

Background: The enzyme generating free oligosaccharides (fOSs) in the lumen of the endoplasmic reticulum (ER) has been unidentified.
Results: Oligosaccharyltransferase (OST), the N-glycosylating enzyme, hydrolyzes dolichol-linked oligosaccharides to release the fOSs.
Conclusion: OST is responsible for the generation of fOSs in the ER lumen.
Significance: This study provides a mechanistic insight into the formation of luminal fOSs in yeast.
Keywords: Carbohydrate Metabolism, Carbohydrate Processing, Endoplasmic Reticulum (ER), Glycobiology, Glycosylation, Glycosyltransferases, Oligosaccharide, N-glycosylation, Free Oligosaccharides, Oligosaccharyltransferase
Abstract
Asparagine (N)-linked glycosylation regulates numerous cellular activities, such as glycoprotein quality control, intracellular trafficking, and cell-cell communications. In eukaryotes, the glycosylation reaction is catalyzed by oligosaccharyltransferase (OST), a multimembrane protein complex that is localized in the endoplasmic reticulum (ER). During N-glycosylation in the ER, the protein-unbound form of oligosaccharides (free oligosaccharides; fOSs), which is structurally related to N-glycan, is released into the ER lumen. However, the enzyme responsible for this process remains unidentified. Here, we demonstrate that eukaryotic OST generates fOSs. Biochemical and genetic analyses using mutant strains of Saccharomyces cerevisiae revealed that the generation of fOSs is tightly correlated with the N-glycosylation activity of OST. Furthermore, we present evidence that the purified OST complex can generate fOSs by hydrolyzing dolichol-linked oligosaccharide, the glycan donor substrate for N-glycosylation. The heterologous expression of a single subunit of OST from the protozoan Leishmania major in S. cerevisiae demonstrated that this enzyme functions both in N-glycosylation and generation of fOSs. This study provides insight into the mechanism of PNGase-independent formation of fOSs.
Introduction
N-Glycosylation is a fundamental and evolutionarily conserved post-translational protein modification that occurs in all domains of life (1, 2). In eukaryotes, the glycosylation often occurs co-translationally on nascent polypeptides emerging from the protein-conducting channel in the endoplasmic reticulum (ER)2 lumen (3, 4). Oligosaccharyltransferase (OST) catalyzes the transfer of the fully assembled glycan, Glc3Man9GlcNAc2, from its dolichylpyrophosphate-linked precursor to the target asparagine residue of polypeptide chains.
During the N-glycosylation process in mammalian cells, free oligosaccharides (fOSs) are generated in the ER lumen by a yet unclarified mechanism (5, 6), but fOSs are thought to be derived from either N-glycosylated proteins or dolichylpyrophosphoryl oligosaccharides (DLOs). The generation of fOSs from glycoproteins suggests that they result from the enzymatic deglycosylation of misfolded glycoproteins destined for proteasomal destruction by a process called ER-associated degradation. This deglycosylation reaction is catalyzed by the cytoplasmic peptide:N-glycanase (PNGase; Png1 in Saccharomyces cerevisiae) (7, 8), which is highly conserved in eukaryotes.
In S. cerevisiae, Png1-mediated cytosolic deglycosylation of glycoproteins is the dominant source for fOSs, as demonstrated by the marked reduction in, but not the complete depletion of, fOSs in png1Δ cells (9, 10). The glycoprotein-derived fOSs are catabolized solely by Ams1, a cytosol-vacuolar α-mannosidase (9, 10). On the other hand, the molecular basis underlying the Png1-independent generation of fOSs in S. cerevisiae remains to be explored.
EXPERIMENTAL PROCEDURES
Yeast Strains
Yeast strains used in this study were listed in Table 1. Various deletion mutants were generated by the PCR-based gene deletion technique (11, 12).
TABLE 1.
Yeast strains used in this study
| Name | Genotypes | Sources |
|---|---|---|
| ams1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 BY4741 | Open Biosystems |
| png1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 png1Δ::kanMX4 BY4741 | Open Biosystems |
| png1Δ ams1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 png1Δ::His3MX6 BY4741 | This study |
| png1Δ ams1Δ ost3Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 png1Δ::His3MX6 ost3Δ::hphNT1 BY4741 | This study |
| png1Δ ams1Δ ost5Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 png1Δ::His3MX6 ost5Δ::hphNT1 BY4741 | This study |
| png1Δ ams1Δ ost6Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 png1Δ::His3MX6 ost6Δ::hphNT1 BY4741 | This study |
| png1Δ ams1Δ ost3Δ ost6Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 png1Δ::His3MX6 ost3Δ::hphNT1 ost6Δ::natNT2 BY4741 | This study |
| png1Δ ams1Δ alg6Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::kanMX4 png1Δ::His3MX6 alg6Δ::hphNT1 BY4741 | This study |
| png1Δ atg19Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 png1Δ::kanMX4 atg19Δ::hphNT1 BY4741 | This study |
| png1Δ ams1Δ gls1Δ mns1Δ htm1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ams1Δ::His3MX6 png1Δ::hphNT1 gls1Δ::LEU2 mns1Δ::natNT2 htm1Δ::kanMX4 BY4741 | This study |
| OST4-FLAG | MAT a leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 OST4–3×FLAG::HisMX6 | This study |
Plasmids
Plasmids used in this study are listed in Table 2. pOST3 and pOST6 were generated using the In-Fusion HD cloning kit (Takara), genomic DNA from BY4741 as a template, and the primers listed in Table 3. Briefly, open reading frame of Ost3 and Ost6 that is flanked by 500 bp upstream of the initiation codon and 200 bp downstream of the stop codon was amplified, using the yeast genomic DNA (100 ng) and primers listed in Table 3, by KOD-Plus DNA polymerase (Toyobo) according to the manufacturer's instructions. YEp352 vector was amplified using the primers listed in Table 3 by PrimeSTAR Max DNA polymerase (Takara) according to the manufacturer's instructions. The PCR product of YEp352 vector was digested with DpnI (0.5 μl, 10 units; Toyobo) for 1 h at 37 °C and terminated by heating for 15 min at 75 °C. All of the PCRs were purified with the NucleoSpin Gel and PCR Clean-Up kit (Clontech) and subjected to recombination with the In-Fusion HD cloning kit. The reaction products were used for transformation of Escherichia coli DH5α strain (Toyobo). pScSTT3 was generated with In-Fusion HD cloning kit as described above, except that their coding sequences were amplified by using primers listed in Table 3. pRS425-GPD vector was amplified, using the primers listed in Table 3, by PrimeSTAR Max DNA polymerase.
TABLE 2.
Plasmids used in this study
| Name | Plasmids | Source/Reference |
|---|---|---|
| pOST3 | YEp352-OST3 | This study |
| pOST6 | YEp352-OST6 | This study |
| pLmSTT3D (WT) | pRS425-GPD-LmSTT3D (WT) | Ref. 36 |
| pLmSTT3D (E102A) | pRS425-GPD-LmSTT3D (E102A) | This study |
| pLmSTT3D (D104A) | pRS425-GPD-LmSTT3D (D104A) | This study |
| pLmSTT3D (D223N) | pRS425-GPD-LmSTT3D (D223N) | This study |
| pLmSTT3D (E225Q) | pRS425-GPD-LmSTT3D (E225Q) | This study |
| pLmSTT3D (D223N E225Q) | pRS425-GPD-LmSTT3D (D223N E225Q) | This study |
| pLmSTT3D (D639A) | pRS425-GPD-LmSTT3D (D639A) | This study |
| pLmSTT3D (D698A) | pRS425-GPD-LmSTT3D (D698A) | This study |
| pLmSTT3D (K701A) | pRS425-GPD-LmSTT3D (K701A) | This study |
| pScSTT3 | pRS425-GPD-ScSTT3 | This study |
TABLE 3.
