

Background: Endocannabinoids can affect pancreatic β cell physiology.
Results: Anandamide and 2-arachidonoylglycerol binding to CB1 receptors induces focal adhesion kinase phosphorylation, which is a prerequisite of insulin release.
Conclusion: Focal adhesion kinase activation downstream from CB1 receptors couples cytoskeletal reorganization to insulin release.
Significance: Identifies the molecular blueprint of 2-arachidonoylglycerol signaling in the endocrine pancreas, and outlines a kinase activation cascade linking endocannabinoid signals to insulin release.
Keywords: Cannabinoid Receptors, Cytoskeleton, Endocannabinoids, Exocytosis, Focal Adhesion Kinase, Insulin Release
Abstract
Endocannabinoid signaling has been implicated in modulating insulin release from β cells of the endocrine pancreas. β Cells express CB1 cannabinoid receptors (CB1Rs), and the enzymatic machinery regulating anandamide and 2-arachidonoylglycerol bioavailability. However, the molecular cascade coupling agonist-induced cannabinoid receptor activation to insulin release remains unknown. By combining molecular pharmacology and genetic tools in INS-1E cells and in vivo, we show that CB1R activation by endocannabinoids (anandamide and 2-arachidonoylglycerol) or synthetic agonists acutely or after prolonged exposure induces insulin hypersecretion. In doing so, CB1Rs recruit Akt/PKB and extracellular signal-regulated kinases 1/2 to phosphorylate focal adhesion kinase (FAK). FAK activation induces the formation of focal adhesion plaques, multimolecular platforms for second-phase insulin release. Inhibition of endocannabinoid synthesis or FAK activity precluded insulin release. We conclude that FAK downstream from CB1Rs mediates endocannabinoid-induced insulin release by allowing cytoskeletal reorganization that is required for the exocytosis of secretory vesicles. These findings suggest a mechanistic link between increased circulating and tissue endocannabinoid levels and hyperinsulinemia in type 2 diabetes.
Introduction
Pancreatic β cells exhibit a remarkable ability to tune their secretory functions in response to altered tissue insulin demands. Glucose, nutrients (amino acids and lipids), neurohormonal signals, and lipid mediators (i.e. free fatty acids, diacylglycerol, phosphatidic acid) all efficaciously modulate insulin secretion (1, 2).
Recently, the presence of a functional endocannabinoid (eCB)5 system (that is, the enzymatic machinery to biosynthesize and degrade 2-arachidonoylglycerol (2-AG) and anandamide (AEA), as well as cannabinoid receptors) was identified in the endocrine pancreas (3–6). This endocannabinoid system is suggested to regulate insulin secretion (7, 8). However, our knowledge of the organization and function of eCB signaling during insulin release is limited. The consensus view on the molecular architecture of 2-AG and AEA signaling is that sn-1-diacylglycerol lipases (DAGLα/β) generate, whereas monoacylglycerol lipase (MGL), and perhaps the serine hydrolase ABHD6, degrade 2-AG. Details of AEA synthesis are more ambiguous with N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) and fatty acid amide hydrolase (FAAH) being implicated in AEA biosynthesis and breakdown, respectively (9, 10). Both 2-AG and AEA act on CB1 and CB2 cannabinoid receptors (CB1R/CB2R) (11, 12). AEA also activates transient receptor potential vanilloid type 1 channels (TRPV1) and G protein-coupled receptor 55 (GPR55) (13–18). Although convincing data suggest that mouse, rat, and human pancreatic β cells express CB1Rs (4, 6, 19), the signaling pathway linking agonist-induced CB1R activation to regulated insulin secretion (3, 5, 20, 21) (but see Ref. 6), beyond the activation of primary kinases, remains unexplored.
The “storage-limited model” of insulin release (22) recognizes two pools of insulin granules that undergo temporally distinct waves of exocytosis. Its first, the Ca2+ transient-dependent phase involves the rapid fusion of the readily releasable pool of granules at the plasma membrane. Its second phase requires the replenishment of released vesicles through the trafficking of reserve insulin granules to positions adjacent to the cell membrane. This replenishment, however, is impeded by the dense web of filamentous actin (F-actin) present in the subplasmalemmal compartment under non-stimulated conditions. Therefore, stimulus-dependent cytoskeletal remodeling is needed to eliminate the F-actin barrier. However, instead of merely disrupting or degrading the pre-existing F-actin barrier (23), actin polymerization in stress fibers is stabilized at focal adhesion (FA) plaques containing e.g. paxilin, talin, and vinculin and serving to route insulin granules toward the plasma membrane (24). This mechanism rests on focal adhesion kinase (FAK) whose activity regulates FA remodeling and turnover (25, 26), and tension signaling (27), facilitating glucose-stimulated insulin secretion (28). This concept is consistent with biphasic insulin release because second phase transport of insulin granules requires F-actin as a motive force (29, 30).
It is appealing to link cannabinoid receptor activation to the second phase of insulin secretion because CB1Rs can activate FAK (31). Moreover, both CB1R and GPR55 in neurons or malignant cells (32–34) can influence Rho family GTPases, which by interacting with FAK (35), furnish a cellular microenvironment facilitating secretory vesicle exocytosis. However, the contribution of FAK signaling downstream from cannabinoid receptors to regulated insulin secretion from pancreatic β cells remains unknown. Here, we hypothesized that agonist activation of cannabinoid receptors can orchestrate a signaling cascade via Akt (or alternatively via extracellular signal-regulated kinases (ERK1/2)) and FAK to trigger F-actin polymerization and FA plaque formation to facilitate second phase insulin release. We have also explored the ligand and receptor specificity and temporal dynamics of eCB-induced insulin release by combining molecular pharmacology and mouse genetics in INS-1E cells and in primary mouse pancreatic islets. Our data suggest that eCBs coordinately activate ERK1/2/Akt and FAK downstream from CB1R to render the cytoskeleton permissive for insulin secretion.
EXPERIMENTAL PROCEDURES
INS-1E Cells and Culture Conditions
INS-1E cells (36) were cultured at 37 °C in RPMI 1640 medium containing glucose (11 mm), HEPES (10 mm), heat-inactivated fetal bovine serum (FBS; 5%), sodium pyruvate (1 mm), β-mercaptoethanol (50 μm), penicillin (50 μg/ml), and streptomycin (100 μg/ml; all from Sigma). Cells were routinely subcultured in 24-well plates up to passage 120 and allowed to reach ∼80% confluence. For immunocytochemistry, INS-1E cells were plated on 12-mm coverslips coated with d-polyornithine (0.001%). The effect of the following drugs on insulin release, alone or in combination, was assessed: ACEA (100 nm), AEA (100 nm-10 μm), AM 251 (100 nm), capsazepine (10 μm), FAKi14 (1 μm), JWH133 (100 nm), JZL 184 (200 nm), O-2050 (100 nm; all from Tocris), and OMDM 188 (100 nm) (37). Insulin release was tested after either 30 min or 24 h of drug exposure. Cytoskeletal reorganization was tested after 30 min of stimulation unless otherwise stated.
Isolation of Pancreatic Islets
Islets from 6-week-old male wild-type and CB1R−/− mice (38) were obtained after perfusion of the pancreas with Hanks' balanced salt solution (Invitrogen) containing collagenase (type I, 0.33 mg/ml; Sigma) and HEPES (200 mm), followed by purification on Histopaque 1077 gradients (Sigma) (39). After repeated washes in Hanks' balanced salt solution containing 10% FBS, isolated islets were maintained in RPMI 1640 medium supplemented as above in humidified atmosphere (5% CO2) at 37 °C overnight.
RNA Isolation and Gene Expression Analysis
Total RNA was isolated from INS-1E cells, rat spleen, cortex, and cerebellum (as tissue-specific positive controls) (38, 40, 41) using the RNeasy Mini Kit (Qiagen) followed by DNase digestion, and verifying RNA integrity on 2% agarose gels (500 ng of RNA). cDNA was prepared by reverse transcription with random primers using the high-capacity cDNA Reverse Transcription Kit (Applied Biosystems) and PCR amplified (30 cycles) by rat-specific primer pairs (Table 1). PCR products were resolved on 2% agarose gels and imaged on a ChemiDoc XRS+ system (Bio-Rad).
TABLE 1.
