 90–100 | 95–100 | 90–100 | |
Total Expression of Single and Double Mutants in HeLa Cells Indicates Defective Maturation of P-gp
Lysates from D164C-, D805C-, and D164C/D805C-transduced HeLa cells were checked by Western blot to analyze the total expression of P-gp. Consistent with the MRK-16 antibody staining, we observed that D164C exhibited near normal total P-gp expression, whereas D805C showed no protein at all (data not shown). The D164C/D805C double mutant shows a very faint mature band but shows immature P-gp levels similar to cysless-WT, indicating that the mutant protein is trapped in the intracellular compartment, possibly in the ER.
Growth at Lower Temperature Corrects the Folding Defect of D164C/D805C Double Mutant
As we knew that the ΔF508 CFTR mutant, which is linked to cystic fibrosis, can be functionally rescued to the cell surface when cells are grown at a lower temperature, we tried to retrieve the surface expression of these P-gp mutants by growing cells for a longer time at 27 °C. The double mutant displayed cell surface expression of up to 70–80% as compared with cysless-WT. On the other hand, neither of the single mutants showed much change when grown at 27 °C (Table 1). Additionally, this temperature-sensitive maturation of the double mutant P-gp results in an active protein being expressed at the surface. The functional status was confirmed by transport assays using fluorescent substrates. It was found that growth at lower (27 °C) temperature rescues the activity of the D164C/D805C mutant up to 70–80% for all tested substrates (Table 1). Furthermore, as there were no changes in the cell surface expression of single mutants when grown at 27 °C, the same was reflected in the transport function.
Because D164C is near normal in function, whereas D805C is drastically defective in cell surface expression, these single mutants were not considered for further studies. The double mutant could be rescued to the cell surface, and this protein at the cell surface was fully functional. We, therefore, decided to characterize the properties of D164C/D805C mutant P-gp expressed in both insect and mammalian cells.
D164C/D805C Mutant P-gp Exhibits Km for ATP Similar to Cysless-WT
To test whether residues Asp-164 or Asp-805 are required for ATP binding and hydrolysis, the D164C/D805C mutant and cysless-WT were expressed in High-Five insect cells. The level of expression for D164C/D805C P-gp was found to be similar to cysless-WT (Fig. 3A), which corroborates with the results exhibited by transduced HeLa cells when grown at 27 °C. Although the surface expression was normal, the basal ATPase activity was found to be <50% (10 nmol Pi/mg of protein/min). Unlike the cysless-WT, this basal activity was not affected by CsA (10 μm) or tariquidar (5 μm) (12 and 12.7 nmol of Pi/min/mg of protein, respectively), which inhibit cysless-WT (Fig. 3B). Verapamil (30 μm), valinomycin (10 μm), and nilotinib (5 μm) stimulate the basal ATPase activity to the same extent as cysless-WT (Fig. 3B).
FIGURE 3.

Expression profile and ATPase activity of D164C/D805C mutant P-gp expressed in insect cells. Crude membranes were prepared from cysless-WT and mutant P-gp-expressing High-Five insect cells. A, expression of cysless-WT and D164C/D805C were checked by separating crude membrane samples on 7% Tris-acetate gel (30 μg of protein/lane) followed by detection with Colloidal blue staining and by Western blot analysis (1 μg of protein/lane) using anti-P-gp antibody C219 at 1:2000 dilution. The arrow shows the position of the P-gp band, and the position of pre-stained molecular weight (111 kDa) is also shown. B, effect of selected compounds on the ATPase activity of cysless-WT and D164C/D805C mutant P-gp. Crude membranes (100 μg/ml) were incubated with DMSO (for basal activity), 10 μm CsA, 5 μm nilotinib, 10 μm valinomycin, or 30 μm verapamil, and ATPase activity was measured as described under “Experimental Procedures.” Values represent the mean ± S.D. (cysless-WT: n = 3; DD: n = 3). C, ATPase activity of cysless-WT and double mutant P-gp measured as a function of varying concentrations (0–10 mm) of ATP. Due to low basal activity of D164C/D805C, the measurements were performed in the presence of 30 μm verapamil. Each data point is the mean ± S.E.M. of three independent experiments. The data were fitted to the Michaelis-Menten curve using GraphPad Prism 5.0.
We further determined the affinity (Km) and rate of hydrolysis (Vmax) of ATP by D164C/D805C mutant P-gp. As this mutant P-gp registers a low basal activity, the ATPase activity was checked in the presence of verapamil (30 μm) with varying concentrations of ATP. We found that the introduction of cysteine residues at both Asp-164 and Asp-805 (D164C/D805C) did not alter the Km of ATP (Cysless-WT, 0.3 ± 0.03 mm; DD, 0.5 ± 0.12 mm) but led to a considerable decrease in Vmax (Cysless-WT, 65.1 ± 1.3; DD, 19.4 ± 1.2) compared with cysless-WT (Fig. 3C). These data clearly demonstrate that Asp-164 and Asp-805 are not critical for ATP binding. The reduced Vmax indicates that these residues might play a role in communication between NBDs and TMDs.
A Pharmacological Chaperone (CsA) and Small Molecules (VRT-325 and Corr-4a) Correct the Folding Defect of D164C/D805C
It is well known that substrates and modulators of P-gp assist in protein folding and rescue the misprocessed mutants to the cell surface (23). Because D164C/D805C showed poor cell surface expression, we incubated transduced HeLa cells with 6.25 μm CsA, a P-gp substrate, for 18 h, as described under “Experimental Procedures”. The percentage change in cell surface expression of D164C/D805C mutant P-gp was observed to be greater than 10-fold in comparison to cysless-WT (Fig. 4A). These results were also corroborated by increased detection of mutant P-gp at the cell surface by UIC2 antibody (data not shown). The rescued mutant protein at the cell surface showed normal transport function (∼80%) for all the tested substrates (Fig. 4D) (Table 1). The single mutants were also grown in the presence of CsA, and it was observed that the cell surface expression of D164C was rescued to ∼80%, whereas for D805C it was rescued to 10–15% only. It should be noted that although 10–15% protein could be detected at the cell surface, we could not detect any protein in the cell lysates by Western blot (data not shown). This may be due to the susceptibility of the cell surface D805C mutant P-gp to degradation by cell machinery. These data are summarized in Table 1.
FIGURE 4.