Primers used in this study
| Primers | 5′ → 3′ | Notes |
|---|---|---|
| E102A-F | gatacctgatccacgcgttcgacccgtggtt | Mutagenesis |
| E102A-R | aaccacgggtcgaacgcgtggatcaggtatc | Mutagenesis |
| D104A-F | ctgatccacgagttcgccccgtggttcaactac | Mutagenesis |
| D104A-R | gtagttgaaccacggggcgaactcgtggatcag | Mutagenesis |
| D223N-F | atggcgggtgagttcaacaacgagtgcatcg | Mutagenesis |
| D223N-R | cgatgcactcgttgttgaactcacccgccat | Mutagenesis |
| E225Q-F | gtgagttcgacaaccagtgcatcgccgtc | Mutagenesis |
| E225Q-R | gacggcgatgcactggttgtcgaactcac | Mutagenesis |
| D639A-F | ttggcctggtgggcctacggctaccag | Mutagenesis |
| D639A-R | ctggtagccgtaggcccaccaggccaa | Mutagenesis |
| D698A-F | gggcagagcggcgccctgatgaagtca | Mutagenesis |
| D698A-R | tgacttcatcagggcgccgctctgccc | Mutagenesis |
| K701A-F | gcggcgacctgatggcgtcaccgcacatgg | Mutagenesis |
| K701A-R | ccatgtgcggtgacgccatcaggtcgccgc | Mutagenesis |
| Ost3-Inf-F | gaattcgagctcggtacccggggatccgatgtcattcccggcggctc | Cloning |
| Ost3-Inf-R | aagcttgcatgcctgcaggtcgactggactaggcagcctcacaaa | Cloning |
| Ost6-Inf-F | gaattcgagctcggtacccggggatcctgtctgagttaggagccaac | Cloning |
| Ost6-Inf-R | aagcttgcatgcctgcaggtcgacttcattcctgtgtgatttaa | Cloning |
| YEp352-Inf-F | gtcgacctgcaggcatgc | Cloning |
| YEp352-Inf-R | ggatccccgggtaccgag | Cloning |
| ScSTT3-Inf-F | gggctgcaggaattcatgggatccgaccggtcg | Cloning |
| ScSTT3-Inf-R | ggtatcgataagcttttagactctcaagcctaa | Cloning |
| p425GPD-Inf-F | aagcttatcgataccgtc | Cloning |
| p425GPD-Inf-R | gaattcctgcagcccggg | Cloning |
Mutagenesis
Mutagenesis of the pLmSTT3D plasmid was carried out using PrimeSTAR Max DNA polymerase (Takara) and the primers listed in Table 3.
Cell Culture
The standard yeast medium was used. The medium and 20% (w/v) glucose were separately autoclaved, and the autoclaved glucose was added to the medium at a concentration of 2% just before use. The yeast cells were spread from the 15% (v/v) glycerol stock onto YPD plates and grown for 3 days at 30 °C. The fresh single colony was inoculated in 50 ml of synthetic complete medium or selective dropout (SD) medium lacking appropriate nutrients and incubated for 24 h at 30 °C. For transformants, a fresh colony was first inoculated in 5 ml of the SD medium lacking appropriate nutrients for 24 h at 30 °C. The preculture was diluted to 0.2 A600 unit in 50 ml of the same medium and grown for 24 h at 30 °C.
Preparation of fOSs
The yeast cells were harvested and washed twice with 10 ml of the ice-cold phosphate-buffered saline (PBS). The washed cells were resuspended in 1 ml of lysis buffer (20 mm Tri-HCl, pH 7.4, and 10 mm EDTA), and then 3 ml of the ice-cold ethanol were added. The cells were homogenized by three 10-s vigorous agitations separated by 5-min cooling periods on ice. The homogenate was centrifuged at 15,000 × g for 15 min, and the supernatant was recovered as the soluble oligosaccharide fraction and evaporated to dryness. The dried soluble oligosaccharide fraction was desalted on the column containing a stack of AG50-X8 (250 μl, 200–400 mesh, H+ form) and AG1-X2 (250 μl, 200–400 mesh, acetate form) (Bio-Rad), followed by an InertSep GC column (150 mg/3 ml) (GL Sciences).
Subcellular Fractionation
The yeast cells were grown in YPD medium for 24 h to reach the cell density at ∼10 A600 units, resuspended in 10 ml of TSD buffer (100 mm Tris sulfate, pH 9.4, and 10 mm DTT), and incubated for 10 min at room temperature. The cells were harvested and resuspended in 5 ml of spheroplasting buffer (0.75× YP, 2% glucose, 1.2 m sorbitol, and 20 mm Tris-HCl, pH 7.4) supplemented with 0.5 mg of zymolyase 100T (Seikagaku Co.). After incubation for 30 min at 30 °C with gentle agitations, the cells were harvested and gently washed twice with 10 ml of the ice-cold PBS containing 1.2 m sorbitol. The cells were resuspended in 300 μl of B88 (20 mm Hepes-KOH, pH 7.4, 300 mm KCl, 5 mm MgCl2, and 200 mm sorbitol) and homogenized in a Potter-Elvehjem homogenizer by 20 strokes on ice. The homogenate was centrifuged at 1,000 × g for 5 min at 4 °C, and the supernatant (S1) was recovered. These homogenization and centrifugation steps were repeated once on the pellet fraction under the same conditions as described above. The combined S1 fraction was centrifuged at 15,000 × g for 5 min at 4 °C, and the supernatant (S15) was recovered. The pellet (P15) was washed once with 300 μl of B88. The S15 fraction was ultracentrifuged at 100,000 × g for 20 min at 4 °C, and the supernatant (S100) was recovered as the cytosol fraction. The P15 and P100 were resuspended in 600 μl of B88. To prepare the soluble oligosaccharide fraction, ice-cold ethanol was added to a final concentration of 75% and incubated for 15 min at 0 °C. After centrifugation at 15,000 × g for 15 min, the supernatant was evaporated to dryness.
Preparation of DLOs
The yeast cells were cultured, harvested, and washed as described above and resuspended in 2 ml of methanol. After the addition of an equal volume of glass beads (0.5 mm, Yasui Kikai, Kyoto, Japan), the cells were homogenized with a bead beater (Yasui Kikai) by three 1-min vigorous agitations separated by 1-min cooling periods on ice. The homogenate was centrifuged at 3,000 × g for 5 min, and the pellet was further washed twice with 5 ml of methanol. The washed pellet was extracted twice with 5 ml of chloroform/methanol (2:1 by volume). The pellet was dried under a nitrogen stream and extracted twice with 5 ml of methanol/water (1:1 by volume) supplemented with 4 mm MgCl2. The DLOs were extracted twice from the pellet by incubation with 5 ml of chloroform/methanol/water (10:10:3 by volume) for 10 min at 37 °C. The supernatant was evaporated to dryness. The residual pellet was re-extracted twice with 1 ml of chloroform/methanol/water (10:10:3 by volume). The resultant supernatant was evaporated to dryness. The pellet was incubated with 1 ml of 20 mm HCl in isopropyl alcohol/water (1:1 by volume) for 30 min at 100 °C. After it was evaporated to dryness, the pellet was extracted three times with 1 ml of water and desalted on an InertSep GC column and then on the AG50-X8 and AG1-X2 column.
Preparation of N-Glycans
The yeast cells were cultured, harvested, and washed twice with 15 ml of water. The cell pellet was resuspended in 1 ml of 10 mm sodium citrate buffer, pH 6.0, and autoclaved for 2 h at 121 °C. After centrifugation at 15,000 × g for 5 min, the supernatant was incubated with 3 ml of ethanol for 15 min on ice. After centrifugation at 15,000 × g for 15 min, the pellet was dried and dissolved in 200 μl of 0.1 m ammonium bicarbonate with 1 mg/ml trypsin. After incubation for 1 h at 37 °C, trypsin was inactivated by heating for 10 min at 100 °C, and PNGase F (5 units; Roche Applied Science) was added. The reaction mixture was further incubated for 16 h at 37 °C. After inactivation by heating for 5 min at 100 °C, the specimen was desalted on the AG50-X8 and AG1-X2 column and then on an InertSep GC column.
Pyridylamination
Fluorescent labeling of the soluble oligosaccharides with 2-aminopyridine was carried out as described previously (10, 13). Briefly, the dried sample was incubated in 20 μl of 2.76 g/ml 2-aminopyridine in acetate for 1 h at 80 °C. After the reaction, 20 μl of 500 mg/ml dimethylamine boran in acetate was added to the mixture and further incubated for 1 h at 80 °C. Excess amounts of 2-aminopyridine was removed by a MonoFas silica gel spin column (GL Sciences) according to the manufacturer's instructions.
Preparation of the Pyridylaminated Standard Glycans
The pyridylaminated Man1GlcNAc2 (PA-Man1GlcNAc2) was prepared from N-glycans released from bovine ribonuclease B (Sigma) by PNGase F (Roche Applied Science), which were further digested with jack bean α-mannosidase (Seikagaku Co.) as described under “Glycosidase Digestions.”
Size Fractionation HPLC
PA-glycans were separated by size fractionation HPLC with a Shodex NH2P-50 4E column (4.6 × 250 mm; Shodex), as reported previously (10). The elution was achieved by two solvent gradients: solvent A (93% acetonitrile in 0.3% acetate (pH adjusted to 7.0 with ammonia)) and solvent B (20% acetonitrile in 0.3% acetate (pH adjusted to 7.0 with ammonia)). The flow rate was 0.8 ml/min. The column temperature was 25 °C. The gradient program was as follows: 0–5 min, isocratic 3% solvent B; 5–8 min, 3–33% solvent B; 8–40 min, 33–71% solvent B; 40–60 min, isocratic 3% solvent B. Fluorescence of the labeled glycans was detected at the excitation wavelength (310 nm) and the emission wavelength (380 nm).