List of PCR primers
PCR were performed with primer pairs amplifying short fragments for each gene. Primer pairs were custom-designed to efficiently anneal to homologous nucleotide sequences from rat.
| Gene | Primer sequence | Product size | Annealing temperature |
|---|---|---|---|
| CB1R | Forward: 5′-CGTCGTTCAAGGAGAATGAGG-3′ | 213 bp | 57 °C |
| Reverse: 5′-TGCCGATGAAGTGGTAGGAAG-3′ | |||
| CB2R | Forward: 5′-GAGTGGAGAGATCCGCTCTG-3′ | 93 bp | 57 °C |
| Reverse: 5′-GGGGCTTCTTCTTTCCCCTC-3′ | |||
| TRPV1 | Forward: 5′-AGCGAGTTCAAAGACCCAGA-3′ | 233 bp | 57 °C |
| Reverse: 5′-TTCTCCACCAAGAGGGTCAC-3′ | |||
| GPR55 | Forward: 5′-ACGGGAGTGTCTTCACCATC-3′ | 173 bp | 57 °C |
| Reverse: 5′-CACTCCCCTGTGGAAAGTGT-3′ | |||
| DAGLα | Forward: 5′-ACCTGCGGCATCGGTTAGAG-3′ | 81 bp | 57 °C |
| Reverse: 5′-TTGTCCGGGTGCAACAGAG-3′ | |||
| DAGLβ | Forward: 5′-CGAGCTGCCCTTCATAGTGG-3′ | 81 bp | 57 °C |
| Reverse: 5′-TCCTGGAGAGACATGGTCCC-3′ | |||
| FAAH | Forward: 5′-TGGAAGTCCTCCAAGAGCCCA-3′ | 197 bp | 57 °C |
| Reverse: 5′-TGTCCATAGACACAGCCCTTCAG-3′ | |||
| MGL | Forward: 5′-ACCAACCCACTTTTCTGGCA-3′ | 170 bp | 57 °C |
| Reverse: 5′-CAACCTCCGACTTGTTCCGA-3′ | |||
| NAPE-PLD | Forward: 5′-CGTGCTCAGATGGCTGATAA-3′ | 192 bp | 57 °C |
| Reverse: 5′-ATGAGCTCGTCCATTTCCAC-3′ |
Cyto- and Histochemistry
Mice were transcardially perfused with ice-cold phosphate buffer (PB, pH 7.4) followed by 4% paraformaldehyde in PB (2 ml/min flow speed). Pancreata were rapidly dissected and postfixed in 4% paraformaldehyde overnight. After equilibrating in 30% sucrose for 48–72 h, tissues were cryosectioned at a thickness of 14 μm and thaw-mounted onto SuperFrost+ glass slides. INS-1E cells were rinsed in 0.1 m PB and immersion fixed in 4% ice-cold paraformaldehyde for 20 min. After rinsing in 0.1 m PB, specimens were exposed to a blocking solution composed of 0.1 m PB, 10% normal donkey serum, 5% bovine serum albumin (BSA), and 0.3% Triton X-100 for 3 h followed by overnight incubation with select combinations of primary antibodies (Table 2). Secondary antibodies were applied in 0.1 m PB supplemented with 2% BSA (2 h, 20–22 °C). Alexa Fluor 488-phalloidin and Alexa Fluor 546-phalloidin (1:1,000, 1 h; Invitrogen) were applied simultaneously with secondary antibodies (1:300, Jackson). Antibody specificities were demonstrated using pancreas and/or brain tissues from CB1R−/− (38), MGL−/− (42), and DAGLα−/− (43) mice (n ≥ 2/genotype, 4–8 weeks of age) and corresponding wild-type littermates (n ≥ 2/genotype) (44). Brain sections were favored because detailed neuroanatomical maps exist on the molecular architecture of the eCB system in this organ (45, 46). Primary antibody (Table 2) binding was revealed by carbocyanine 2-, 3-, or 5-conjugated secondary antibodies (1:300, Jackson). Nuclei were routinely counterstained by Hoechst 33,342 (1:20,000; Sigma).
TABLE 2.
List of markers used for immunofluorescence labeling and Western blotting
Panel of antibodies applied to study the molecular composition and cell-type specificity of endocannabinoid signaling in INS-1E cells, native β cells and adult mouse brains. Antibodies have been previously characterized (44, 62).
| Antibody | Source | Concentration | Supplier |
|---|---|---|---|
| ABHD6 | Rabbit | 1:500 | Dr. K. Mackie |
| pAkt (Ser-473) | Rabbit | 1:500 | Cell Signaling |
| Akt | Goat | 1:500 | Cell Signaling |
| β-Actin HRP-conjugated | Mouse | 1:20,000 | Santa Cruz |
| CB1R | Rabbit | 1:1,000 | Abcam |
| CB1R | Guinea pig | 1:500 | Dr. K. Mackie |
| DAGLα | Guinea pig | 1:500 | Dr. K. Mackie |
| DAGLβ | Rabbit | 1:500 | Dr. K. Mackie |
| pERK1/2 (Thr-202/Tyr-204) | Rabbit | 1:1,000 | Cell Signaling |
| ERK1/2 | Mouse | 1:1,000 | Cell Signaling |
| FAAH | Rabbit | 1:1,000 | Dr. K. Mackie |
| pFAK | Rabbit | 1:500 | Cell Signaling |
| FAK | Rabbit | 1:500 | Cell Signaling |
| Glucagon | Mouse | 1:2,000 | Sigma |
| Insulin | Mouse | 1:500 | Cell Signaling |
| Insulin | Rabbit | 1:400 | Cell Signaling |
| MGL | Rabbit | 1:500 (histochemistry) | Dr. K. Mackie |
| MGL | Rabbit | 1:1,000 (Western) | Dr. K. Mackie |
| NAPE-PLD | Rabbit | 1:500 | Dr. K. Mackie |
| Pancreatic polypeptide | Guinea pig | 1:10,000 | Linco Research |
| Somatostatin | Rat | 1:200 | Dr. A. C. Cuello |
| TRPV1 | Rabbit | 1:200 | Santa Cruz |
| Vinculin | Mouse | 1:500 | Sigma |
INS-1E cells, pancreas, and brain sections were inspected on a Zeiss LSM700 laser-scanning microscope equipped with a ×40 water immersion objective using ×1.0–2.5 optical zoom and differential interference contrast when required (Fig. 2). Comparison of null and wild-type tissues was performed when keeping threshold and intensity settings identical. Images were acquired in the ZEN2010 software package. Cytoskeletal remodeling, defined as the CB1R-dependent formation of stress fibers and FA plaques, was quantified at ×40 primary magnification in n = 30–80 INS-1E cells/condition. Data from triplicate experiments were statistically analyzed (see below). Multi-panel images were assembled in CorelDraw X5 (Corel Corp.).
FIGURE 2.

Cannabinoid receptor stimulation induces the molecular rearrangement of 2-AG metabolism and signaling in INS-1E cells. A–C, INS-1E cells were stimulated with anandamide (AEA, 10 μm) for 2 or 24 h. Subsequently, the integrated immunofluorescence intensity (mean intensity/surface area excluding nuclei) for MGL (A), CB1R (B), and DAGLα (C) in cell cohorts was determined. D and D2, representative photomicrographs upon AEA exposure acquired by differential interference contrast (DIC)-laser scanning microscopy using uniform parameters of image acquisition. E, JZL 184 (200 and 500 nm) inhibited 4-nitrophenylacetate hydrolysis in INS-1E cell extracts, suggesting partial MGL inhibition in vitro (47). F, JZL 184 (200 nm) rapidly increased (30 min) and maintained (24 h) 2-AG partitioning in the culture supernatant of INS-1E cells, as determined by mass spectrometry. Equal volumes of lipid extracts were processed. G–I, experiments as in A–C in the presence of JZL 184 (200 nm) for 24 h in vitro. Integrated immunofluorescence intensity of MGL, CB1R, and DAGL at the single cell level was assayed using the ZEN2010 software package. J, cell viability assay after exposure of INS-1E cells to AEA (10 μm, 24 h) or vehicle. K, cell proliferation measured by BrdU incorporation in INS-1E cells after AEA (10 μm), JZL 184 (200 nm), or ACEA (100 nm) treatment alone or in combination with O-2050 (100 nm). Data were expressed as mean ± S.D., n ≥ 10 cells/group were analyzed in triplicate experiments. Scale bar = 10 μm (D). **, p < 0.01; *, p < 0.05 (one-way ANOVA).