Cyclosporine A or small molecule correctors VRT-325 (C3) and corr-4a (C4) rescue the expression of D164C/D805C mutant P-gp at the cell surface. HeLa cells transduced with the D164C/D805C BacMam baculovirus were grown in the presence of CsA (6.25 μm) or CFTR correctors C3 or C4 (20 μm) for 16–18 h as described under “Experimental Procedures.” Cell surface localization of cysless-WT and D164C/D805C was determined for cells incubated in the presence of CsA (A), corrector C3 (B), and corrector C4 (C) using MRK-16 antibody as described under “Experimental Procedures.” The increase in surface expression in the presence of CsA (WT, n = 13; DD, n = 13; ****, p < 0.0001) or small molecules C3 (cysless-WT, n = 3; DD, n = 3; ***, p < 0.001) and C4 (cysless-WT, n = 3; DD, n = 3; *, p < 0.05) is represented as percent change in cell surface expression. Values are mean ± S.E. Rescue of transport function by treatment with CsA (n = 13), corrector C3 (n = 3), or C4 (n = 3) is shown in panels D–F, which represent typical histograms for the accumulation of daunorubicin in HeLa cells expressing D164C/D805C in the presence of CsA, corrector C3 and C4.
CFTR correctors have been shown to improve the processing of non-CFTR proteins including P-gp (24) and ABCG2 (25). We tested VRT-325 (C3) and corr-4a (C4) for rescue of the misprocessed double mutant. Although these correctors are known to rescue the misprocessed P-gp mutants, they could not completely rescue D164C/D805C to the plasma membrane. The percentage changes in cell surface expression of D164C/D805C mutant P-gp were much less in the presence of VRT-325 (200%) or corr-4a (70%) as opposed to CsA (500%) (Fig. 4, A–C). It was also observed that C3 could better rescue the expression (Fig. 4B) than C4 (Fig. 4C). The rescue of function was comparable to the cell surface expression in both cases. Treatment with C3 and C4 rescued the transport of daunorubicin to 70–75 and 50–55%, respectively (Fig. 4, E and F).
Detection of Cell Surface Expression of D164C/D805C Mutant P-gp in HeLa Cells by Immunofluorescence Confocal Microscopy
The cell surface expression of mutant P-gp was also monitored using immunofluorescence, as described under “Experimental Procedures.” MRK-16 antibody was used for detection of P-gp, and the extent of labeling was monitored using anti-mouse FITC IgG2a antibody. Labeling with MRK-16 and FITC-secondary antibody was quantified as the average intensity/number of pixels of the green fluorescence (Fig. 5, bar graph). We observed very low fluorescence at cell surface in mutant P-gp-expressing cells grown at 37 °C in contrast to cysless-WT, whereas a comparable level (70–75%) of cysless-WT and mutant-Pgp was seen when HeLa cells were grown at 27 °C (Fig. 5). The level of fluorescence at the cell surface was also checked in cells grown for 18 h in the presence of the P-gp substrate CsA and was found to be similar (90–100%) to cysless-WT (Fig. 5).
FIGURE 5.
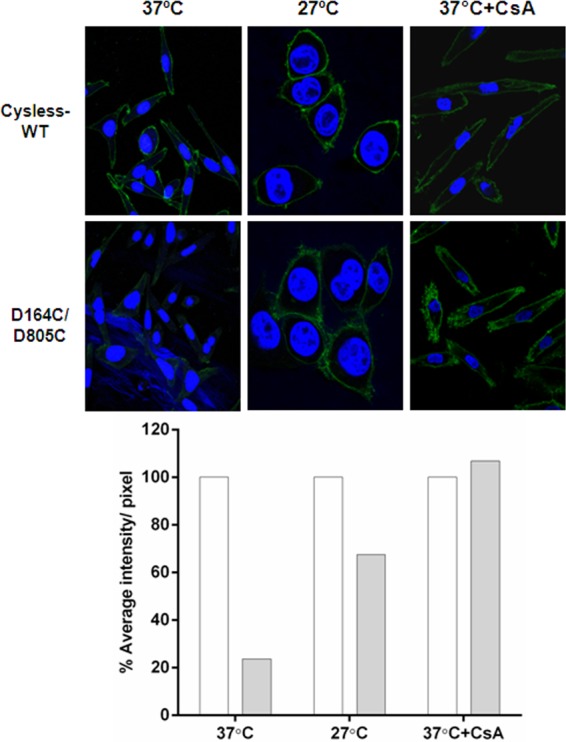
Detection of cysless-WT and D164C/D805C mutant P-gp at the cell surface by immunofluorescence. HeLa cells transduced with cysless-WT or D164C/D805C BacMam virus were fixed and stained using MRK-16 antibody followed by FITC-labeled IgG2a anti-mouse secondary antibody, and nuclei were stained with DAPI. Cells were visualized using a Leica TCS 4D CLSM confocal microscope equipped with an argon-krypton laser; 488-nm light was used for excitation of the FITC-labeled antibody, and images were collected using a 100× oil immersion objective (Leica Lasertechnik). The left panels show expression of cysless-WT and D164C/D805C in HeLa cells grown at 37 °C. The middle panels show cells grown at 27 °C for 39 h, and the right panels show level of cysless-WT and D164C/D805C in HeLa cells incubated in the presence of 6.25 μm CsA for 18 h at 37 °C. The intensity of green fluorescence in all panels was quantified as average intensity/number of pixel using Volocity 6.3. In the bar graph, the percentage average intensity/number of pixels for the double mutant (solid bars) is calculated with respect to cysless-WT, which was taken as 100% (empty bars). The values are represented as an average of two independent experiments.
D164C/D805C Transports Rhodamine 123 but at a Significantly Lower Efflux Rate
HeLa cells transduced with double (D164C/D805C) mutant P-gp expressed only 10–20% protein at the cell surface, thus resulting in decreased transport of almost all the substrates. The exception to this was Rh123, which showed a normal steady state transport despite remarkably low cell surface expression. To investigate further, we determined the rate of Rh123 efflux by cysless-WT and double mutant (D164C/D805C) P-gp-transduced cells when grown in the absence or presence of 6.25 μm CsA, as described under “Experimental Procedures.” De-energized cells were loaded with Rh123 or NBD-CsA, and the efflux was initiated at 37 °C by the addition of PBS with 50 mm glucose. In the absence of CsA, the t½ for Rh123 efflux by cysless-WT was 1.8 min versus 15 min for D164C/D805C mutant P-gp (Fig. 6). Furthermore, the rate of efflux for Rh123 in double mutant P-gp when rescued with CsA was restored almost to the level observed with cysless-WT with t½ of 3.6 min. We also studied the kinetics of efflux of NBD-CsA, a substrate that was poorly effluxed by the mutant P-gp. We observed that mutant P-gp was not able to efflux NBD-CsA beyond 40%, but when it was rescued by CsA, the rate of efflux was restored comparable to cysless-WT (Fig. 6).