For Figs. 2E, 3 (B and D), 4 (B and C), 5 (A, B, and F), and 6 (A–C) and Table 5, the amounts of PA-oligosaccharides were estimated by size fractionation HPLC, based on the peak area of PA-glucose hexamer (PA-Glc6) included in the PA-glucose oligomer (degree of polymerization = 3–15; Takara). To determine the sizes of the fOSs generated in png1Δ atg19Δ cells (Table 5), the isolated peaks in the size fractionation HPLC were digested with jack bean α-mannosidase, as described under “Glycosidase Digestions.” The digests were reanalyzed by size fractionation HPLC under the same conditions. As a control, the standard Man1GlcNAc2, the expected product generated by jack bean α-mannosidase, was also analyzed in parallel to make sure that the fOSs are structurally related to the N-glycans.
FIGURE 2.

fOSs are generated in the ER lumen and processed by the ER glycosidases. A, models for the generation of fOSs by the Png1-dependent (left) and independent (right) pathways. OST transfers the oligosaccharide from Glc3Man9GlcNAc2-PP-Dol to proteins in the ER lumen (left). The N-glycans are sequentially processed by the ER glucosidase I (Gls1), glucosidase II (Gls2), and mannosidase I (Mns1). If the glycoproteins are properly folded, they are destined for the target organelle. When misfolded, the glycoproteins are further processed by another ER mannosidase, Htm1. The misfolded glycoproteins are retrotranslocated to the cytosol, where Png1 releases the glycans. In the cytosol, the glycoprotein-derived fOSs are catabolized by Ams1. In the right panel, it is totally unknown whether fOSs are generated independently from the action of Png1 in the ER lumen in S. cerevisiae (i); whether the fOSs, if any, are processed by the ER glycosidases (ii); and whether they are transported and catabolized (iii). B, a size fractionation HPLC profile of fOSs from png1Δ ams1Δ gls1Δ mns1Δ htm1Δ cells. The major glycan structures in peaks a–c are indicated in the HPLC chart. PA-glucose oligomers were used as a reference, and the glucose units (GU) are indicated on the HPLC chart. C, quantitation of fOSs in png1Δ ams1Δ gls1Δ mns1Δ htm1Δ cells. D, quantitation of DLOs in png1Δ ams1Δ gls1Δ mns1Δ htm1Δ cells. Structural determination and quantitation of fOSs and DLOs were carried out by dual-gradient, reversed-phase HPLC (14). Note that several unusual DLO intermediates (M7D, M8A, G3M8B, and G3M8C) were generated as minor biosynthetic products in the ER glycosidase-deficient cells. E, subcellular fractionation experiments using png1Δ ams1Δ cells. The cells were fractionated into S100, P15, and P100 fractions by differential centrifugation. Total fOSs were set to 100%. N.D., not detected. F, Western blot analysis of the S100, P15, and P100 fractions. Top, anti-Kar2 antibody; bottom, anti-3-phosphoglycerate kinase antibody. Percentages of the recovery of each protein are indicated in parentheses.
FIGURE 3.

OST is involved in the generation of fOSs. A, N-glycosylation status of carboxypeptidase Y (CPY) in png1Δ ams1Δ cells and those lacking OST3, OST5, OST6, both OST3 and OST6, or ALG6. Whole cell lysate was analyzed by Western blot with anti-CPY antibody. The number of N-glycans (Gly) on CPY is indicated by arrows. The average number of N-glycans on CPY was calculated by quantification of each CPY band. B, relative amounts of the total PA-fOSs in yeast mutant cells used in A. The total fOSs in png1Δ ams1Δ cells were set to 1.0. C, N-glycosylation status of CPY in png1Δ ams1Δ ost3Δ cells carrying empty vector (vector) or the plasmid encoding either OST3 (pOST3) or OST6 (pOST6). D, relative amounts of the total PA-fOSs in yeast mutant cells used in C. The total fOSs in png1Δ ams1Δ cells carrying pOST3 were set to 1.0. *, p = 0.03 (Student's t test). Error bars, S.D. from three independent experiments.
FIGURE 4.
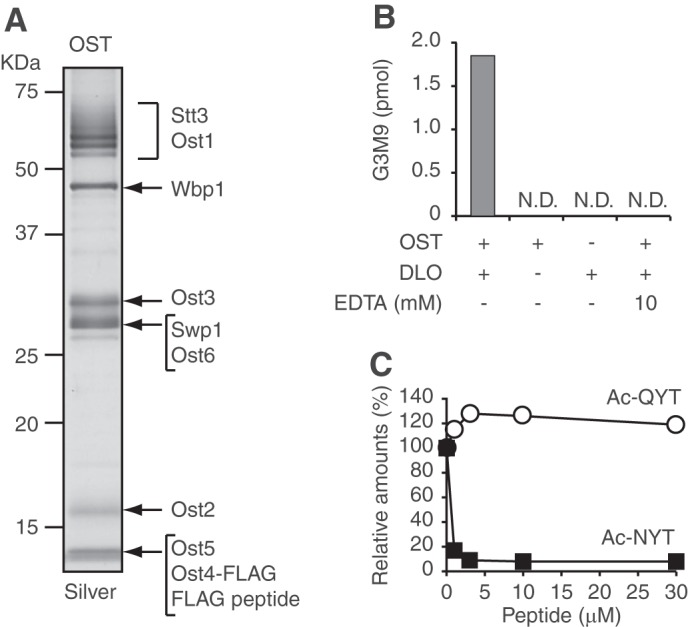
The purified OST complex generates fOSs directly from DLOs. A, SDS-PAGE of the purified OST complex (1.5 pmol), followed by silver staining. The migration positions of OST subunits were predicted based on previous reports (42, 43). B, in vitro fOS generation assay. OST, DLOs, and EDTA were incubated for 24 h in combinations as indicated at the bottom of the graph. After the reaction, the generated Glc3Man9GlcNAc2 oligosaccharide (G3M9) was quantitated. N.D., not detected. C, competition assay using acceptor and non-acceptor peptides. OST was incubated with DLOs in the presence or absence of various concentrations of Ac-NYT and Ac-QYT for 24 h. The amounts of G3M9 generated in the absence of the peptide were set to 100%. Closed squares, Ac-NYT; open circles, Ac-QYT.
FIGURE 5.

Stt3 is responsible for the generation of fOSs. A, quantitation of fOSs generated in png1Δ ams1Δ cells carrying either empty vector (vector) or plasmid encoding L. major STT3D (pLmSTT3D). The total fOSs from png1Δ ams1Δ cells carrying empty vector were set to 1.0. Error bars, S.D. from three independent experiments. B, quantitation of fOSs generated in png1Δ ams1Δ cells or png1Δ ams1Δ alg6Δ cells carrying either empty vector (vector) or plasmid encoding S. cerevisiae STT3 (pScSTT3). The total fOSs from png1Δ ams1Δ cells carrying empty vector were set to 1.0. Error bars, S.D. from three independent experiments. C, Western blot of whole cell lysates from png1Δ ams1Δ alg6Δ cells carrying either empty vector (vector) or plasmids encoding the wild type L. major STT3D (Lm (WT)) and the mutants. Top, anti-CPY; middle, anti-HA (for LmStt3D-HA); bottom, anti-DPM1 antibodies. The number of N-glycans (Gly) on CPY is indicated on the right. The average number of N-glycans on CPY was calculated by quantification of each CPY band. D, size fractionation HPLC profiles of the PA-fOSs from png1Δ ams1Δ alg6Δ cells carrying either empty vector (vector) or the wild type pLmSTT3D (Lm (WT)). Asterisks indicate the nonspecific peak derived from the labeling reagents. E, plasmid shuffling assay using stt3Δ cells carrying the empty vector (vector) or the plasmid encoding LmStt3D used in C. 5-Fold serial dilutions of the yeast cells were spotted on the agar plate lacking leucine and uracil (left) and one lacking leucine supplemented with 5-fluoroorotic acid (5-FOA) (right) and then grown for 6 days at 23 °C. F, relative amounts of the total PA-fOSs in png1Δ ams1Δ alg6Δ cells carrying either the empty vector (vector) or the plasmid encoding the wild type (WT) or mutant LmStt3D. The total fOSs from png1Δ ams1Δ alg6Δ cells carrying pLmSTT3D (WT) were set to 1.0. Error bars, S.D. from three independent experiments.
FIGURE 6.
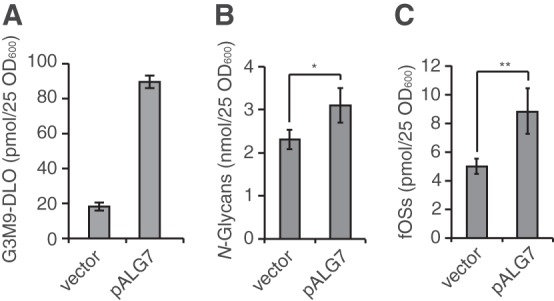
The generation of fOSs by OST is a tightly regulated process. Quantitation of Glc3Man9GlcNAc2-PP-Dol (G3M9-DLO) (A), total N-glycans (B), and total fOSs (C) in png1Δ ams1Δ cells carrying either the empty vector (vector) or the plasmid encoding ALG7 (pALG7). Error bars indicate the range of two independent experiments (A) and S.D. from three (B) or five (C) independent experiments. *, p = 0.04; **, p = 0.001 (Student's t test).