Measurement of Monoacylglycerol Lipase-like Activity by 4-Nitrophenylacetate
INS-1E cells were treated with increasing concentrations of JZL 184 or vehicle for 24 h. Then, cells were washed, scraped, and sonicated in 50 mm sodium phosphate buffer (pH 8.0) containing 0.3 m sucrose (∼40 s) at 4 °C. Samples were then centrifuged at 100,000 × g at 4 °C for 50 min. Pellets containing the cell membrane fraction were collected, re-suspended in 10 mm Tris-HCl (pH 7.4), and stored at −80 °C until use. MGL-like activity was assayed in a 96-well plate format (microtiter plates, 200 μl total volume) as described (47). Samples (4 μg of protein/well in 150 μl of Tris-HCl (pH 7.4) also containing 0.1% fatty acid-free BSA (Sigma)) were aliquoted and mixed with JZL 184 (200 or 500 nm) or vehicle, the latter used as controls. 4-Nitrophenylacetate (Sigma), as substrate, was dissolved in 250 mm Tris-HCl (pH 7.4) and added to each well. MGL-like activity was determined by measuring the rate of 4-nitrophenylacetate hydrolysis at 10-min intervals in a colorimetric assay on an Infinite M1000 PRO microplate reader (Tecan) tuned at 405 nm at 37 °C. Samples containing buffer only were used as blanks.
Liquid Chromatography-Atmospheric Pressure Chemical Ionization-Mass Spectrometry
INS-1E cells were treated with JZL 184 (200 nm) for 30 min or 24 h (Fig. 2F). Cell-free supernatants were separated and flash-frozen in liquid N2. Cells were scraped in 2 ml of ice-cold methanol and kept at −20 °C until processing for liquid chromatography-atmospheric pressure chemical ionization-mass spectrometry. Extraction, purification, and quantification of 2-AG and AEA followed published protocols (5, 48). After lipid extraction and pre-purification on silica gel columns, endocannabinoid levels from 2–4 independent samples/condition were analyzed by isotope dilution using liquid chromatography-atmospheric pressure chemical ionization-mass spectrometry (Shimadzu LCMS-2020).
Cell Viability and Proliferation Assays
To assess cell viability, INS-1E cells were stimulated with AEA (10 μm) or vehicle for 24 h. Cell Titer Blue Reagent (Promega), diluted 1:10 in the culture medium, was added during the last 3 h of the incubation period. Media were then transferred onto 96-well plates and their λ = 570/590 nm absorbance ratio measured on an Infinite M1000 PRO microplate reader (Tecan). The absorbance of the culture medium (background) was subtracted. INS-1E cell proliferation was determined 24 h after exposure to AEA (10 μm), ACEA (100 nm), or JZL 184 (200 nm) with/without the presence of O-2050 (100 nm). Cell proliferation was determined by measuring the rate of bromodeoxyuridine (BrdU) incorporation with a colorimetric Cell Proliferation ELISA (Roche Applied Science).
Insulin Secretion from INS-1E Cells and Pancreatic Islets
INS-1E cells were washed with phosphate-buffered saline (PBS, 0.1 m, pH 7.4) and equilibrated in a modified Krebs-Ringer bicarbonate HEPES buffer containing NaCl (135 mm), KCl (3.6 mm), CaCl2 (1.5 mm), MgCl2 (0.5 mm), NaH2PO4 (1.5 mm), NaHCO3 (5 mm), HEPES (10 mm, pH 7.4), and BSA (0.1%) for 1 h (hereafter referred to as “KRBH”). Next, the cells were incubated in KRBH containing AEA (100 nm-10 μm), ACEA (100 nm), or JWH133 (100 nm) in the presence of low (2.5 mm) or high (15 mm) glucose at 37 °C for 30 min. Ca2+-free conditions were established by using KRBH lacking CaCl2. O-2050, capsazepine, or FAKi14 application preceded agonist exposure by 10 min. For 24 h stimulation, AEA (10 μm), JZL 184 (200 nm), or OMDM 188 (100 nm) was added to standard culture media for 23 h, followed by equilibration in KRBH for 1 h and glucose challenge in the continued presence of either or both drugs for 30 min.
CB1R−/− and wild-type pancreatic islets were plated in 24-well plates (10 islets/well) and incubated in KRBH for 1 h. Next, islets were incubated in KRBH supplemented with either 2.75 or 16.5 mm glucose to which ACEA (100 nm) or vehicle had been added at 37 °C for 30 min.
KRBH buffers from basal conditions and after stimulation were recovered to determine secreted insulin levels. The amount of insulin in the incubation buffers was measured by a rat/mouse Insulin ELISA Kit (Millipore) as per the manufacturer's recommendations. Insulin concentrations were normalized to the total protein content of INS-1E samples or to the number of stimulated islets.
Protein Measurement
Protein concentrations were determined by the Bradford method (Bio-Rad) with optical density of the total protein content measured at 595 nm. Protein concentrations were calculated against BSA standards.
Analysis of G-actin and F-actin Contents
INS-1E cells were preincubated in KRBH for 1 h followed by exposure to AEA (10 μm) or vehicle for 15 min. After treatment, cells were washed with ice-cold PBS. G-actin and F-actin fractions were obtained (49) by keeping INS-1E cells in actin stabilization buffer (PIPES (pH 6.9, 0.1 m), glycerol (30%), DMSO (5%), MgSO4 (1 mm), EGTA (1 mm), Triton X-100 (1%), ATP (1 mm), and protease inhibitors (Roche Applied Science)) on ice for 10 min. Cells were then scraped and centrifuged at 16,000 × g at 4 °C for 75 min. The supernatant containing G-actin was recovered, and the pellet containing F-actin was solubilized with actin depolymerization buffer consisting of PIPES (pH 6.9, 0.1 m), MgSO4 (1 mm), CaCl2 (10 mm), and cytochalasin D (5 μm). Aliquots were separated on 12% SDS-PAGE, blotted with HRP-conjugated anti-β-actin antibody (Promega), and signals were detected by the enhanced chemiluminescence (ECL) method (Pierce). Optical densitometry was performed in Image J to calculate the F/G-actin ratio.
Western Blotting
INS-1E cells were washed, scraped, and extracted using a radioimmunoprecipitation assay buffer (44) containing: Tris (pH 7.4, 50 mm), NaCl (150 mm), NaF (10 mm), EDTA (pH 8.0, 5 mm), Triton X-100 (1%), Na3VO4 (1 mm), PMSF (1 mm), pepstatin A (5 μg/μl), leupeptin (10 μg/μl), and aprotinine (2 μg/μl). Protein lysates were centrifuged at 12,000 × g at 4 °C for 10 min. Samples were denatured in Laemmli buffer and boiled at 95 °C for 5 min. Proteins were probed by loading 20-μg aliquots (8–10% gels) under denaturing conditions on SDS-PAGE, followed by wet transfer onto PVDF membranes (Millipore). Primary antibodies were listed in Table 2. β-Actin was used as loading control. After exposure to HRP-conjugated secondary antibodies (Bio-Rad, 1:10,000, 2 h), target proteins were visualized by the ECL method. ImageJ 1.45 with appropriate plug-ins was deployed to perform quantitative densitometry.
Statistics
Phosphoprotein levels were normalized to those of the respective non-phosphorylated (“total”) proteins. Experiments were performed in triplicate unless stated otherwise. Data were expressed as mean ± S.D. p < 0.05 was considered statistically significant. Data were analyzed using one-way ANOVA followed by pairwise comparisons when appropriate. Student's t test (independent group design) was used to statistically analyze data on immunofluorescence intensities. Correlation coefficients were calculated by Pearson's test.
RESULTS
Molecular Organization of Endocannabinoid Signaling Networks in INS-1E Cells
For rat β cell-derived INS-1E cells to serve as adequate tools to study the relationship of eCB signals and insulin release, they must recapitulate signaling arrangements seen in vivo (4). Particularly, because the complexity of eCB signals in whole islets (50) may be vastly different from that seen in functionally segregated cell types. Therefore, we first profiled the presence of relevant mRNAs by PCR, and validated our results against reference tissues (38, 41, 46).
We found CB1R, TRPV1, and CB2R (Fig. 1A) but not GPR55 mRNA in INS-1E cells. The mRNAs for metabolic enzyme components of 2-AG signaling (DAGLα, DAGLβ, MGL; Fig. 1A1) were also expressed. Western blotting confirmed the presence of CB1R, DAGLα, MGL, and ABHD6, the “prototypic” 2-AG signaling cassette, as well as of TRPV1 receptors (Fig. 1B).
FIGURE 1.