FIGURE 6.

Rate of efflux of rhodamine 123 or NBD-cyclosporine A by D164C/D805C mutant P-gp was restored when grown in the presence of CsA. P-gp virus-transduced HeLa cells grown in the presence or absence of 6.25 μm CsA were studied for rate of Rh123 or NBD-CsA efflux. 300,000 cells were de-energized before loading with Rh123 or NBD-CsA as described under “Experimental Procedures”. The efflux of the substrates was initiated by resuspending cells in PBS with 50 mm glucose at 37 °C and was monitored over time by flow cytometry. The accumulation of Rh123 at 0 min is taken as 100%, and the values are calculated as the percentage of remaining fluorescence inside the cells. These values represent the average from two independent experiments. The t½ of Rh123 efflux for cysless-WT is 1.8 min versus 15 min for D164C/D805C (absence of CsA), which is restored to levels similar to cysless-WT when the cells were grown in the presence of CsA. The right panel shows efflux of NBD-CsA; t½ of NBD-CsA efflux for cysless-WT is 4.2 min, whereas the rate cannot be determined for D164C/D805C as the total efflux was <50% (in the absence of CsA). The t½ was restored to levels similar to cysless-WT when the cells were grown in the presence of CsA.
Short Term Treatment with CsA Is Sufficient to Rescue Intracellularly Trapped D164C/D805C Mutant P-gp
We observed that the misprocessed mutant protein was localized to the cell surface when grown in the presence of CsA for 18 h (Figs. 4A and 5). To determine whether the presence of CsA is required during biosynthesis or whether it can rescue pre-formed mutant P-gp from the ER to the cell surface, we performed time-course experiments. BacMam baculovirus-transduced HeLa cells were grown for 24 h at 37 °C, and then 15 μm CsA was added. The cell surface expression of D164C/D805C was checked at the indicated time points. As shown in Fig. 7A, the rescue of mislocalized D164C/D805C to the cell surface starts as early as 30 min. Furthermore, in about 4–6 h 70–80% of rescue was observed compared with the extent of rescue after 18 h of growth in the presence of CsA (Fig. 7A). To check whether pre-formed or newly synthesized mutant protein from ER was rescued, after 24 h the transduced cells were treated with 5 μm cycloheximide (CYH) for 2 h. 15 μm CsA was then added, and rescue to the cell surface was monitored. Cell surface expression was rescued to 60–70% compared with CsA treatment alone for 18 h (Fig. 7B). Notably, no rescue was seen in the presence of 5 μm CYH only. These results were corroborated with the total level of mutant P-gp in cell lysates by Western blotting (data not shown).
FIGURE 7.

Short term treatment with P-gp substrates or modulators is sufficient to rescue the mutant protein to the cell surface. A, time course of recovery of cell surface expression of D164C/D805C mutant P-gp upon short term treatment with CsA is plotted. The transduced cells were grown for 24 h after which the medium was replaced by fresh DMEM with 15 μm CsA. The cell surface expression of D164C/D805C was monitored over regular time intervals for 8 h. The expression of double (D164C/D805C) mutant at the cell surface after 18 h treatment with CsA during growth was taken as 100%. The values represent the average of two independent experiments. B, recovery of cell surface expression of D164C/D805C mutant-Pgp by CsA in the presence of CYH. Transduced HeLa cells were grown for 24 h, and then the medium was replaced by DMEM with CYH (5 μm), a de novo protein synthesis inhibitor. After 2 h the rescue was initiated by the addition of 15 μm CsA, and the cells were allowed to grow for another 4 h. The recovery by 15 μm CsA at 4 h in the absence of CYH was taken as 100%. The values are plotted as an average from two independent experiments. C, rescue of D164C/D805C to the cell surface by incubation with 15–25 μm of several substrates and modulators of P-gp was measured at 4 h. The values are the average of two independent experiments. XR, tariquidar; VBL, vinblastine; Nilo, nilotinib; Val, valinomycin; Thaps, thapsigargin. D, rescue of D164C/D805C when treated with lower concentrations (25, 250, 350 nm) of either CsA (filled bars) or FK506 (empty bars) for 4 h. The level of rescue in the presence of 15 μm CsA or FK506, respectively, for 4 h was taken as 100%. Data points are plotted as the mean ± S.E. (n = 4).
Short Term Treatment with Various P-gp Substrates and Modulators Rescues D164C/D805C Mutant P-gp to the Cell Surface
Because CsA, a well known substrate of P-gp, was able to rescue the misprocessed mutant to the cell surface, we further tested several other substrates and modulators for their ability to rescue this misfolded mutant. The rescue of the mutant protein in HeLa cells incubated in the presence of these compounds for 4 h was compared with the extent of rescue obtained in the presence of CsA. It was seen that various substrates/modulators at 15–25 μm concentration were able to rescue the D164C/D805C mutant to an extent ranging from 40 to 100% (Fig. 7C). Interestingly, compounds like cisplatin and camptothecin, which are not substrates or modulators of P-gp, were not able to rescue D164C/D805C mutant P-gp.
FK506 Rescues the Misfolded Double Mutant P-gp via the Immunophilin-independent Pathway
Immunophilins such as cyclosphilins and FK506-binding proteins (FKBP) are known to function as peptidyl prolyl cis-trans isomerases, thus accelerating folding of some proteins by catalyzing steps in initial folding and rearrangement. At lower (nM) concentrations, CsA and FK506 are known to inhibit the activity of cyclosphilins and FKBPs, respectively, thus inhibiting their peptidyl prolyl cis-trans isomerase activity. To show that the rescue of D164C/D805C was via an immunophilin-independent pathway, HeLa cells were transduced for 24 h with cysless-WT and D164C/D805C mutant P-gp after which the virus was removed and the cells were grown in the presence of varying concentrations of CsA or FK506 for 4 h. We observed that similar to CsA, this short term treatment with 15 μm FK506 was sufficient to rescue the trapped protein from the ER to the cell surface (Fig. 7D). Lower concentrations of either CsA (25 nm, 250 nm) or FK506 (25 nm, 350 nm) were not able to correct the folding defect of this double mutant (Fig. 7D). These results substantiate the fact that the rescue of D164C/D805C mutant P-gp in the presence of CsA/FK506 is not mediated via immunophilin-dependent pathway.