TABLE 5.
Quantitation of the PA-fOSs from png1Δ ams1Δ, png1Δ, and png1Δ atg19Δ cells
| Glycan structures | Amounts (relative amounts)a |
||
|---|---|---|---|
| png1Δ ams1Δ | png1Δ | png1Δ atg19Δ | |
| pmol/25 A600 (%) | |||
| Man1GlcNAc2 | NDb | 0.4 (3.9) | 39.4 (46.1) |
| Man2GlcNAc2 | NDb | 1.4 (15.5) | 23.2 (27.2) |
| Man3GlcNAc2 | NDb | 0.5 (5.9) | 6.9 (8.0) |
| Man4GlcNAc2 | NDb | 0.4 (4.1) | 2.1 (2.5) |
| Man5GlcNAc2 | NDb | 1.1 (11.5) | 2.2 (2.5) |
| Man6GlcNAc2 | NDb | 1.0 (10.5) | 4.0 (4.7) |
| Man7GlcNAc2 | 1.1 (16.5) | 1.3 (14.2) | 4.4 (5.2) |
| Man8GlcNAc2 | 5.0 (75.3) | 2.9 (31.7) | 3.1 (3.7) |
| Man9GlcNAc2 | 0.5 (8.2) | 0.2 (2.7) | 0.1 (0.1) |
| Total amount | 6.6 (100) | 9.2 (100) | 85.4 (100) |
a The amounts of fOSs were estimated from peak area of PA-Glc6 in size fractionation HPLC.
b ND, not detected.
Dual-gradient, Reversed-phase HPLC
Glycan isomers were separated by dual-gradient, reversed-phase HPLC with an Inertsil ODS-3 column (2.1-mm inner diameter × 150 mm; GL Sciences) (14). Elution was achieved by two solvent gradients: solvent A (0.1 m ammonium acetate buffer, pH 6.4) and solvent B (0.1 m ammonium acetate buffer, pH 4.0, and 0.5% 1-butanol). The flow rate was 200 μl/min. The column temperature was 25 °C. The gradient program was as follows: 0–10 min, isocratic 99% solvent A; 10–110 min, 99–30% solvent A; 110–150 min, isocratic 99% solvent A. Fluorescence of the labeled glycans was detected at the excitation wavelength (320 nm) and the emission wavelength (400 nm). Glucose units and amounts of each PA-fOS were determined as described previously (14). For Table 4 and Fig. 2 (C and D), fOSs and DLOs were quantitated by dual-gradient, reversed-phase HPLC.
TABLE 4.
Quantitation of the PA-labeled fOSs from png1Δ ams1Δ cells
| Glycan IDa | Amountsb |
Number of hexoses shifted after jack bean α-mannosidase digestionc | Number of hexoses shifted after α1,2-mannosidase digestiond | Number of hexoses shifted after α1,2-mannosidase and α1,6-mannosidase double digestione | Glucose unitg | |
|---|---|---|---|---|---|---|
| png1Δ ams1Δ | ams1Δ | |||||
| pmol/100 A600 | ||||||
| a | 2.0 | 134.8 | 6 | 2 | NDf | 5.76 |
| b-1 | 9.1 | 167.4 | 7 | 3 | ND | 4.91 |
| b-2 | 1.1 | 45.1 | 7 | 2 | 3 | 5.75 |
| c | 1.6 | 28.6 | 8 | 3 | 4 | 4.86 |
| Total amounts | 13.8 | 375.9 | ||||
a Glycan ID was based on Fig. 1B.
b Amounts of the fOSs were estimated from peak area of PA-Glc6 in dual-gradient, reversed-phase HPLC (14).
c PA-fOSs were isolated in dual-gradient, reversed-phase HPLC (14) and digested with jack bean α-mannosidase. The digests were analyzed by size fractionation HPLC.
d PA-fOSs were isolated in dual-gradient, reversed-phase HPLC (14) and digested with α1,2-mannosidase. The digests were analyzed by size fractionation HPLC.
e PA-fOSs were isolated in dual-gradient, reversed-phase HPLC (14) and double digested with α1,2-mannosidase and α1,6-mannosidase. The digests were analyzed by size fractionation HPLC.
f ND, not done.
g Glucose units of the fOSs were determined by dual-gradient, reversed-phase HPLC (14).
To unambiguously determine the structure of Man1GlcNAc2 in Table 5, the isolated corresponding peaks in the size fractionation HPLC were analyzed by the dual-gradient, reversed-phase HPLC in parallel with the standard PA-Man1GlcNAc2. The structural identity was judged by their identical elution position.
Glycosidase Digestions
The PA-glycans were incubated with 5 milliunits of endoglycosidase H (Roche Applied Science) in 20 μl of 10 mm sodium acetate buffer, pH 5.5, for 16 h at 37 °C. The numbers of mannose residues of the labeled glycans were determined by exoglycosidase digestions with the Jack bean α-mannosidase (40 milliunits; Seikagaku Co.) in 20 μl of 10 mm sodium citrate buffer, pH 4.5, or Aspergillus saitoi α1,2-mannosidase (0.5 milliunit; Seikagaku Co.) in 20 μl of 10 mm sodium acetate buffer, pH 5.5, for 16 h at 37 °C. For the enzyme digestion with Xanthomonas manihotis α1,6-mannosidase (New England Biolabs), the PA-glycans were incubated with 40 units of X. manihotis α1,6-mannosidase together with 40 milliunits of A. saitoi α1,2-mannosidase in 20 μl of 50 mm sodium citrate buffer, pH 4.5, supplemented with 0.1 mg/ml bovine serum albumin for 16 h at 37 °C. The reaction was terminated by the addition of 80 μl of water, followed by the addition of 300 μl of ethanol. After centrifugation at 15,000 × g for 15 min, the supernatant was evaporated to dryness and dissolved in a small volume of water.
Western Blot
Yeast cells (10 A600 units) were harvested and incubated in 200 μl of 0.1 m NaOH for 5 min at room temperature. After harvesting the cells, the pellet was resuspended in 100 μl of water, and then 100 μl of 2× SDS sample buffer (125 mm Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 0.01% bromphenol blue, and 10% β-mercaptoethanol) were added. The cell suspension was heated for 5 min at 100 °C. After a centrifugation at 15,000 × g for 5 min, the supernatant (equivalent to 0.5 A600 unit) was separated by SDS-PAGE and analyzed by Western blot with anti-carboxypeptidase Y (CPY) (10A5, Invitrogen), anti-Kar2 (y-115, Santa Cruz Biotechnology, Inc.), and anti-3-phosphoglycerate kinase (22C5, Invitrogen) antibodies. Visualization was performed with a LAS-3000 mini (Fujifilm).
For the detection of LmStt3D-HA, the yeast cells (500 A600 units) were resuspended in 1 ml of PBS, and an equal volume of the glass beads (0.5 mm) was added. The cells were lysed by three 1-min periods of bead beating separated by 1-min cooling period. The lysate was centrifuged at 1,000 × g for 5 min, and the supernatant was further centrifuged at 6,000 × g for 5 min. The supernatant was ultracentrifuged at 100,000 × g for 20 min. The pellet (microsomes) was resuspended in 100 μl each of a lysis buffer (20 mm Tris-HCl, pH 7.4, and 10 mm EDTA) and 2× SDS sample buffer. To denature, the sample was incubated for 1 h at 37 °C. 20 μl of the sample were analyzed by SDS-PAGE, followed by Western blot with anti-HA (F-7, Santa Cruz Biotechnology, Inc.) and anti-dolichol phosphate mannose synthase 1 (DPM1) (5C5, Invitrogen) antibodies.
Plasmid Shuffling Assay
stt3Δ cells (MATa met15 lys2 stt3Δ::KanMX4) that have been complemented with YEp352-GPD-LmSTT3D-HA were transformed with pLmSTT3Ds or p425GPD empty vector and selected on the SD plate lacking leucine and uracil for 2 days at 30 °C. The transformants were grown in SD medium lacking leucine and uracil for 2 days at 30 °C. The cells (15 A600 units) were serially diluted 5-fold in water, and 5 μl of the cell suspension were spotted on the SD plate lacking leucine and uracil and one lacking leucine supplemented with 1 mg/ml 5-fluoroortic acid. The cells were grown for 6 days at 23 °C.