Molecular determinants of endocannabinoid signaling in INS-1E cells. A and A1, reverse transcription PCR products (arrowheads) of select receptor (A) and enzyme components of 2-AG (A1) signaling. Brain and spleen samples served as positive controls. Samples without reverse transcriptase (RT−) were used as negative controls. Note that INS-1E cells did not express GPR55 receptor mRNA at detectable levels (open arrowhead). B, Western analysis of antibodies used to reveal minimal requirements of 2-AG signaling. C–C2, CB1R, MGL, and DAGLα localization in INS-1E cells combining fluorescence and differential interference contrast (DIC) microscopy. D–G1, histochemical controls demonstrating the specificity of rabbit anti-CB1R (D and D1), guinea pig anti-CB1R (E and E1), anti-MGL (F and F1), and guinea pig anti-DAGLα (G and G1) in the pancreas (D and D1), hippocampus (E and F1), or cerebellum (G and G1) of adult wild-type and respective null mice (38, 42, 43). The lack of residual immunolabeling in knock-out tissues unequivocally supports the specificity of the antibodies used. Hoechst 33,342 was used as nuclear counterstain. mRNA (H) and protein (I) detection for NAPE-PLD (N-PLD) and fatty acid amide hydrolase (FAAH), involved in anandamide metabolism (4, 9), in INS-1E cells. Arrows in B and I point to immunoreactive protein bands at the calculated molecular weight of each target. Abbreviations: mol, molecular layer of the cerebellum; PC, Purkinje cell layer; pyr, pyramidal layer of the CA1 hippocampal subfield. Scale bars = 100 μm (D1), 15 μm (C–C2), and 10 μm (E1 and F1).
In INS-1E cells, both CB1R and DAGLα were abundant at the plasmalemma, and also filled most of the cytoplasm and processes (Fig. 1, C and C1). MGL immunoreactivity appeared punctate in both the perisomatic compartment and processes (Fig. 1C2). Given our primary focus on 2-AG signaling, we also verified the specificity of CB1R, DAGLα, and MGL immunosignals in pancreas and/or nervous tissues from respective null mice (Fig. 1, D–G1).
We suggest that AEA signaling might also operate in INS-1E cells because both mRNA (Fig. 1H) and protein (Fig. 1I) of the enzymes NAPE-PLD and FAAH, partaking in AEA biosynthesis and degradation, respectively (9, 52), were detected.
CB1R Activation Promotes DAGLα Expression in INS-1E Cells
Presence of the molecular constituents required for both 2-AG and AEA signaling prompted us to test whether agonist stimulation of cannabinoid receptors modulates the “2-AG signaling cassette” (that is, metabolic enzymes and receptors). In testing cell- or tissue-specific eCB signal specificity we focused on 2-AG because the enzymes required for its metabolism have been more unequivocally identified (10, 53). We exploited direct and indirect stimulus protocols: first, in pharmacological protocols we applied exogenous AEA for 30 min or 24 h, whose (∼12 h) half-life is compatible with prolonged stimulation in vitro (54). Second, we inhibited MGL activity by JZL 184 (55) for 30 min or 24 h to increase endogenous 2-monoacylglycerol (i.e. 2-AG) availability.
If cannabinoid receptor activation cell autonomously regulates 2-AG metabolism in INS-1E cells then stimulation by an eCB could modify the coordinated expression of synthetic and degrading enzymes. AEA (10 μm) exposure of INS-1E cells significantly decreased MGL availability after 2 and 24 h (p < 0.05; Fig. 2, A and D), as measured by quantitative immunocytochemistry. Simultaneous detection of CB1Rs revealed the removal of this receptor from the cell surface and accumulation in the perinuclear cytoplasm (Fig. 2D1). This coincided with decreased CB1R content (Fig. 2B) suggesting intracellular degradation (56). In contrast, the DAGLα protein, which is present at greater levels than DAGLβ in the endocrine pancreas (57), significantly increased (Fig. 2, C and D2).
Next, we inhibited MGL (by JZL 184, 200 nm) to increase 2-AG availability (55), and to test whether the above molecular rearrangements were selective to AEA. First, we confirmed that JZL 184 rapidly inhibited MGL-like enzyme activity (Fig. 2E), defined as the significantly reduced hydrolysis of 4-nitrophenylacetate in JZL 184-exposed INS-1E cells. Subsequently, we measured 2-AG and AEA concentrations in supernatants and cell pellets of INS-1E cells exposed to JZL 184 for 30 min or 24 h. We found a substantial JZL184-induced increase in extracellular 2-AG content that persisted up to 24 h (“supernatant,” 30 min: 1.70 ± 0.99 (control) versus 7.20 ± 4.63 (JZL 184) pmol/ml; 24 h, 1.22 ± 0.38 (control) versus 4.74 ± 2.89 (JZL 184) pmol/ml). In contrast, intracellular (“pellet”) 2-AG content did not change (Fig. 2F). Likewise, AEA content, whether extracellular (supernatant, 30 min, 0.12 ± 0.06 (control) versus 0.83 ± 0.96 (JZL 184) pmol/ml; 24 h, 0.73 ± 0.46 (control) versus 0.59 ± 0.76 (JZL 184) pmol/ml) or intracellular remained unchanged. These data are significant because they highlight that signal-competent, extracellular 2-AG (58) becomes selectively elevated upon pharmacological manipulation of MGL activity.
Upon 1 h of JZL 184 application, MGL levels in INS-1E cells significantly decreased, followed by full recovery by 24 h (Fig. 2G). CB1R levels remained reduced after 1 and 24 h of JZL 184 treatment (Fig. 2H). Likewise, the increased DAGLα level upon JZL 184 exposure recapitulated AEA effects (Fig. 2I). These data suggest that both “direct” and “indirect” CB1R agonism might drive 2-AG synthesis to maintain the efficacy of eCB signaling even if CB1Rs are desensitized or degraded.
CB1R activity can induce apoptosis in pancreatic β cells (57). Therefore, we tested whether AEA affected INS-1E cell survival. AEA did not decrease INS-1E cell viability (Fig. 2J). Instead, AEA and JZL 184 induced cell proliferation by 1.62 ± 0.06- and 1.35 ± 0.05-fold, respectively. This effect was CB1R independent because it was not blocked by O-2050, a CB1R antagonist (1.55 ± 0.06 (AEA + O-2050) and 1.25 ± 0.08 (JZL 184 + O-2050), fold) and ACEA, a selective CB1R agonist, failed to induce INS-1E cell proliferation (Fig. 2K).
Agonist-induced CB1R Activation Drives Insulin Release from INS-1E Cells
Endocannabinoid signals have been suggested to regulate insulin secretion (5, 6, 59). However, the precise molecular mechanism of eCB action, including receptor identity and signal transduction sequence, on insulin secretion remains uncertain. AEA rapidly increased basal insulin secretion (2.1 ± 0.25-fold, p = 0.002 (100 nm); 1.34 ± 0.18-fold, p = 0.038 (10 μm), 30 min; Fig. 3A). Moreover, AEA (≥1 μm) acutely augmented glucose-induced insulin release (reaching 3.19 ± 0.44-fold, p = 0.008; (10 μm); Fig. 3A). The effect of AEA (10 μm) on basal insulin secretion under glucose-free conditions was time-dependent, reaching quasi-saturation by 30 min (Fig. 3B). Therefore, we used 30-min stimuli throughout. AEA is a pleiotropic ligand, being an agonist at CB1R, CB2R, and TRPV1 and other receptors and channels (60). Therefore, we first dissected receptor requirements of AEA-induced insulin release. O-2050, a silent CB1R antagonist (F(1,12) = 20.43, p < 0.001), but not capsazepine, a TRPV1 antagonist, reduced AEA-induced basal insulin release (1.64 ± 0.2 (AEA) versus 1.17 ± 0.18 (AEA + O-2050); p = 0.04). Likewise, O-2050, but not capsazepine, blocked the augmentation of glucose-induced insulin secretion of AEA (3.33 ± 0.29 (AEA) versus 2.11 ± 0.08 (AEA + O-2050); p = 0.014; Fig. 3C). We confirmed these data by showing that O-2050 (F(1,8) = 52.31, p < 0.001) also inhibited insulin release evoked or modulated by ACEA (F(1,8) = 185.39, p < 0.01), a high-efficacy CB1R agonist (5.39 ± 0.50 (ACEA) versus 3.21 ± 0.82-fold (ACEA + O-2050), p = 0.017; Fig. 3E). Although being statistically ineffective on its own (p > 0.2), AM 251, a CB1R inverse agonist, also decreased AEA-induced insulin hypersecretion under both basal and glucose stimulatory conditions (F(1,12) = 13.73, p < 0.001; Fig. 3F), corroborating earlier findings with SR141716 (7). These data, together with the inability of JWH133 (a CB2R agonist) to affect insulin release (Fig. 3D), suggest that CB1Rs drives the AEA-induced insulin secretion from INS-1E cells and augments the immediacy of insulin release upon stimulation with high glucose.