Enzymatic Digestion Shows That the Misfolded Double (D164C/D805C) Mutant Is Core-glycosylated and Is Trapped in the ER
The flow cytometry results were further supported by Western blot analysis of the total cell lysates from cysless-WT and D164C/D805C-expressing cells. The rescue of fully glycosylated mature protein at the cell surface in the presence of CsA or CFTR correctors C3 and C4 is evident from Fig. 8A. Enzymatic digestion of cysless-WT or the D164C/D805C by PNGase F or Endo H showed that the mature protein (Band A) was susceptible to PNGase F digestion and thus corresponds to the complex glycosylated mature protein present on the plasma membrane. Endo H cleaves early high mannose forms but not the forms containing complex carbohydrates (26) that are added to the protein in the Golgi. When digested with Endo H, Band A was found to be resistant, as seen in the cysless-WT as well as double (D164C/D805C) mutant samples, although the band is faint in the double mutant because of very low expression of this mature protein. The immature protein (Band B) seen in cysless-WT and also the major form for mutant P-gp is Endo H-sensitive (Fig. 8B). The PNGase F and Endo H digestion pattern thus indicates that the double mutant is trapped in the ER. We further observed that Band B of the double mutant corresponds to the same size as the P-gp expressed in High-Five insect cells, which is unglycosylated. Treatment with PNGase F or Endo H had no effect on the size of cysless-WT or double D164C/D805C mutant P-gp expressed in insect cells (Fig. 8C).
FIGURE 8.

Susceptibility of mature and immature forms of cysless-WT and D164C/D805C mutant P-gp to digestion by PNGase F and Endo H. A, Western blot analysis of total cell lysates of HeLa cells transduced with cysless-WT or D164C/D805C mutant P-gp when grown in the absence or presence of 6.25 μm CsA or CFTR correctors C3 and C4 (20 μm). Cell lysate proteins (3 μg) were loaded in each lane, and immunoblotting with C219 antibody was performed as described under “Experimental Procedures.” Lane 1, cysless-WT; lane 2, cysless-WT treated with 6.25 μm CsA; lane 3, cysless-WT treated with 20 μm corrector C3, lane 4, cysless-WT treated with 20 μm corrector C4; lane 5, D164C/D805C; lane 6, D164C/D805C treated with 6.25 μm CsA; lane 7, D164C/D805C treated with 20 μm C3; lane 8, D164C/D805C treated with 20 μm corrector C4. Levels of β-actin are shown as loading controls for each lane in the immunoblot. Arrows show the position of mature P-gp and β-actin, whereas an arrowhead marks the position of the immature P-gp. B, glycosidase digestion of cell lysates (2 μg of protein) from cysless-WT and double mutant P-gp was loaded in each lane. Cell lysates from HeLa cells transduced with cysless-WT (lanes 1, 2, and 3) and D164C/D805C (lanes 4, 5, and 6) and grown in the absence of CsA were subjected to Endo H or PNGase F digestion as described under “Experimental Procedures” and processed for immunoblotting using C219 antibody. The complex glycosylated (Band A, mature protein) and deglycosylated (Band B, immature protein) forms are indicated. C, glycosidase digestion of crude membranes from High-Five insect cells (0.5 μg of protein) expressing cysless-WT (lanes 1, 2, and 3) and D164C/D805C (lanes 4, 5, and 6) P-gp. In both cases the digest was resolved on 7% Tris-acetate gel and subjected to Western blotting using C219 antibody. The details of treatments are given in the figure, and blots from a representative experiment are shown. Similar results were obtained from three independent experiments.
The D164C/D805C Mutant Co-immunoprecipitates More with Hsp70 but Not with Hsc70 or Calnexin Antibodies
Some of the chaperones, including calnexin, Hsp70, and Hsc70 have been found to associate with P-gp (27). To determine whether any of these chaperones associate more with the double mutant, cysless-WT and double mutant P-gp were immunoprecipitated using the P-gp-specific C219 antibody, and the immunoprecipitates were checked for levels of calnexin, Hsp70, and Hsc70. It was seen that all three chaperones co-immunoprecipitated with P-gp. Interestingly, we found that Hsp70 co-precipitates more (200–300%) with the trapped double mutant as compared with the cysless-WT (Fig. 9A). However, the level of Hsp70 co-precipitated in the CsA-treated samples was similar to that observed with cysless-WT. This suggests that Hsp70 associates more with protein having folding defects and treatment with CsA results in dissociation of Hsp70 from the double mutant, allowing it to be trafficked to the cell membrane. Importantly, in both cysless-WT and D164C/D805C, we observed less association of Hsp70 with immature P-gp when cells were treated with CsA (Fig. 9A). We also checked for Hsc70, a member of Hsp70 family, but failed to observe any difference in its level of association with misfolded and rescued double mutant protein (Fig. 9B). Additionally, no difference was seen in the levels of calnexin coimmunoprecipitated with either cysless-WT or the double mutant P-gp (Fig. 9B).
FIGURE 9.
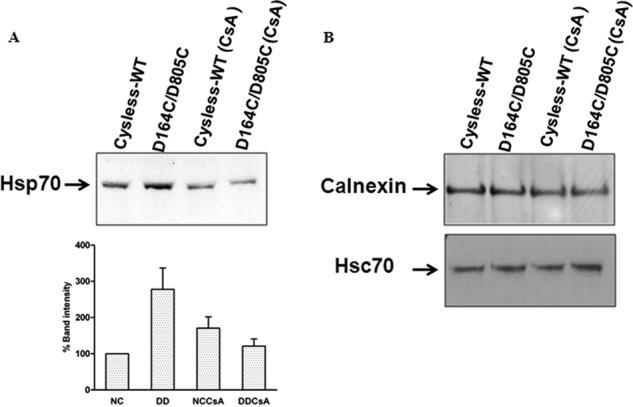
Association of D164C/D805C mutant P-gp with chaperones calnexin, Hsp70, and Hsc70. A, lysates from 2 × 106 cells transduced with cysless-WT and double mutant P-gp, grown in the absence and presence of 6.25 μm CsA, were immunoprecipitated with P-gp antibody C219. Western blot analysis of the co-immunoprecipitate was carried out with anti-Hsp70 antibody at 1:2500 dilution. The relative percentage band intensity is represented as a bar graph, and intensity of cysless-WT (untreated) is taken as 100% (n = 3, error bars denote S.E.). NC, cysless-WT; DD, D164C/D805C; NCCsA, cysless-WT treated with 6.25 μm CsA for 18 h; DDCsA, D164C/D805C treated with 6.25 μm CsA for 18 h. B, the co-immunoprecipitate was also probed with anti-Hsc70 (a member of Hsp70) and anti-calnexin antibody at 1:1000 and 1:500 dilutions, respectively. Similar results were obtained in two independent experiments.