Purification of the OST Complex Containing Ost4-FLAG
The yeast OST complex containing Ost4-FLAG was purified as described previously (15) with slight modifications. The microsomes (16,000 eq) (44) that were prepared from yeast strain W303-1A expressing Ost4-FLAG (MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 OST4-3×FLAG::HisMX6) were solubilized for 20 min at 0 °C in 10 ml of buffer A (20 mm Tris-HCl, pH 7.4, 500 mm NaCl, 1 mm MnCl2, 1 mm MgCl2, and 10% (v/v) glycerol) supplemented with 1.5% (w/v) digitonin (Calbiochem). After ultracentrifugation at 100,000 × g for 20 min at 4 °C, the supernatant was diluted by the addition of 20 ml of buffer A to reduce the concentration of digitonin. To immunoprecipitate the OST complex containing Ost4-FLAG, the diluted supernatant was incubated with 500 μl of anti-FLAG M2 beads (Sigma-Aldrich) for 2 h at 4 °C. After washing the beads three times with 3 ml of buffer B (20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm MnCl2, 1 mm MgCl2, and 0.2% (w/v) digitonin), the bound materials were eluted by incubation of the washed beads twice with 750 μl of buffer B supplemented with 0.2 mg/ml 3× FLAG peptide (Sigma-Aldrich) for 30 min at 4 °C.
To further purify the OST complex, the eluate (500 μl) from anti-FLAG M2 beads was loaded onto the top of a glycerol density step gradient that consisted of 1 ml each of 10, 20, 30, and 40% (v/v) glycerol in buffer B. After ultracentrifugation at 200,000 × g for 2 h at 4 °C, nine fractions (500 μl each) were manually taken from the top to the bottom of the glycerol density gradient. All fractions were analyzed in parallel with bovine serum albumin as an external standard for the quantitation of OST by SDS-PAGE, followed by silver staining. The amounts of the OST complex were estimated as those of the Wbp1 subunit. The OST complex was enriched in the third and fourth fractions from the top of the glycerol density gradient.
Large Scale Preparation of DLOs
A large scale preparation of DLOs was carried out as described previously (16) with slight modifications. Yeast haploid cells BY4741 (Open Biosystems) that were transformed with pALG7 were inoculated in 6 liters of SD medium lacking leucine and incubated for 24 h at 30 °C. The cells (30,000 A600 units) were harvested, washed twice with 500 ml of water, and resuspended in 150 ml of lysis buffer (10 mm Tris-HCl, pH 7.4, 50 mm potassium acetate, 200 mm sorbitol, 1 mm DTT, and 2 mm EDTA). The resuspended cells were added to an equal volume of glass beads and lysed with a bead beater by three 1-min homogenizations separated by 1-min cooling periods. After centrifugation at 1,000 × g for 5 min, the supernatant was recovered and further centrifuged at 6,000 × g for 5 min. The resultant supernatant was ultracentrifuged at 100,000 × g for 30 min, and the microsomal pellet was resuspended in 10 ml of 20 mm Tris-HCl, pH 7.4. The microsomes were added to 50 ml of chloroform/methanol (3:2, v/v), and the mixture was vigorously agitated. After centrifugation at 2,000 × g for 5 min, the upper and lower liquid phases were discarded so as not to disturb the wafer at the interface between the two liquid phases. The wafer was extracted three times with 30 ml of chloroform/methanol (2:1, v/v) containing 4 mm MgCl2. After centrifugation under the same conditions as described above, the pellet was dried with nitrogen gas and further extracted three times with 30 ml of methanol/water (1:1) containing 4 mm MgCl2. After the centrifugation, the pellet was extracted twice with 10 ml of chloroform/methanol/water (10:10:3, v/v/v). After the centrifugation, the supernatant that contains DLOs was dried with a rotary evaporator and dissolved in 8 ml of chloroform/methanol/water (10:10:3, v/v/v). To quantitate Glc3Man9GlcNAc2-PP-Dol (G3M9-DLO) by size fractionation HPLC, the oligosaccharide moiety was released from DLOs by mild acid hydrolysis, desalted, and labeled with 2-aminopyridine as described under “Preparation of DLOs” and “Pyridylamination.”
fOS Generation Assay
The purified OST complex (1.5 pmol) was incubated for 24 h at 30 °C in 100 μl of reaction buffer (20 mm Tris-HCl, pH 7.4, 5 mm MnCl2, 5 mm MgCl2, 0.3 μm G3M9-DLO, 0.1% (v/v) Triton X-100, and 0.25 mg/ml phosphatidylcholine) (3). For competition assays, various concentrations of Nα-acetyl-Asn-Tyr-Thr (Ac-NYT) or Nα-acetyl-Gln-Tyr-Thr (Ac-QYT) peptide (RIKEN Research Resource Center) were added to the reaction mixture. The reaction was terminated by the addition of 300 μl of ethanol, followed by incubation for 15 min at 0 °C. After centrifugation at 15,000 × g for 15 min, the supernatant was evaporated to dryness. The resultant pellet was dissolved in 1 ml of water and desalted on InertSep GC column. The fOS fraction was labeled with PA as described under “Pyridylamination” and analyzed by size fractionation HPLC.
RESULTS
fOSs Are Generated in the ER Lumen of png1Δ ams1Δ Cells
To clarify the origin and turnover mechanism of fOSs generated via the Png1-independent process, fOSs were prepared from png1Δ ams1Δ double mutant cells of S. cerevisiae and were analyzed by size fractionation HPLC (Fig. 1A, top). The glycans that eluted as peaks a–c in Fig. 1A (top) were susceptible to endoglycosidase H treatment (data not shown), suggesting the presence of high mannose type glycans. Consistent with a previous report (9, 10), the amount of fOSs in png1Δ ams1Δ cells was ∼4% of that in ams1Δ cells (Fig. 1A and Table 4). The main structures of fOSs generated in png1Δ ams1Δ cells were determined by dual-gradient, reversed-phase HPLC (14) and various glycosidase digestions and were found to consist of Man7–9GlcNAc2 (Fig. 1B (for the results of quantification, see Table 4)).
FIGURE 1.

Characterization of fOSs in yeast mutant cells. A, comparison of size fractionation HPLC profiles of the PA-fOSs from png1Δ ams1Δ cells (25 A600 units; top) and ams1Δ cells (2.5 A600 units; bottom). The endoglycosidase H-sensitive peaks a–c in png1Δ ams1Δ cells are indicated by arrowheads. The major structures of fOSs in ams1Δ cells are indicated in the HPLC chart. PA-glucose oligomers (degree of polymerization = 3–15) were used as a reference, and the glucose units (GU) are indicated on the HPLC chart. An asterisk indicates the nonspecific peak derived from the labeling reagents. B, the major glycan structures of the PA-fOSs found in peaks a–c in A. The glycan structures were determined by their elution positions in dual-gradient, reversed-phase HPLC (14) and various α-mannosidase digestions (Table 4). Orientations of the α-mannosidic linkages are indicated on the right.
We next determined if these glycan isoforms were formed as a result of the action of ER glycosidases (Fig. 2A), particularly α-glucosidases (Gls1 and Gls2) and α-mannosidases (Mns1 and Htm1) (17). To this end, fOSs were prepared from a quintuple deletion mutant, png1Δ ams1Δ gls1Δ mns1Δ htm1Δ, of S. cerevisiae. Glc3Man9GlcNAc2 oligosaccharide was identified as the major fOS (peak a in Fig. 2, B and C), suggesting that in png1Δ ams1Δ cells, the fOSs are first generated as Glc3Man9GlcNAc2, which is then processed by glycosidases in the ER. Glc3Man8GlcNAc2 oligosaccharide was also detected as a minor peak (peak b in Fig. 2, B and C). When the DLO fraction was extracted from png1Δ ams1Δ gls1Δ mns1Δ htm1Δ cells and analyzed by dual-gradient, reversed-phase HPLC, the identical structure was also observed (Fig. 2D). Because the ratio of Glc3Man9GlcNAc2 and Glc3Man8GlcNAc2 was similar between the fOS and DLO fractions, the truncated oligosaccharide was probably derived from the truncated DLO rather than processing by an unknown α-mannosidase in the ER glycosidase-deficient cells. We also noted that the glycans corresponding to peaks b-2 and c in Fig. 1B were modified by the Golgi α1,6-mannosyltransferase Och1 (18, 19). Consistent with this result, a minor peak corresponding to Glc3Man10GlcNAc2 was detected in the fOS fraction obtained from png1Δ ams1Δ gls1Δ mns1Δ htm1Δ cells (peak c in Fig. 2B).
Subcellular fractionation experiments using png1Δ ams1Δ cells revealed that over 60% of fOSs were found in the P15 fraction (Fig. 2E), which contains ER proteins, as judged by the enrichment of the ER chaperone Kar2 (Fig. 2F). This result supported our idea that fOSs produced in png1Δ ams1Δ cells are generated in the ER lumen.
fOSs Generated in the ER Lumen Are Transported to the Cytosol and Degraded by Ams1
We next examined how fOSs generated in png1Δ ams1Δ cells are catabolized. Because fOSs were not detected in the culture medium of png1Δ ams1Δ cells (data not shown), we hypothesized that they are catabolized intracellularly. To determine if Ams1 is involved in their catabolism, as is the case for the glycoprotein-derived fOSs, we first compared the structures of fOSs between png1Δ ams1Δ cells and png1Δ cells. As shown in Table 5, the demannosylation of fOSs occurred in png1Δ cells, as judged by an accumulation of Man1–6GlcNAc2, indicating that Ams1 is involved in the catabolism of fOSs.