FIGURE 3.
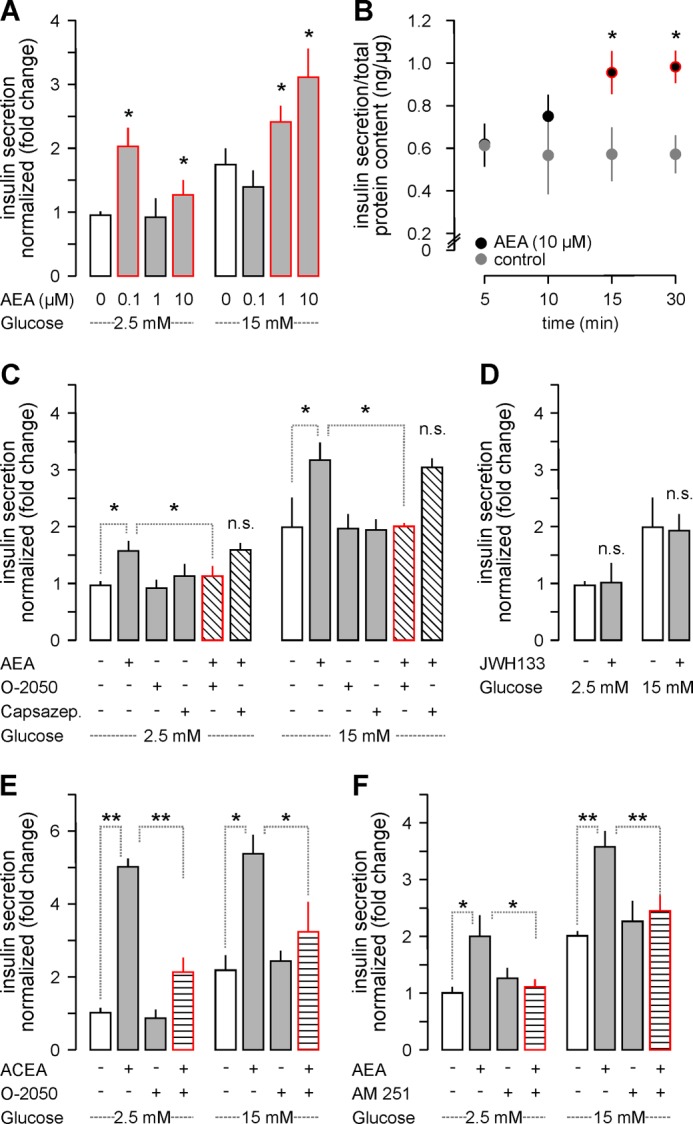
CB1R activation induces insulin secretion from INS-1E cells. A, AEA increased basal (2.5 mm glucose) and glucose (15 mm)-induced insulin release after 30 min. B, temporal dynamics of insulin release (within 10–30 min) evoked by AEA (10 μm) in glucose-free medium. C, O-2050 (100 nm) but not capsazepine (10 μm) pre-treatment (10 min) occluded AEA-induced insulin release irrespective of the glucose concentration. D, JWH133 (100 nm) also failed to affect insulin release. E, ACEA, a CB1R agonist, recapitulated the effect of AEA on insulin secretion. O-2050 attenuated the ACEA response. F, similarly, AM 251, a CB1R inverse agonist, rendered AEA ineffective to induce insulin release from INS-1E cells. Data in A and C–F were normalized to control in low glucose, and expressed as mean ± S.D. fold values from triplicate experiments. **, p < 0.01; *, p < 0.05 versus control (pairwise comparisons after one-way ANOVA); n.s., non-significant.
Vesicular insulin exocytosis is ubiquitously dependent on intracellular Ca2+ signals (22). Therefore, we tested the reliance of CB1R-dependent insulin secretion from INS-1E cells on extracellular (bath-applied) Ca2+. Both basal and glucose-stimulated insulin secretion were inhibited in Ca2+-free extracellular medium. In contrast, the ability of AEA to significantly augment glucose-induced insulin release remained unaffected (Fig. 4). Because cytoplasmic Ca2+ underpins insulin secretion (61), our data suggest that intracellular Ca2+ mobilization is a candidate mechanism to couple CB1R activation to insulin release (5, 59).
FIGURE 4.
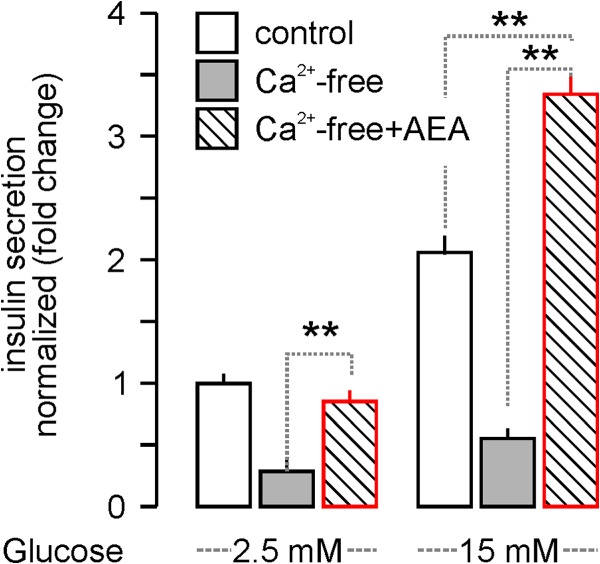
AEA-stimulated acute insulin release from INS-1E cells persists in Ca2+-free extracellular medium. AEA (30 min), but not glucose-induced, insulin secretion from INS-1E cells persisted in Ca2+-free extracellular medium, suggesting AEA-induced Ca2+ release from intracellular stores (5). Data were normalized to control in low glucose, and expressed as mean ± S.D.; fold-values from triplicate experiments are shown. **, p < 0.01 (one-way ANOVA).
Prolonged Endocannabinoid Exposure Maintains the Readiness of CB1R-dependent Insulin Release
Receptor recycling and desensitization, dampening of signal transduction efficacy or metabolic checkpoints of ligand availability can modify the impact of prolonged endocannabinoid exposure on insulin release. Therefore, we first tested whether AEA exposure for 24 h followed by 30 min stimulation with low or high glucose in the presence of AEA could increase insulin release. We showed that AEA significantly augmented both basal (1.41 ± 0.12-fold of control; p < 0.05) and high glucose-induced insulin release (2.4 ± 0.25-fold of control; p < 0.05; Fig. 5A) even if present for 24 h prior to probing insulin release. Second, we addressed whether modifying endogenous 2-monoacylglycerol availability by MGL inhibition affected insulin secretion. JZL 184 treatment for 24 h significantly increased both basal and glucose-stimulated insulin release (F(1,8) = 22.94, p < 0.001), which was antagonized, particularly at 2.5 mm glucose concentrations, by O-2050 (overall effect: F(1,8) = 5.39, p = 0.034; Fig. 5B). Besides inhibiting MGL, JZL 184 dose dependently can affect “off targets.” Therefore, we used OMDM 188, a DAGL inhibitor (37), to test 2-AG involvement. OMDM 188 application for 24 h alone did not affect basal insulin release (Fig. 5C). In contrast, OMDM 188 alone (24 h) occluded insulin secretion brought about by high glucose (overall effect: F(1,8) = 57.77, p < 0.001), suggesting that DAGL activity is required for insulin release. Moreover, OMDM 188 also counteracted the effect of JZL 184 on glucose-induced insulin release (F(1,8) = 13.06, p = 0.002; Fig. 5C), with combined treatment decreasing insulin secretion to the level observed in low glucose. These data suggest that high glucose increases 2-AG synthesis in β cells to tonically drive insulin release, which agrees with a previous finding in rat RIN-m5F cells (5).
FIGURE 5.