CsA Rescues Misfolded Transporter by Altering Its Association with Hsp70
Because less association of Hsp70 with immature P-gp was seen when the cells were grown in the presence of CsA (Fig. 9A), we further investigated by immunoprecipitating Hsp70 from cell lysates with Hsp70-specific antibody and measuring the levels of associated P-gp both in the presence and absence of CsA. The level of associated P-gp was checked using the P-gp-specific antibody C219 in both CsA-treated and untreated cysless-WT and D164C/D805C mutant P-gp. As expected, a decreased association of immature P-gp with Hsp70 was observed in the presence of CsA for both cysless-WT and D164C/D805C mutant P-gp (see weaker immature (lower) P-gp band in the presence of CsA in both control and mutant P-gp; left immunoblot in Fig. 10).
FIGURE 10.
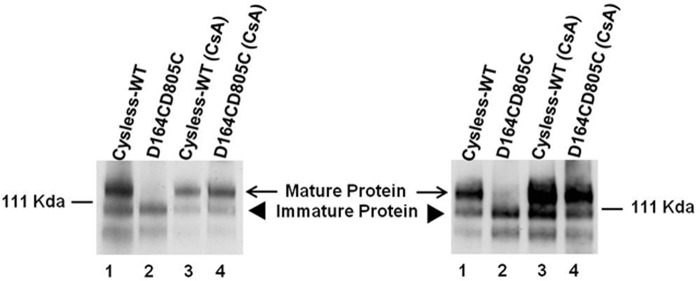
Treatment with cyclosporine A decreases the association of P-gp with Hsp70. Lysates from 2 × 106 cells transduced with cysless-WT and double mutant P-gp, grown in the absence and presence of 6.25 μm CsA for 18 h, were immunoprecipitated using rabbit polyclonal Hsp70 antibody (ab79852, Abcam), and the immunoprecipitate was probed with anti-P-gp antibody C219 at 1:2000 dilution. Lane 1, cysless-WT; lane 2, D164C/D805C; lane 3, cysless-WT (CsA); lane 4, D164C/D805C (CsA). Lanes 1 and 2 show associated P-gp when cells were grown in the absence of CsA, whereas lanes 3 and 4 depict these levels in the presence of CsA. The immunoblot on the right was probed with C219 antibody to show the level of P-gp in cell lysates (3 μg/lane), which were used for immunoprecipitation with Hsp70 antibody.
DISCUSSION
Recently, contacts at the atomic level between residues in NBDs and ICLs in the inward-open homology model of human P-gp have been analyzed using in silico methods (5, 20, 28). These studies have proposed that the residues in ICL1 and ICL4 interact with those in NBD1, whereas the residues in ICL2 and ICL3 exhibit interactions with those in NBD2. They have further pinpointed Asp-164 and Asp-805 among the key contact locations in ICL1 and ICL3, suggesting a hydrogen bond between Tyr-444 and Asp-164, thus pointing to the direct contact between NBD1 and the coupling helix of ICL1. Similar interactions are seen in the other half of the protein, where Asp-805 directly interacts with Tyr-1087, a residue in NBD2 (28). Previous reports on P-gp predicted that ICL1 and ICL3 contact both the NBDs (20). Furthermore, the computational models suggest that residues Asp-164 and Asp-805 in ICL1 and ICL3, respectively, interact with bound ATP molecules in the active site of NBDs (5). Taken together, if the interactions between Asp-164 and Asp-805 are critical for binding and hydrolysis of ATP, then any change in the electrostatic charge at these sites is expected to greatly perturb this interaction and result in changes in ATP binding/hydrolysis. However, our data clearly show that these residues are not critical for ATP binding or hydrolysis. The reduced maximal velocity of ATP hydrolysis indicates that these residues (and ICL1 and -3) may be involved in communication between substrate binding site(s) in TMDs and NBDs (Fig. 3B).
We expressed D164C, D805C, and D164C/D805C P-gp in HeLa cells for characterization of expression and function of these mutant P-gps. We observed that the D164C mutation leads to lower cell surface expression, but the transport at steady state is normal. The D805C mutation drastically affects the protein expression (although the level of mRNA is normal) (data not shown), whereas the D164C/D805C double mutation results in retention of the predominantly immature protein in the ER; only 10–15% of double mutant protein is present at the cell surface (Fig. 2, Table 1). However, this misprocessing in the case of D164C and D164C/D805C could be completely corrected when the cells were grown either at lower temperature (27 °C) or in the presence of pharmacological chaperones or small molecule correctors at 37 °C. The D805C mutation is detrimental to the protein expression, and its expression could not be rescued. We also substituted these two aspartates paired with either Ala (D164A/D805A) or Asn (D164N/D805N) and observed lower cell surface expression in both cases. Single substitution of Asp-164 with Ala (D164A) or Asn (D164N) led to similar expression and function as D164C.3 The substitution of Asp-805 with Ala (D805A) or Asn (D805N) is less drastic as compared with D805C, but the protein still does not localize to the cell surface.3 All these substitutions with either Ala or Asn could be rescued to the cell surface in the presence of CsA (data not shown). Pajeva et al. (28) also reported the asymmetry in interaction between the two halves of the protein, which is confirmed by our observed differences upon single mutation of Asp-164 and Asp-805. A homology model of human P-gp based on mouse P-gp structure predicts that the distance between these two cysteine-substituted positions is ∼55 Å in the apo conformation, and it is reduced to ∼25.5 Å in the closed conformation, which rules out the plausibility of formation of a disulfide bond. We were unable to cross-link these two substituted cysteines in the presence of ATP and vanadate (to trap protein in ADP-vanadate, closed conformation) using a 25 Å cross-linker (M17M), suggesting that these residues are either farther apart even in the closed conformation (data not shown) or not properly oriented for chemical cross-linking.
Although only 10–25% of the D164C/D805C mutant P-gp was localized to the plasma membrane at 37 °C, at steady state (45 min) it could efflux Rh123 to almost the same extent as the cysless-WT, whereas the other substrates were transported only 10–40% (Table 1). This discrepancy in efflux appears to be due to the measurements at steady state (45 min) rather than the initial rate. This is supported by the fact that the rate of efflux was actually 7–9-fold less than cysless-WT (t½ 15 min versus 1.8 min; Fig. 6). This rate of efflux was also recovered to a level similar to cysless-WT when the cell surface expression was restored by treatment with CsA (Fig. 6).