Ams1 is an α-mannosidase synthesized in the cytosol and transported to the vacuole via the cytoplasm-to-vacuole targeting pathway (20). We next addressed the question of which cellular compartment is the site for the catabolism of fOSs generated in the ER lumen. We analyzed the structures of fOSs in png1Δ atg19Δ cells. Atg19 is involved in the cytoplasm-to-vacuole targeting pathway, and in yeast cells lacking ATG19, Ams1 accumulates in the cytosol (20). Interestingly, when fOSs from png1Δ atg19Δ cells and png1Δ cells were compared, we found accumulation of Man1–7GlcNAc2 in png1Δ atg19Δ cells (Table 5). This result indicated that fOSs generated in the ER lumen are transported to the cytosol and efficiently degraded by the cytoplasmic Ams1. It is of note that a similar transport mechanism has been found in mammalian cells, which is mediated by a putative oligosaccharide transporter in the ER membrane (21, 22). It should also be noted that the total amounts of fOSs are greatly increased in png1Δ atg19Δ cells by unknown mechanisms. Further studies will be needed to clarify this issue.
OST Is Involved in the Generation of fOSs in png1Δ ams1Δ Cells
In mammals, the fully assembled DLO substrate is thought to be hydrolyzed by unidentified mechanisms that lead to the release of the Glc3Man9GlcNAc2 oligosaccharide (23). Previous biochemical analyses on the hydrolytic activity of calf thyroid microsomes or tissue slices (24) have suggested that OST may hydrolyze DLOs. However, no direct experimental evidence for the involvement of OST has been presented. To test whether OST is involved in the generation of fOSs in S. cerevisiae, the N-glycosylation activity of OST was impaired by gene disruptions in png1Δ ams1Δ cells. In S. cerevisiae, OST is a multimembrane protein complex composed of five essential gene products (Stt3p, Wbp1p, Swp1p, Ost1p, and Ost2p) and three non-essential gene products (Ost4p, Ost5p, and either Ost3p or Ost6p), which are required for optimal glycosylation activity (1). For example, a deletion of OST5 is known to cause the mild N-glycosylation defects (25). Here, png1Δ ams1Δ ost5Δ cells showed a very weak hypoglycosylation of CPY, as visualized by the numerous bands of CPY signals in the Western blot analysis (Fig. 3A). Notably, the level of fOSs was also reduced by approximately half in the OST5 deletion strain (Fig. 3B). A deletion of the OST3 resulted in a more severe hypoglycosylation of CPY (Fig. 3A) and in a marked reduction of fOSs (Fig. 3B). Taken together, these data suggested that OST is involved in the generation of fOSs. In addition, we noticed that deletion of OST6, a paralog of OST3, had less effect on both N-glycosylation and the generation of fOSs (Fig. 3, A and B). Because it has been estimated that the Ost3-containing OST complex consists of ∼80% of the total OST in S. cerevisiae (26), the effects of OST6 overexpression in png1Δ ams1Δ ost3Δ cells on the generation of fOSs were examined. Interestingly, we found that OST6 overexpression partially rescued the hypoglycosylation of CPY (Fig. 3C), whereas the level of fOSs was only slightly increased (Fig. 3D). By comparing the average number of N-glycans on CPY (Fig. 3, A and C) and the amount of fOSs (Fig. 3, B and D), it was revealed that a positive correlation exists between the glycosylation efficiency and fOS level and that the efficient generation of fOSs required a high glycosylation efficiency (>3.44 N-glycans/CPY molecule).
The Fully Assembled DLO Substrate Is Required for the Efficient Generation of fOSs by OST
In S. cerevisiae, OST has a preferred glycan specificity toward the completely assembled Glc3Man9GlcNAc2-PP-Dol, whereas the incompletely assembled DLOs are suboptimal donor substrates for N-glycosylation (27). Consistent with the previous reports, a deletion of ALG6, which leads to the generation of DLO lacking three glucose residues, resulted in the hypoglycosylation of CPY (Fig. 3A) (28). Interestingly, the amount of fOSs was markedly reduced in png1Δ ams1Δ alg6Δ cells (Fig. 3B), consistent with the in vivo OST activity. Taken together, these data showed that OST is involved in the generation of fOSs in S. cerevisiae lacking Png1.
The Purified OST Complex Generates fOSs Directly from DLOs
Although we showed that the N-glycosylation activity of OST is important for the generation of fOSs, it was unclear whether fOSs are directly released from DLOs because the release of fOSs can occur as a post-transfer reaction by an unclarified enzyme from glycoproteins. To address this issue, we performed an in vitro fOS generation assay using the immunopurified OST complex containing the FLAG-tagged Ost4 (Fig. 4A) and a DLO preparation. Interestingly, the incubation of OST and DLOs resulted in the formation of Glc3Man9GlcNAc2 oligosaccharide (Fig. 4B), which was observed only when both OST and DLOs were present in the reaction mixture (Fig. 4B). Under our experimental conditions, the reaction proceeded linearly up to 24 h (data not shown). These results clearly indicated that in vitro, OST can hydrolyze Glc3Man9GlcNAc2-DLO, resulting in the generation of the fOS. Next, to clarify whether the catalytic activity of OST is required for the generation of fOSs in vitro, the effect of EDTA, an inhibitor for the N-glycosylation reaction (29), was examined. Upon the addition of EDTA, the generation of fOSs by OST was completely abolished (Fig. 4B), supporting our findings that the N-glycosylation activity of OST is highly correlated with the generation of fOSs. Furthermore, co-incubation with an acceptor NYT peptide, but not with a non-acceptor QYT peptide, also reduced the generation of Glc3Man9GlcNAc2 oligosaccharide (Fig. 4C), strongly indicating that N-glycosylation and fOS generation reactions compete the same DLO pool. Collectively, these results indicated that OST has a hydrolytic activity to DLOs in vitro. The specific activity of the hydrolysis by the purified OST was calculated to be 3.5 pmol/min/mg protein.
Stt3, the Catalytic Subunit of OST, Functions in the Generation of fOSs in png1Δ ams1Δ Cells
To study the role of OST in the generation of fOSs in more detail in vivo, we performed analyses with a single-subunit form of OST found in kinetoplastids. The catalytic subunit of OST, Stt3, is evolutionarily conserved in all domains of life (30–32). The protozoan Leishmania major encodes four STT3 paralogs (STT3A–D) but does not encode any other OST subunits (33–35). The expression of LmStt3D alone in S. cerevisiae that lacks the essential OST genes is sufficient for N-glycosylation activity (31, 36). We found that the expression of LmStt3D in png1Δ ams1Δ cells increases the level of fOSs (Fig. 5A), albeit in the presence of the fully active, endogenous OST complex. This finding demonstrated that the Stt3 protein from L. major mediates the generation of fOSs. Interestingly, overexpression of S. cerevisiae Stt3 (ScStt3) in png1Δ ams1Δ cells, but not in png1Δ ams1Δ alg6Δ cells, also resulted in an increase in fOSs (Fig. 5B). Compared with S. cerevisiae OST, LmStt3 has a relaxed-glycan specificity to DLOs and can efficiently utilize the incompletely assembled, non-glucosylated substrates, such as Man9GlcNAc2-PP-Dol, for N-glycosylation (31, 34, 36), probably due to the fact that their natural glycan donor substrate is Man7GlcNAc2-PP-Dol (34). Accordingly, a hypoglycosylation of CPY in png1Δ ams1Δ alg6Δ cells was partially suppressed upon the overexpression of LmStt3D (Fig. 5C). Notably, LmStt3D also generated fOSs efficiently in this mutant strain (Fig. 5D), further supporting the hypothesis that LmStt3D utilizes Man9GlcNAc2 glycans for both N-glycosylation and the generation of fOS.
To evaluate the functional relationship between the N-glycosylation and glycan-releasing activity of OST, various mutants of LmStt3D were expressed in png1Δ ams1Δ alg6Δ cells. In Stt3 proteins, three evolutionarily conserved motifs (DXD-like, WWD, and DXXK) have been implicated in N-glycosylation (30–32). Mutations of these motifs in LmStt3D resulted in a failure to improve the hypoglycosylation of CPY in png1Δ ams1Δ alg6Δ cells (Fig. 5C). As a control, it was confirmed that all of the tested LmStt3D mutations could not rescue the lethal phenotype of yeast cells lacking STT3 (Fig. 5E), indicating that these motifs are critical for the function of OST. Importantly, the mutations that altered the N-glycosylation activity of LmStt3D also reduced the generation of fOSs (Fig. 5F), further supporting the idea that the N-glycosylation activity and fOS generation by OST are tightly linked.