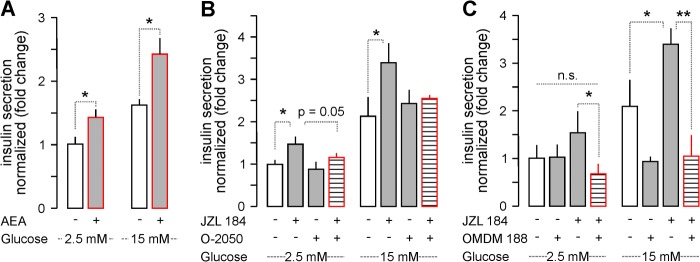
Prolonged AEA stimulation and JZL 184-induced 2-AG signaling trigger insulin release in vitro. A, AEA exposure for 24-h induced insulin release upon stimulation with low (2.5 mm) and high (15 mm) glucose. B, the effect of JZL 184 (24 h) alone or in combination with O-2050 on insulin release from INS-1E cells. C, OMDM 188, a DAGLα inhibitor (37), alone (24 h) occluded insulin release under conditions of high glucose. OMDM 188 also significantly reduced JZL 184-induced insulin release. Data were normalized to control in low glucose, and expressed as mean ± S.D. fold-values from triplicate experiments. **, p < 0.01; *, p < 0.05 versus control (pairwise comparisons after one-way ANOVA); n.s., non-significant.
2-AG Signaling Networks in and CB1R-dependent Insulin Release from Pancreatic Islets
We affirmed the physiological relevance of the above findings by histochemical profiling of the distribution and cell type-specificity of DAGL, MGL, and ABHD6 in relationship to CB1R localization and cell type diversity in the adult mouse pancreas. First, we confirmed (4) that mouse insulin+ (β) cells expressed CB1Rs, which often appeared membrane-bound (Fig. 6A). In contrast, glucagon+ (α) cells remained mostly unlabeled (Fig. 6, A1 and A2). Both insulin+ and glucagon+ cells expressed DAGLα (Fig. 6, B and B2). Unexpectedly, MGL concentrated in glucagon+ cells with only low to moderate MGL immunoreactivity seen in glucagon− cells, including β cells (Fig. 6C). Instead, ABHD6 appeared as a major 2-AG-degrading enzyme in glucagon− cells (Fig. 6D) including β cells (Fig. 6D1) and pancreatic polypeptide+ F cells (62) (Fig. 6D2). In contrast, somatostatin+ δ cells (62) appeared as ABHD6− (Fig. 6D3). MGL is highly up-regulated in malignant cells (63). Therefore, immortalized INS-1E cells might favor MGL expression. Taken together, these data suggest that both autocrine and paracrine 2-AG signaling can occur in β cells via CB1Rs. In fact, genetic disruption of CB1R expression prevented ACEA-stimulated insulin secretion from native pancreatic islets (p < 0.05; Fig. 6E).
FIGURE 6.

CB1R, DAGLα, MGL, and ABHD6 distribution in the endocrine pancreas of mouse. A and A2, CB1R immunoreactivity was seen in insulin+ β cells, but not glucagon+ α cells, in pancreatic islets. Arrows point to membrane-associated CB1Rs. B and B2, both insulin+ (β) and glucagon+ (α) cells harbored DAGLα immunoreactivity. Arrows in B1, inset, denote DAGL in glucagon+ α cells. C and D, generally, MGL and ABHD6 immunoreactivity was moderate in pancreatic islets of adult mice. C, MGL preferentially localized to glucagon+ α cells (arrows). Inset depicts the co-existence of cytoplasmic MGL and insulin immunoeactivities (arrow). D, ABHD6 was found in both glucagon+ α cells (arrows) and glucagon− cells (open arrowheads). Of the glucagon− cells, insulin+ β cells (D1 and D1′) and pancreatic polypeptide+ F-cells (D2) but not somatostatin+ δ cells (D3) harbored ABHD6 immunosignal. E, pancreatic islets isolated from CB1R−/− and wild-type mice were challenged with ACEA (30 min) to release insulin. Note that ACEA failed to augment glucose-induced insulin release in CB1R−/− islets. Hoechst 33,342 was used as nuclear counterstain. n.s., non-significant. Scale bars = 60 μm (A2 and B2), 18 μm (C, D, and D3), 9 μm (C/inset, D1′, and D2). Data in E were normalized to control in low glucose, and expressed as mean ± S.D. fold-values. *, p < 0.05 versus control (pairwise comparisons after one-way ANOVA are shown).
CB1R Activation Induces Akt and ERK1/2 Phosphorylation in INS-1E Cells
Akt and/or ERK1/2 activation and phosphorylation are key consequences of CB1R activation in many signaling contexts (64, 65). In INS-1E cells, AEA (10 μm) induced rapid Akt phosphorylation, peaking at 5 min (Fig. 7A). Escalating AEA concentrations revealed dose-dependent ERK1/2 phosphorylation (Fig. 7B). Similarly, ACEA (100 nm) increased ERK1/2 phosphorylation (Fig. 7C). O-2050 blocked both ACEA- and AEA-induced ERK1/2 phosphorylation, supporting CB1R involvement (Fig. 7, C and C1). The magnitude of CB1R-dependent ERK1/2 phosphorylation was largely equivalent to that seen in the presence of high glucose (Fig. 7C1). Thus, we hypothesized that simultaneous Akt/ERK1/2 activation downstream from CB1R activation can coordinately interact with and activate FAK, which, by inducing cytoskeletal remodeling (Fig. 7D), might modulate Ca2+-dependent insulin secretion (66).
FIGURE 7.
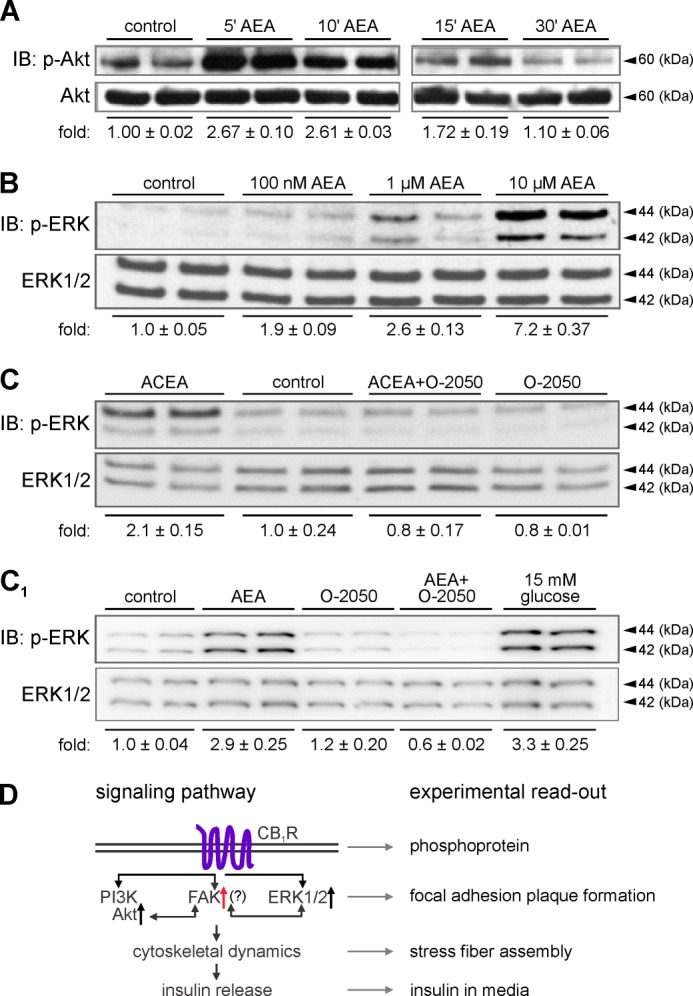
CB1R activation induces Akt and ERK1/2 phosphorylation in INS-1E cells. A, treatment of INS-1E cells with AEA (10 μm) for the periods indicated induced Akt kinase phosphorylation (p-Akt), as revealed by immunoblotting (IB). B, dose-response relationship of ERK1/2 phosphorylation in INS-1E cells exposed to escalating AEA concentrations (10 min). C, likewise, ACEA (100 nm, 10 min) induced ERK1/2 phosphorylation, which was O-2050 (100 nm) sensitive. C1, AEA-induced (10 μm) ERK1/2 phosphorylation was also inhibited by O-2050, reinforcing CB1R involvement. Note the largely equivalent ERK1/2 responses in the presence of high glucose concentrations used as positive control. Integrated optical intensities of the immunoreactive bands were determined and normalized to the mean intensity of total levels with control being 1.0. Representative blots are shown. Data were expressed as mean ± S.D. from triplicate experiments. D, scheme of the proposed signaling cascade including experimental read-outs. Note that FAK is poised to link phosphoinositide 3-kinase (PI3K)-Akt/ERK1/2 activation with cytoskeletal remodeling permissive for insulin release.