Studies with human CFTR and Arabidopsis ABCB19 demonstrate the role of FKBPs in facilitating protein folding by virtue of their peptidyl prolyl cis-trans isomerase activity and interaction with Hsp90 chaperone via an immunophilin-dependent pathway. FKBP38 regulates both synthesis and post-translational folding of CFTR (7), whereas ABCB19 has an interaction with FKBP42 for its proper localization and hence enhanced auxin transport in Arabidopsis (29). These FKBPs are known targets of the immunosuppressant FK506. Our results show that in the presence of a higher concentration of FK506 (15 μm), the double mutant P-gp was rescued to the cell surface in the short term (4–6 h) treatment assay similar to CsA. This could be attributed to the fact that both CsA and FK506 are known substrates of P-gp (30), and thus the rescue occurs via an immunophilin-independent pathway. We demonstrate that the rescue of mutant P-gp by CsA or FK506 does not involve de novo synthesis (Fig. 7B). Also, lower concentrations of these compounds (25–350 nm) do not affect the rescue of the misprocessed double mutant (Fig. 7D). Taken together, these data strongly suggest that CsA and FK506 rescue this double mutant P-gp by an immunophilin-independent pathway. The specific substrates or modulators of P-gp serve as pharmacological chaperones (Fig. 7C) and are predicted to act as scaffolds that stabilize the folding of the protein in the native conformation, thus protecting it from the degradation machinery of the cell (31). Studies with CFTR show that the NBDs fold co-translationally, whereas post-translational folding involves only TM domains (32). Known mutations such as ΔY490 in P-gp (equivalent to ΔF508 in CFTR) appear to interfere with post-translational folding of the proteins (33). Similarly, the D164C and D805C mutations might interfere with the proper packing of a mature protein by disrupting NBD-TMD interactions via ICLs.
Previous studies reported that during the early stages of biosynthesis when P-gp is only core-glycosylated, wild-type P-gp associates only transiently with calnexin (34). As the protein matures, it is dissociated from calnexin and is targeted to the plasma membrane. Calnexin is known to be a chaperone that promotes folding of the protein by preventing aggregation of folding intermediates (34). It recognizes glycoproteins only when they are misfolded and is known to be responsible for retention of misfolded proteins. The misprocessed mutants of P-gp remain associated with calnexin throughout their lifetime and are thus never targeted to the plasma membrane (34). P-gp is known to associate with calnexin through TMDs, whereas association with Hsc70 is through the NBDs (27). However, our results with immunoprecipitation of P-gp with C219 antibody do not show any changes in association of calnexin as well as Hsc70 with double mutant P-gp. Rather, they show increased association of the D164C/D805C mutant protein with other members of the Hsp70 family (Fig. 9). The ER lumen also possesses GRP78/BiP, a member of Hsp70 family. BiP has been shown to play an important role in ER-associated protein degradation (ERAD) (35). GRP78/BiP is known to interact with hydrophobic regions of the protein with relatively low affinity and shields misfolded protein from aggregation. The release of this Hsp70 family member from the mutant protein in the presence of CsA might be driving the delivery of mature and properly folded protein through the Golgi complex to the plasma membrane (36). Failure to ever reach its properly folded state likely results in continued interaction with and retention by Hsp70, which acts as a quality control system. We do not observe overexpression of Hsp70 in HeLa cells expressing this mutant (data not shown); only an increased association with mutant transporter was noted. It was observed that association of the immature P-gp with Hsp70 decreases when grown in the presence of CsA in both cysless-WT and D164C/D805C double mutant P-gp (Fig. 10). This confirms that CsA rescues the trapped double mutant by reducing the retention of the immature protein with Hsp70. Furthermore, even though D164C/D805C is misfolded, it does not seem to degrade rapidly. It is also known that mannose residues protect folding-defective polypeptides from ER-associated protein degradation (35).
The region of the ICL3, which lies in close proximity to Asp-805 in P-gp, is particularly significant as it is surrounded by a number of single-nucleotide polymorphisms (SNPs). The homology model of human P-gp based on the crystal structure of mouse P-gp shows that SNPs D800N, V801M, and D1088N are located spatially close to Asp-805, and these residues together form a tight cluster (Fig. 11). In ICL1, Val-168, which is close to Asp-164, is reported as a SNP site (37). Furthermore, processing mutations in P-gp are known to disrupt domain-domain interactions and has been found in other regions besides the TMD-NBD interface (37). The majority of the mutation sites participate in tight interhelical interactions. Hence, it is reasonable to expect that the mutations at such critical contact points result in protein misfolding. There is more than one site close to both of these two aspartates that when mutated results in a misprocessed protein (37). Also, it is reported that mutations at the two consecutive preceding residues, Trp-803 and Phe-804, cause folding defects in P-gp (31, 38). Also G251V, which is in ICL1 and lies spatially close to Asp-164, is a well known processing mutant of P-gp (37). Recent studies by Cheepala et al. (39) show that the ICL3:NBD1 contacts are important for maturation of ABCC4 (MRP4). These data are consistent with our results demonstrating that the residues in this region (ICL3) are critical for proper folding of P-gp.
FIGURE 11.
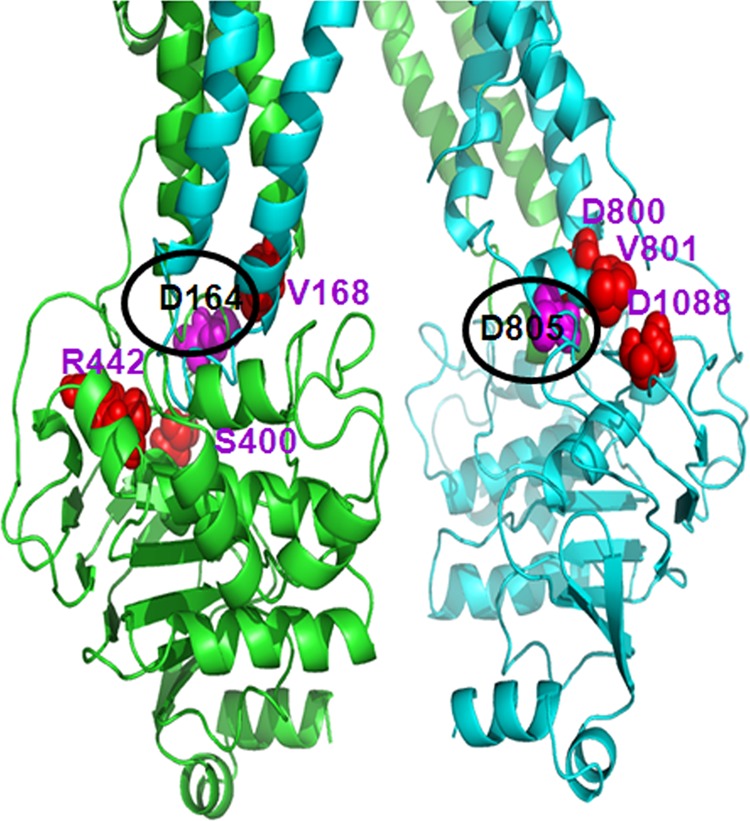
Known SNPs are located spatially close to Asp-164 and Asp-805. The location of known non-synonymous SNPs (34) (marked as red spheres) in the vicinity of residues Asp-164 and Asp-805 (marked as pink spheres) are shown to emphasize the importance of these loops for proper folding and assembly of this transporter. In addition, mutations of Trp-803 and Phe-804 also have been shown to cause misfolding of P-gp (28, 35); these residues are not shown in the figure.