The Generation of fOSs by OST Is Not Enhanced by Overproduction of the DLO Substrates
To ensure the efficient N-glycosylation, cells produce excess DLO substrates. One of the potential roles for the generation of fOSs is to fine tune the levels of DLO substrates by the hydrolysis. To test whether the generation of fOSs in png1Δ ams1Δ cells is regulated by the biosynthetic flux of DLO substrates, we examined the effect of an overproduction of DLO substrates. To this end, Alg7, the enzyme initiating the DLO biosynthesis, was overexpressed in png1Δ ams1Δ cells, resulting in an ∼5-fold increase in the amounts of Glc3Man9GlcNAc2-PP-Dol (Fig. 6A). In response to the overproduction of the DLO substrates, however, N-glycans and fOSs were increased only 1.4-fold (Fig. 6B) and 1.8-fold, respectively (Fig. 6C). This result indicated that overproduction of the fully assembled DLO does not result in a proportional increase of N-glycans and fOSs. These data suggested that, as with the case for N-glycosylation, the generation of fOSs by OST is not a stochastic process but is rather a highly regulated reaction.
DISCUSSION
In the present study, we provided conclusive evidence that eukaryotic OSTs generate fOSs as a consequence of the hydrolysis of DLOs in the ER lumen. Among OST subunits, we identified the catalytic Stt3 subunit as the enzyme responsible for the generation of fOSs. It has been shown that in the food-borne bacterial pathogen Campylobacter jejuni, the Stt3 ortholog, PglB, is responsible for the generation of fOSs (37). Periplasmic fOSs are thought to be important for the modulation of osmolarity, whereas the functional importance of fOSs in the ER remains unclear. Irrespective of function, however, our results suggested that the OST-mediated generation of fOSs may be a general feature of Stt3 proteins throughout evolution.
Comparison of specific activity between hydrolysis (3.5 pmol/min/mg protein) and N-glycosylation (300 pmol/min/mg protein) (3) for the purified OST complex showed that the efficiency of the hydrolytic reaction is ∼ that of the N-glycosylation reaction, indicating that the hydrolysis efficiency of OST is markedly lower than that for the N-glycosylation of nascent polypeptides. This result is in good agreement with the fact that PNGase-independent fOSs only account for ∼4% of total fOSs. In mammalian cells, a UDP-[3H]glucose labeling experiment revealed that the efficiency of fOS release was ∼35% that of N-glycosylation after 30 min (38). The release of fOSs in mammalian cells appears to be mediated almost exclusively by PNGase-independent processes, because the knockdown of PNGase did not reduce the amount of fOSs (39). These findings imply that the efficiency of hydrolytic reaction by mammalian OST is higher than that of yeast OST.
Positive correlation between the level of fOSs and the average glycans per CPY in yeast cells lacking various OST subunits revealed a requirement of the maximum N-glycosylation activity of OST for the efficient generation of fOSs. This result may explain, at least in part, the reason why the overexpression of Ost6 in png1Δ ams1Δ ost3Δ cells barely accumulated fOSs because it resulted in an average number of glycans per CPY (3.26) below the threshold (3.41–3.44; see Fig. 3A) required for the efficient generation of fOSs (Fig. 3B). Alternatively, it is also possible that the generation of fOSs is exclusively dependent on the action of the Ost3-containing OST complex. In either case, our data clearly indicated that the presence of Ost3 or Ost6 defines the fOS generation activity of yeast OST.
Identification of fOSs modified with an additional α1,6-linked Man residue in png1Δ ams1Δ cells raised an important question for their origin. This modification mediated by the Golgi α1,6-mannosyltransferase Och1 has been shown to occur not only on N-glycosylated glycoproteins destined for the cell wall compartment (18), but also on the fluorescently labeled oligosaccharides in vitro (40), raising the possibility that fOSs may also serve as a substrate for this glycosyltransferase. Because Och1 is localized in the Golgi (19, 41), our result may imply that a portion of fOSs may reach the Golgi and receive the modification. Alternatively, the ER-localized minor Och1 may be responsible for the modification of fOSs. In either case, because we could not detect the Och1-modified DLOs, it is unlikely that Och1 directly modifies the DLOs.
The ER-to-cytosol transport of fOSs was evident in S. cerevisiae. Surprisingly, an impairment of cytosol-to-vacuole transport of Ams1 promoted the demannosylation of fOSs generated in the ER, leading us to speculate that cytosolic Ams1 may have regulatory roles in the ER-to-cytosol transport of fOSs via yet unidentified mechanisms. Notably, similar ER-to-cytosol transport of fOSs has been identified in mammals, and this process has been shown to require ATP in streptolysin O-permeabilized cultured cells (21) or both ATP and the cytosol fraction in reconstituted mouse liver microsomes (22). Identification of the gene encoding the fOS transporter on the ER membrane will clarify the mechanism for the transport of fOSs as well as the function of Ams1 in this process.
We presented evidence that the reaction to generate fOSs is not regulated by the DLO pool. Interestingly, however, our in vitro fOS generation assay revealed that the OST complex has an ability to release fOSs directly from DLOs, implying that the binding of OST to acceptor peptides is not a prerequisite for the fOS generation reaction. Site-directed mutagenesis of LmStt3D revealed that substitution of an aspartate residue at position 223 of LmStt3D, which constitutes a potential DXD-like motif important for binding to divalent metal ions, to an Asn residue resulted in a loss of function of this enzyme to improve hypoglycosylation of CPY in png1Δ ams1Δ alg6Δ cells, but the amounts of fOSs were reduced only partially. This observation suggests that the Asp-223 residue in the DXD-like motif is indispensable for the N-glycosylation activity of OST, whereas this amino acid residue is not critical for the fOS-generation activity. Further studies, especially with an in vitro experiment using the purified OST enzyme, will be required to fully understand the regulatory mechanism of the OST-mediated generation of fOSs.
Irrespective of mechanism involved, the OST-mediated generation of fOSs has to be a strictly regulated reaction to ensure the efficiency and fidelity of the N-glycosylation. Notably, although the generation of fOSs by OST is much less active than that by Png1 in S. cerevisiae, the opposite situation is found in mammalian cells; PNGase-independent generation of fOSs is dominant (39). Clarification of how such regulation is achieved must await future studies.
Acknowledgments
We thank all members of the Glycometabolome Team and Dr. Shinya Hanashima (Osaka University, Japan) for fruitful discussions.
This study was partly supported by Grants-in-aid for Scientific Research 24770134 (to Y. H.) and 25291030 (to T. S.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Global Center of Excellence Program (to T. S.); and Swiss National Science Foundation Grant 31003A_127098 (to M. A.).
- ER
- endoplasmic reticulum
- PNGase
- peptide:N-glycanase
- PA-
- pyridylaminated
- OST
- oligosaccharyltransferase
- fOS
- free oligosaccharide
- DLO
- dolichylpyrophosphoryl oligosaccharide
- SD
- selective dropout
- CPY
- carboxypeptidase Y
- G3M9
- Glc3Man9GlcNAc2.