CB1R Activation in INS-1E Cells Promotes Cytoskeletal Remodeling
Cytoskeletal remodeling is a prerequisite of the trafficking and release of insulin granules (29). Here, returning to INS-1E cells, we first assessed CB1R-mediated actin polymerization in stress fibers, and FA plaque formation. Stimulation by AEA for 30 min (76 ± 18% (AEA) versus 13 ± 9% (control); p < 0.01 (post hoc; Fig. 8, A and B) and 24 h (41 ± 12% (AEA) versus 17 ± 5% (control); p < 0.05; Fig. 8C), as well as by JZL 184 for 24 h (63 ± 12% (JZL 184) versus 13 ± 9% (control); p < 0.01 (post hoc); Fig. 8B) led to stress fiber formation. O-2050 significantly reduced the abundance of stress fiber-bearing cells (21 ± 17% (AEA + O-2050) or 22 ± 18% (JZL 184 + O-2050); F(1,21) = 15.26, p = 0.002; Fig. 8B), confirming CB1R involvement in both AEA and JZL 184-induced cytoskeletal remodeling.
FIGURE 8.

CB1R activation induces cytoskeletal remodeling. A, exposure to glucose (15 mm) or AEA (10 μm, 10 min) induced stress fiber and FA plaque formation in INS-1E cells. O-2050 reversed AEA-induced cytoskeletal modifications. A1, FA plaques localized at the plus ends of polymerized F-actin, providing cytoskeletal anchor points. B, quantitative assessment of the percentage of INS-1E cells with stress fibers upon treatment with AEA (30 min) or JZL 184 (24 h) stimuli alone or in combination with O-2050 (100 nm as 10 min pre-treatment). C, INS-1E cells were exposed to AEA (10 μm) for 24 h to verify that the induction of insulin release by both endocannabinoids upon prolonged activation (see also Fig. 5, A and B) involves stress fiber formation. D, OMDM 188, a DAGL inhibitor (24 h), increased CB1R immunoreactivity in INS-1E cells. E, high glucose (15 mm for 10 min) induced stress fibers in INS-1E cells, as revealed by staining for F-actin (phalloidin). This effect was not reversed by O-2050, a CB1R antagonist. F, quantitative assessment of the percentage of INS-1E cells with FA plaques from experiments as above. High glucose (15 mm) was used as positive control. Data were expressed as mean ± S.D.; n = 30–80 cells/condition were analyzed in triplicate experiments. **, p < 0.01; *, p < 0.05 (pairwise comparisons following one-way ANOVA). Scale bars = 20 μm (F), 10 μm (A, C, and E), and 3 μm (A1).
Exposure to high glucose served as positive control (64 ± 10%, F(1,21) = 13.86, p = 0.002; Fig. 8, A and B). However, glucose-induced stress fiber formation was not affected by O-2050 (Fig. 8E), suggesting a non-eCB-mediated mechanism of glucose action.
Coincident with F-actin remodeling, we observed the glucose-induced formation of vinculin+ FA plaques at the tips of actin filaments (61 ± 10% (glucose) versus 10 ± 9% (control); p < 0.01; Fig. 8, A and A1). AEA (57 ± 12%; p < 0.05) or JZL 184 (51 ± 15%; p < 0.05) increased the amount of FA plaques (Fig. 8F) in an O-2050-sensitive manner (14 ± 4% (AEA + O-2050) and 14 ± 5% (JZL 184 + O-2050), both p < 0.01 versus drugs alone; Fig. 8F). Inhibition of 2-AG biosynthesis upon OMDM 188 application (24 h) led to heterogeneous cell morphologies, largely without an effect on cytoskeletal integrity, and a robust increase in CB1R content (Fig. 8D). Cumulatively, these data show that CB1R signaling can alter cytoskeletal dynamics, including the formation of FA plaques, and that endogenous 2-AG participates in this phenomenon.
Focal Adhesion Kinase Activity Links Endocannabinoid Signaling to Insulin Release
AEA (15 min) did not impair actin polymerization and/or stability per se, as indicated by the unaltered G/F-actin ratio in AEA-exposed INS-1E (Fig. 9A). Thus, and considering the formation of vinculin+ FA plaques upon CB1R activation, we hypothesized that FAK, a tyrosine kinase, might be poised to mediate the interaction between integrins and actin cytoskeleton in response to CB1R activation (31). FAK, also activated by growth factors (27) and glucose (26), participates in the multimolecular complex making up focal contact sites (FA plaques) to transduce extracellular signals to i.e. initiate the exocytosis of insulin granules (26). Therefore, FA plaque formation upon CB1R stimulation (Fig. 7D) is well suited to link cytoskeletal remodeling to insulin release. FAK phosphorylation (>10-fold increase versus control) occurred within 30 min of AEA stimulation (Fig. 9B), was O-2050 sensitive, and mimicked the effect of high glucose (Fig. 9B). This FAK phosphorylation predominantly occurred at the tips of processes protruding from INS-1E cells, which provided adhesion points as judged by them being vinculin+ (Fig. 9, C and C1′). The AEA-induced concentration of vinculin+/phospho-FAK+ FA plaques served to anchor the stress fibers (Fig. 9C1′). The overall phospho-FAK immunoreactivity of AEA-treated INS-1E cells markedly increased (Fig. 9C1), recapitulating the results of our Western analysis on total FAK levels upon AEA exposure (Fig. 9B). O-2050 reduced phospho-FAK immunoreactivity, the density of vinculin+ FA plaques and their association with stress fibers (Fig. 9, C2 and C2′).
FIGURE 9.

CB1R-mediated insulin release requires focal adhesion kinase activity. A, comparative assessment of G- and F-actin concentrations upon AEA challenge. Optical density of G- and F-actin was measured, and expressed as the “G/F-actin ratio.” B, treatment of INS-1E cells with AEA (10 μm) for 30 min induced FAK kinase phosphorylation (p-FAK). Fold-changes were normalized to the phosphorylation level in control, and relative to actin. A representative Western blot is shown. C–C2, immunofluorescence detection of increased FAK phosphorylation in INS-1E cells exposed to AEA (30 min). Open rectangles denote the positions of insets. Hoechst 33,432 nuclear localization signal was color-coded in yellow. C′-C1′, insets demonstrate the accumulation of p-FAK at vinculin+ focal adhesion points, which also anchor actin filaments (stress fibers, solid arrowheads) upon AEA application. O-2050 prevented stress fiber assembly (open arrowheads). D–D3, FAK inhibitor 14 (FAKi14; 1 μm, 10 min), a small molecule inhibitor of FAK (67), prevented AEA or high glucose-induced vinculin+ FA plaque (C2), and stress fiber assembly (C3). E, media were collected and insulin concentrations determined by ELISA. FAKi14 abrogated AEA-induced insulin secretion. Scale bars = 10 μm (B–B2 and C) and 3 μm (B′-B2′, and C1). Data were expressed as mean ± S.D.; n = 80 cells/group; **, p < 0.01; *, p < 0.05 (pairwise comparisons after one-way ANOVA). F and F1, correlation analysis revealed a positive relationship between the number of INS1-E cells containing FA plaques (F) or stress fibers (F1) and the amount of insulin secreted (ng/μg protein). Note that particularly close correlation was seen in the presence of high glucose. Pearson's correlation co-efficient are shown (dashed lines: linear regression plots).
Next, we asked whether pharmacological inhibition of FAK activity impacts AEA-induced cytoskeletal remodeling of INS-1E cells. Pre-treatment with FAKi14, a small molecule inhibitor that prevents FAK autophosphorylation at Tyr-397 (67), significantly reduced the number of FA plaque-bearing INS-1E cells (F(1,12) = 24.89, p < 0.001; Fig. 9, D and D2). This effect was particularly notable upon AEA treatment in 15 mm glucose (27 ± 13% (AEA + FAKi14) versus 77 ± 5% (AEA), p = 0.004; Fig. 9, D and D2). FAKi14 also antagonized stress fiber assembly (F(1,12) = 131.75, p < 0.001, Fig. 9D3), reinforcing that the formation of FA plaques is central to anchoring actin polymers. In particular, FAKi14 was efficacious to reduce the number of stress fiber-bearing cells in high glucose (53 ± 4% (AEA) versus 13 ± 3% (AEA + FAKi14); p < 0.001).