Many human diseases are caused as a result of loss of function of ABC transporters due to single point mutations/deletions (40). In several cases the mutant transporters are retained in the ER due to misfolding or improper processing. Cystic fibrosis is a classic example, where the deletion of a single amino acid (ΔF508) results in abnormal trafficking of CFTR to the plasma membrane, thus leading to clinical manifestations of this disease. Notably, the deletion of the equivalent residue in ABCB1, Tyr-490, which lies at the interfacial region, also results in misfolded P-gp (24). Other examples include point mutations in ABCB11 and ABCG2, which are linked to Dubin Johnson syndrome (41) and Gout (42), respectively. In the present study we show that residues Asp-164 and Asp-805 in ICL1 and ICL3, respectively, of human P-gp play a role in proper folding and maturation of P-gp. These two negatively charged residues are conserved among P-gps of various species such as human MDR2 (ABCB4), rat mdr1 and mdr2, hamster mdr1, mdr2, and mdr3, etc. These two residues, when mutated individually or together to cysteine, lead to changes in protein folding and cell surface expression.
In summary, our results demonstrate the role of residues in ICLs in proper folding of P-gp. Importantly, like ΔF508-CFTR (43), once the D164C/D805C mutant P-gp is rescued and reaches the cell surface, it is functional to the same levels as cysless-WT. Undoubtedly the interaction between Asp-164 and Asp-805 in ICL1 and -3 plays a crucial role in stabilizing domain associations during protein targeting. Pharmacological chaperones (CsA, FK506, and small molecule CFTR correctors) rescue the misfolded double mutant P-gp via an immunophilin-independent pathway by decreasing its association with the Hsp70 chaperone and thus allow the protein to be trafficked to the Golgi for maturation and then localize to the cell surface. Our results strongly suggest that a similar mechanism may be operative in the rescue of misfolded mutant CFTR (ABCC7), ABCG2 (25), and other ABC transporters (19, 40) associated with disease conditions, which should be further investigated.
Acknowledgments
We thank Dr. Robert Bridges at Rosalind Franklin University and the Cystic Fibrosis Foundation for providing CFTR correctors VRT-325 (C3) and corr-4a (C4) and Dr. Hong May Sim for the cross-linking studies. We thank Drs. Anika Hartz and Bjorn Bauer for providing NBD-cyclosporine A. We thank Dr. Indu Ambudkar (NIDCR, National Institutes of Health (NIH)) and Dr. William Swaim (NIDCR, NIH) for help with confocal microscopy and George Leiman for editorial assistance.
This work was supported, in whole or in part, by the Research Program of the National Institutes of Health, Center for Cancer Research, NCI Grant ZIA BC010030-13.
K. Kapoor, E. E. Chufan, and S. V. Ambudkar, unpublished data.
- ABC
- ATP binding cassette
- P-gp
- P-glycoprotein
- CsA
- cyclosporine A
- CYH
- cycloheximide
- ER
- endoplasmic reticulum
- ICL
- intracellular loop
- NBD
- nucleotide binding domain
- NBD-CsA
- NBD-cyclosporine A
- Rh123
- rhodamine 123
- SNP
- single-nucleotide polymorphism
- TMD
- transmembrane domain
- Endo H
- endo-β-N-acetylglucosaminidase H
- PNGase F
- peptide N-glycosidase F
- FKBP
- FK506-binding protein
- DD
- D164C/D805C.
REFERENCES
- 1. Sauna Z. E., Ambudkar S. V. (2007) About a switch. How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 6, 13–23 [DOI] [PubMed] [Google Scholar]
- 2. Sharom F. J. (2011) The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178 [DOI] [PubMed] [Google Scholar]
- 3. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Flexibility in the ABC transporter MsbA. Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 5. Becker J. P., Depret G., Van Bambeke F., Tulkens P. M., Prévost M. (2009) Molecular models of human P-glycoprotein in two different catalytic states. BMC Struct. Biol. 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin M. S., Oldham M. L., Zhang Q., Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banasavadi-Siddegowda Y. K., Mai J., Fan Y., Bhattacharya S., Giovannucci D. R., Sanchez E. R., Fischer G., Wang X. (2011) FKBP38 peptidylprolyl isomerase promotes the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum. J. Biol. Chem. 286, 43071–43080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramachandra M., Ambudkar S. V., Chen D., Hrycyna C. A., Dey S., Gottesman M. M., Pastan I. (1998) Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry 37, 5010–5019 [DOI] [PubMed] [Google Scholar]
- 9. Kerr K. M., Sauna Z. E., Ambudkar S. V. (2001) Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J. Biol. Chem. 276, 8657–8664 [DOI] [PubMed] [Google Scholar]
- 10. Sauna Z. E., Ambudkar S. V. (2000) Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 97, 2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shukla S., Schwartz C., Kapoor K., Kouanda A., Ambudkar S. V. (2012) Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos. 40, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamada H., Tsuruo T. (1986) Functional role for the 170-kDa to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc. Natl. Acad. Sci. U.S.A. 83, 7785–7789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ozvegy-Laczka C., Várady G., Köblös G., Ujhelly O., Cervenak J., Schuetz J. D., Sorrentino B. P., Koomen G. J., Váradi A., Német K., Sarkadi B. (2005) Function-dependent conformational changes of the ABCG2 multidrug transporter modify its interaction with a monoclonal antibody on the cell surface. J. Biol. Chem. 280, 4219–4227 [DOI] [PubMed] [Google Scholar]
- 14. Mechetner E. B., Schott B., Morse B. S., Stein W. D., Druley T., Davis K. A., Tsuruo T., Roninson I. B. (1997) P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl. Acad. Sci. U.S.A. 94, 12908–12913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiberghien F., Loor F. (1996) Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs 7, 568–578 [DOI] [PubMed] [Google Scholar]