REFERENCES
- 1. Kelleher D. J., Gilmore R. (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R [DOI] [PubMed] [Google Scholar]
- 2. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 3. Harada Y., Li H., Li H., Lennarz W. J. (2009) Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. U.S.A. 106, 6945–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruiz-Canada C., Kelleher D. J., Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cacan R., Hoflack B., Verbert A. (1980) Fate of oligosaccharide-lipid intermediates synthesized by resting rat-spleen lymphocytes. Eur. J. Biochem. 106, 473–479 [DOI] [PubMed] [Google Scholar]
- 6. Hanover J. A., Lennarz W. J. (1981) Transmembrane assembly of membrane and secretory glycoproteins. Arch. Biochem. Biophys. 211, 1–19 [DOI] [PubMed] [Google Scholar]
- 7. Suzuki T., Park H., Hollingsworth N. M., Sternglanz R., Lennarz W. J. (2000) PNG1, a yeast gene encoding a highly conserved peptide:N-glycanase. J. Cell Biol. 149, 1039–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suzuki T., Seko A., Kitajima K., Inoue Y., Inoue S. (1994) Purification and enzymatic properties of peptide:N-glycanase from C3H mouse-derived L-929 fibroblast cells. Possible widespread occurrence of post-translational remodification of proteins by N-deglycosylation. J. Biol. Chem. 269, 17611–17618 [PubMed] [Google Scholar]
- 9. Chantret I., Frénoy J. P., Moore S. E. (2003) Free-oligosaccharide control in the yeast Saccharomyces cerevisiae. Roles for peptide:N-glycanase (Png1p) and vacuolar mannosidase (Ams1p). Biochem. J. 373, 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirayama H., Seino J., Kitajima T., Jigami Y., Suzuki T. (2010) Free oligosaccharides to monitor glycoprotein endoplasmic reticulum-associated degradation in Saccharomyces cerevisiae. J. Biol. Chem. 285, 12390–12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 12. Goldstein A. L., McCusker J. H. (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553 [DOI] [PubMed] [Google Scholar]
- 13. Hase S., Ikenaka T., Matsushima Y. (1979) Analyses of oligosaccharides by tagging the reducing end with a fluorescent compound. I. Application to glycoproteins. J Biochem. 85, 989–994 [DOI] [PubMed] [Google Scholar]
- 14. Suzuki T., Matsuo I., Totani K., Funayama S., Seino J., Taniguchi N., Ito Y., Hase S. (2008) Dual-gradient high-performance liquid chromatography for identification of cytosolic high-mannose-type free glycans. Anal. Biochem. 381, 224–232 [DOI] [PubMed] [Google Scholar]
- 15. Chavan M., Chen Z., Li G., Schindelin H., Lennarz W. J., Li H. (2006) Dimeric organization of the yeast oligosaccharyl transferase complex. Proc. Natl. Acad. Sci. U.S.A. 103, 8947–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelleher D. J., Karaoglu D., Gilmore R. (2001) Large-scale isolation of dolichol-linked oligosaccharides with homogeneous oligosaccharide structures. Determination of steady-state dolichol-linked oligosaccharide compositions. Glycobiology 11, 321–333 [DOI] [PubMed] [Google Scholar]
- 17. Aebi M., Bernasconi R., Clerc S., Molinari M. (2010) N-Glycan structures. Recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82 [DOI] [PubMed] [Google Scholar]
- 18. Nakayama K., Nagasu T., Shimma Y., Kuromitsu J., Jigami Y. (1992) OCH1 encodes a novel membrane bound mannosyltransferase. Outer chain elongation of asparagine-linked oligosaccharides. EMBO J. 11, 2511–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaynor E. C., te Heesen S., Graham T. R., Aebi M., Emr S. D. (1994) Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell Biol. 127, 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shintani T., Huang W. P., Stromhaug P. E., Klionsky D. J. (2002) Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev. Cell 3, 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moore S. E., Bauvy C., Codogno P. (1995) Endoplasmic reticulum-to-cytosol transport of free polymannose oligosaccharides in permeabilized HepG2 cells. EMBO J. 14, 6034–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haga Y., Totani K., Ito Y., Suzuki T. (2009) Establishment of a real-time analytical method for free oligosaccharide transport from the ER to the cytosol. Glycobiology 19, 987–994 [DOI] [PubMed] [Google Scholar]
- 23. Gao N., Shang J., Lehrman M. A. (2005) Analysis of glycosylation in CDG-Ia fibroblasts by fluorophore-assisted carbohydrate electrophoresis. Implications for extracellular glucose and intracellular mannose 6-phosphate. J. Biol. Chem. 280, 17901–17909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anumula K. R., Spiro R. G. (1983) Release of glucose-containing polymannose oligosaccharides during glycoprotein biosynthesis. Studies with thyroid microsomal enzymes and slices. J. Biol. Chem. 258, 15274–15282 [PubMed] [Google Scholar]
- 25. Reiss G., te Heesen S., Gilmore R., Zufferey R., Aebi M. (1997) A specific screen for oligosaccharyltransferase mutations identifies the 9 kDa OST5 protein required for optimal activity in vivo and in vitro. EMBO J. 16, 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spirig U., Bodmer D., Wacker M., Burda P., Aebi M. (2005) The 3.4-kDa Ost4 protein is required for the assembly of two distinct oligosaccharyltransferase complexes in yeast. Glycobiology 15, 1396–1406 [DOI] [PubMed] [Google Scholar]
- 27. Lehle L., Bause E. (1984) Primary structural requirements for N- and O-glycosylation of yeast mannoproteins. Biochim. Biophys. Acta 799, 246–251 [Google Scholar]
- 28. Reiss G., te Heesen S., Zimmerman J., Robbins P. W., Aebi M. (1996) Isolation of the ALG6 locus of Saccharomyces cerevisiae required for glucosylation in the N-linked glycosylation pathway. Glycobiology 6, 493–498 [DOI] [PubMed] [Google Scholar]
- 29. Welply J. K., Shenbagamurthi P., Lennarz W. J., Naider F. (1983) Substrate recognition by oligosaccharyltransferase. Studies on glycosylation of modified Asn-X-Thr/Ser tripeptides. J. Biol. Chem. 258, 11856–11863 [PubMed] [Google Scholar]
- 30. Lizak C., Gerber S., Numao S., Aebi M., Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 [DOI] [PubMed] [Google Scholar]
- 31. Hese K., Otto C., Routier F. H., Lehle L. (2009) The yeast oligosaccharyltransferase complex can be replaced by STT3 from Leishmania major. Glycobiology 19, 160–171 [DOI] [PubMed] [Google Scholar]
- 32. Igura M., Maita N., Kamishikiryo J., Yamada M., Obita T., Maenaka K., Kohda D. (2008) Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J. 27, 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McConville M. J., Mullin K. A., Ilgoutz S. C., Teasdale R. D. (2002) Secretory pathway of trypanosomatid parasites. Microbiol. Mol. Biol. Rev. 66, 122–154; table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Samuelson J., Banerjee S., Magnelli P., Cui J., Kelleher D. J., Gilmore R., Robbins P. W. (2005) The diversity of dolichol-linked precursors to Asn-linked glycans likely results from secondary loss of sets of glycosyltransferases. Proc. Natl. Acad. Sci. U.S.A. 102, 1548–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ivens A. C., Peacock C. S., Worthey E. A., Murphy L., Aggarwal G., Berriman M., Sisk E., Rajandream M. A., Adlem E., Aert R., Anupama A., Apostolou Z., Attipoe P., Bason N., Bauser C., Beck A., Beverley S. M., Bianchettin G., Borzym K., Bothe G., Bruschi C. V., Collins M., Cadag E., Ciarloni L., Clayton C., Coulson R. M., Cronin A., Cruz A. K., Davies R. M., De Gaudenzi J., Dobson D. E., Duesterhoeft A., Fazelina G., Fosker N., Frasch A. C., Fraser A., Fuchs M., Gabel C., Goble A., Goffeau A., Harris D., Hertz-Fowler C., Hilbert H., Horn D., Huang Y., Klages S., Knights A., Kube M., Larke N., Litvin L., Lord A., Louie T., Marra M., Masuy D., Matthews K., Michaeli S., Mottram J. C., Muller-Auer S., Munden H., Nelson S., Norbertczak H., Oliver K., O'Neil S., Pentony M., Pohl T. M., Price C., Purnelle B., Quail M. A., Rabbinowitsch E., Reinhardt R., Rieger M., Rinta J., Robben J., Robertson L., Ruiz J. C., Rutter S., Saunders D., Schafer M., Schein J., Schwartz D. C., Seeger K., Seyler A., Sharp S., Shin H., Sivam D., Squares R., Squares S., Tosato V., Vogt C., Volckaert G., Wambutt R., Warren T., Wedler H., Woodward J., Zhou S., Zimmermann W., Smith D. F., Blackwell J. M., Stuart K. D., Barrell B., Myler P. J. (2005) The genome of the kinetoplastid parasite, Leishmania major. Science 309, 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nasab F. P., Schulz B. L., Gamarro F., Parodi A. J., Aebi M. (2008) All in one. Leishmania major STT3 proteins substitute for the whole oligosaccharyltransferase complex in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 3758–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nothaft H., Liu X., McNally D. J., Li J., Szymanski C. M. (2009) Study of free oligosaccharides derived from the bacterial N-glycosylation pathway. Proc. Natl. Acad. Sci. U.S.A. 106, 15019–15024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spiro M. J., Spiro R. G. (1991) Potential regulation of N-glycosylation precursor through oligosaccharide-lipid hydrolase action and glucosyltransferase-glucosidase shuttle. J. Biol. Chem. 266, 5311–5317 [PubMed] [Google Scholar]
- 39. Chantret I., Fasseu M., Zaoui K., Le Bizec C., Yayé H. S., Dupre T., Moore S. E. (2010) Identification of roles for peptide:N-glycanase and endo-β-N-acetylglucosaminidase (Engase1p) during protein N-glycosylation in human HepG2 cells. PLoS One 5, e11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kitajima T., Chiba Y., Jigami Y. (2006) Saccharomyces cerevisiae α1,6-mannosyltransferase has a catalytic potential to transfer a second mannose molecule. FEBS J. 273, 5074–5085 [DOI] [PubMed] [Google Scholar]
- 41. Harris S. L., Waters M. G. (1996) Localization of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J. Cell Biol. 132, 985–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karaoglu D., Kelleher D. J., Gilmore R. (1997) The highly conserved Stt3 protein is a subunit of the yeast oligosaccharyltransferase and forms a subcomplex with Ost3p and Ost4p. J. Biol. Chem. 272, 32513–32520 [DOI] [PubMed] [Google Scholar]
- 43. Knauer R., Lehle L. (1999) The oligosaccharyltransferase complex from Saccharomyces cerevisiae. Isolation of the OST6 gene, its synthetic interaction with OST3, and analysis of the native complex. J. Biol. Chem. 274, 17249–17256 [DOI] [PubMed] [Google Scholar]
- 44. Kelleher D. J., Gilmore R. (1994) The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J. Biol. Chem. 269, 12908–129017 [PubMed] [Google Scholar]