- 16. Ambudkar S. V. (1998) Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 292, 504–514 [DOI] [PubMed] [Google Scholar]
- 17. Kartner N., Evernden-Porelle D., Bradley G., Ling V. (1985) Detection of P-glycoprotein in multidrug-resistant cell lines by monoclonal antibodies. Nature 316, 820–823 [DOI] [PubMed] [Google Scholar]
- 18. Tanaka S., Currier S. J., Bruggemann E. P., Ueda K., Germann U. A., Pastan I., Gottesman M. M. (1990) Use of recombinant P-glycoprotein fragments to produce antibodies to the multidrug transporter. Biochem. Biophys. Res. Commun. 166, 180–186 [DOI] [PubMed] [Google Scholar]
- 19. Gautherot J., Durand-Schneider A. M., Delautier D., Delaunay J. L., Rada A., Gabillet J., Housset C., Maurice M., Aït-Slimane T. (2012) Effects of cellular, chemical, and pharmacological chaperones on the rescue of a trafficking-defective mutant of the ATP-binding cassette transporter proteins ABCB1/ABCB4. J. Biol. Chem. 287, 5070–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zolnerciks J. K., Wooding C., Linton K. J. (2007) Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 21, 3937–3948 [DOI] [PubMed] [Google Scholar]
- 21. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sauna Z. E., Müller M., Peng X. H., Ambudkar S. V. (2002) Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1). Biochemistry 41, 13989–14000 [DOI] [PubMed] [Google Scholar]
- 23. Loo T. W., Bartlett M. C., Clarke D. M. (2005) Rescue of folding defects in ABC transporters using pharmacological chaperones. J. Bioenerg. Biomembr. 37, 501–507 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y., Loo T. W., Bartlett M. C., Clarke D. M. (2007) Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol. Pharmacol. 71, 751–758 [DOI] [PubMed] [Google Scholar]
- 25. Woodward O. M., Tukaye D. N., Cui J., Greenwell P., Constantoulakis L. M., Parker B. S., Rao A., Köttgen M., Maloney P. C., Guggino W. B. (2013) Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. U.S.A. 110, 5223–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maley F., Trimble R. B., Tarentino A. L., Plummer T. H. (1989) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180, 195–204 [DOI] [PubMed] [Google Scholar]
- 27. Loo T. W., Clarke D. M. (1995) P-glycoprotein. Associations between domains and between domains and molecular chaperones. J. Biol. Chem. 270, 21839–21844 [DOI] [PubMed] [Google Scholar]
- 28. Pajeva I. K., Hanl M., Wiese M. (2013) Protein contacts and ligand binding in the inward-facing model of human P-glycoprotein. ChemMedChem 8, 748–762 [DOI] [PubMed] [Google Scholar]
- 29. Wu G., Otegui M. S., Spalding E. P. (2010) The ER-localized TWD1 immunophilin is necessary for localization of multidrug resistance-like proteins required for polar auxin transport in Arabidopsis roots. Plant Cell 22, 3295–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saeki T., Ueda K., Tanigawara Y., Hori R., Komano T. (1993) Human P-glycoprotein transports cyclosporine-a and Fk506. J. Biol. Chem. 268, 6077–6080 [PubMed] [Google Scholar]
- 31. Loo T. W., Clarke D. M. (1997) Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem. 272, 709–712 [DOI] [PubMed] [Google Scholar]
- 32. Kleizen B., van Vlijmen T., de Jonge H. R., Braakman I. (2005) Folding of CFTR is predominantly cotranslational. Mol. Cell 20, 277–287 [DOI] [PubMed] [Google Scholar]
- 33. Lewis H. A., Zhao X., Wang C., Sauder J. M., Rooney I., Noland B. W., Lorimer D., Kearins M. C., Conners K., Condon B., Maloney P. C., Guggino W. B., Hunt J. F., Emtage S. (2005) Impact of the ΔF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J. Biol. Chem. 280, 1346–1353 [DOI] [PubMed] [Google Scholar]
- 34. Loo T. W., Clarke D. M. (1994) Prolonged association of temperature-sensitive mutants of human P-glycoprotein with calnexin during biogenesis. J. Biol. Chem. 269, 28683–28689 [PubMed] [Google Scholar]
- 35. Hebert D. N., Molinari M. (2007) In and out of the ER. Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377–1408 [DOI] [PubMed] [Google Scholar]
- 36. Brown C. R., Hong-Brown L. Q., Biwersi J., Verkman A. S., Welch W. J. (1996) Chemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wolf S. J., Bachtiar M., Wang J., Sim T. S., Chong S. S., Lee C. G. (2011) An update on ABCB1 pharmacogenetics. Insights from a 3D model into the location and evolutionary conservation of residues corresponding to SNPs associated with drug pharmacokinetics. Pharmacogenomics J. 11, 315–325 [DOI] [PubMed] [Google Scholar]
- 38. Loo T. W., Bartlett M. C., Clarke D. M. (2006) Insertion of an arginine residue into the transmembrane segments corrects protein misfolding. J. Biol. Chem. 281, 29436–29440 [DOI] [PubMed] [Google Scholar]
- 39. Cheepala S. B., Bao J., Nachagari D., Sun D., Wang Y., Zhong T. P., Zhong T., Naren A. P., Zheng J., Schuetz J. D. (2013) Crucial role for phylogenetically conserved cytoplasmic loop 3 in ABCC4 protein expression. J. Biol. Chem. 288, 22207–22218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Silverton L., Dean M., Moitra K. (2011) Variation and evolution of the ABC transporter genes ABCB1, ABCC1, ABCG2, ABCG5. and ABCG8. Implication for pharmacogenetics and disease. Drug Metabol. Drug Interact. 26, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Z. S., Tiwari A. K. (2011) Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 278, 3226–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Woodward O. M., Köttgen A., Coresh J., Boerwinkle E., Guggino W. B., Köttgen M. (2009) Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. U.S.A. 106, 10338–10342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gee H. Y., Noh S. H., Tang B. L., Kim K. H., Lee M. G. (2011) Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760 [DOI] [PubMed] [Google Scholar]