If FAK activity drives eCB-induced insulin release then its inhibition must result in the loss of secreted insulin upon CB1R stimulation. Indeed, FAK inhibition counteracted AEA-induced insulin hypersecretion under basal and glucose-stimulated conditions (F(1,12) = 78.25, p < 0.001; low glucose: 1.27 ± 0.09 (AEA) versus 0.75 ± 0.14 (AEA + FAKi14); high glucose: 3.02 ± 0.36 (AEA) versus 1.27 ± 0.25 (AEA + FAKi14); both p < 0.01; Fig. 9E). Regression analysis showed that the formation of FA plaques (R2 = 0.92; Fig. 9F) and stress fibers (R2 = 0.90, Fig. 9F1) correlated with glucose-stimulated, but less so with basal, insulin release. Cumulatively, we show that agonist stimulated the CB1Rs signal through Akt/ERK1/2/FAK to induce cytoskeletal remodeling, including FA plaque formation, underscoring eCB-stimulated second phase insulin release.
DISCUSSION
Our report identifies CB1R-mediated signaling events facilitating insulin release from INS-1E cells, immortalized rat β cells, and native mouse β cells. These findings highlight FAK activation as a key step in eCB-induced insulin release. Moreover, and although INS-1E and native pancreatic β cells reportedly express a variety of (endo-) cannabinoid-sensing receptors (3, 16, 59), eCB-stimulated insulin release appears to rely, in a large part, on CB1Rs.
Impaired energy balance is considered to be the main cause for obesity and type 2 diabetes (68, 69). For energy balance to be kept within a physiologically neutral range, the tight regulatory interplay of major centers, including the nervous system, gastrointestinal tract, pancreas, adipose tissue, liver, and skeletal muscles has evolved to continuously monitor nutrient and energy status at the periphery. Integration of vital peripheral information requires long-range communication among various organ systems. eCB signaling at CB1Rs has recently emerged as a particularly efficacious signaling network to control central aspects of food intake and energy homeostasis (68). Similarly, eCB signaling in insulin-sensitive peripheral tissues impacts glucose utilization and lipid homeostasis (70, 71). Despite mounting interest (3, 4, 6, 59, 68), the molecular link between eCB signaling and insulin release from the endocrine pancreas remain poorly understood.
Here, by using INS-1E cells and pancreatic islets from CB1R−/− mice, we demonstrated the upstream role of eCB signals to regulated insulin secretion. We show that INS-1E cells, like native β cells, can co-express CB1Rs, CB2Rs, and TRPV1 to sense and integrate eCB signals. Yet data from direct (AEA, ACEA) and indirect (JZL 184) agonist pharmacology and CB1R−/− mice cumulatively demonstrate that CB1R activation dominates eCB-stimulated insulin secretion in a dose- and time-dependent manner. eCB action is relatively fast, and promotes adaptive and long-lasting reorganization of eCB metabolism with opposing changes in MGL and DAGLα levels in β cells. Decreased CB1R levels suggest rapid receptor recycling and degradation. The rapid loss of MGL also suggests protein degradation, which reportedly involves CB1R-dependent activation of breast cancer-associated protein 1 (BRCA1) (72, 73), an E3 ubiquitin ligase. In contrast, increased DAGLα expression suggests compensatory eCB synthesis in response to disrupted second messenger signaling upon CB1R desensitization. This notion is supported by increased CB1R content in INS-1E cells treated with the DAGL inhibitor OMDM 188, which may compensate for decreased 2-AG availability by potentially conferring receptor hypersensitivity.
Both immortalized INS-1E cells and native mouse β cells express the metabolic enzymes and receptors sufficient for autocrine 2-AG signaling. Nevertheless, differences in experimental designs (e.g. α cells can contribute to 2-AG production/degradation in isolated pancreatic islets) have limited our ability to determine whether autocrine or paracrine eCB signals regulate insulin secretion. They might also explain differences between AEA and JZL 184-stimulated insulin release from INS-1E cells reported here and largely non-CB1R-mediated inhibition of insulin secretion reported elsewhere (6, 59). Inconsistencies as to the cellular sites of CB1R expression in pancreatic islets exist in the literature (4, 6), primarily due to the lack of appropriate genetic controls. We find mouse β cells endowed with CB1Rs, confirming data reported by Starowicz et al. (4) using immunoperoxidase methods in rat and mouse pancreas. Notably, α cells do not harbor detectable CB1R levels in vivo, suggesting that glucagon secretion might either be insensitive to endocannabinoid signals or modulated via non-CB1R-mediated mechanisms. To maintain glucose utilization and homeostasis, however, eCB actions on glucagon levels in blood might be indirect because eCB-driven insulin release from β cells could per se serve to prime glucagon secretion. α Cells, lining the outer border of pancreatic islets, appear to express both MGL and ABHD6 at levels exceeding those of β cells. This raises the possibility that α cells can restrict the spatial spread of eCBs, and isolate the endocrine pancreas from surrounding tissues. This hypothesis is supported by pronounced ABHD6-like immunoreactivity in pancreatic polypeptide-containing F cells. Unexpectedly, we detected marked ABHD6-like immunosignals in β cells. Although the robustness of immunoreactivity cannot be taken indicative of enzyme activity, our histochemical results suggest that MGL and ABHD6 might cooperate in terminating eCB signaling in the endocrine pancreas, including β cells. Moreover, our molecular identification of the coexistence of the biosynthesis and degradation machinery for 2-AG (and probably also AEA) highlights that both autocrine and (juxta-)paracrine eCB signaling must be considered in specific cellular contexts when assessing the functionality and influences of these signaling networks in the pancreas (4, 70). The notion that DAGL inactivation occludes insulin release, particularly when glucose concentrations are high, suggest the unexpectedly broad metabolic significance of these enzymes, perhaps beyond regulating 2-AG signaling at CB1Rs.
AEA in glucose-free conditions induced insulin release (Fig. 3B). This finding supports that glucose and AEA action are additive on insulin release, corroborating earlier reports (6, 59). CB1R agonists significantly increased insulin release from INS-1E and β cells, by a mechanism involving intracellular Ca2+ mobilization. These data in conjunction with high glucose-induced AEA and 2-AG synthesis (5) suggest a tight feed forward coupling between eCB production and signaling and glucose availability. eCBs can rapidly mobilize intracellular Ca2+ through inositol 1,4,5-trisphosphate signaling (74, 75) or by inositol 1,3,4-phosphate receptors on the endoplasmic reticulum (8). Such rapid Ca2+ transients, as seen in insulinomas cells upon AEA stimulation (8), are compatible with a role in first phase insulin release.
A rise of intracellular Ca2+ being a prerequisite of insulin release is also compatible with FAK and FAK-related kinase (76, 77) activation and cytoskeletal rearrangements during the second phase of insulin secretion, particularly because vesicle docking during exocytosis is reliant on the Ca2+-driven assembly of the SNARE machinery. Exocytosis involves a series of highly coordinated and sequential steps. Among these, FAK interacts with integrins, vinculin, paxillin, talin, and α-actinin to link FA plaques to filamentous actin. FAK in neurons is a key effector of CB1R-induced cytoarchitectural reorganization (31). FAK is an appealing target because it can suppress Rho GTPase activity, thereby promoting FA turnover (35). Considering that CB1Rs can modulate the activity of RhoA (and other small GTPases) (34, 76), FAK is poised to orchestrate cystoskeletal remodeling, particularly FA plaque assembly, required for insulin release. CB1R activation establishes maximal FAK activity upon cooperative signaling with cell surface tyrosine kinase receptors and the sequential phosphorylation of Tyr-397 and Tyr-576/577 FAK residues (76). Here, we showed FAK phosphorylation at Tyr-397, which is considered a permissive step for the recruitment of Src-family kinases, whose second-phase phosphorylation of an additional five Tyr residues in the activation loop of the kinase, confers maximal FAK activity (76). Because CB1R-dependent Tyr-397 phosphorylation persists for >1 h, requires integrin activation, and exhibits adhesion dependence (76), and is abated by FAKi14 (67), we conclude that FAK activation is an essential transducer and integrator of eCB and integrin signaling in pancreatic β cells.
In response to diverse microenvironmental stimuli, ERK1/2, Akt, and FAK were shown to be bidirectionally activated with a putative ERK1/2 and FAK interaction involved in glucose-dependent insulin secretion (25, 66, 78–80). FAK-induced dynamic cytoskeletal remodeling enables trafficking of insulin granules (25, 66). Stress-fiber formation in INS-1E cells observed upon CB1R stimulation was reminiscent of that seen in FAK−/− cells, particularly keratinocytes (27). Yet this phenocopy is unlikely to confer loss-of-function. Instead, our data show functionally permissive “structural gain-of-function” assuming increased temporal dynamics (28) but not overt cellular rigidity (i.e. tension signaling) (27). In fact, FAK activity is also crucial for adequate insulin signaling in peripheral tissues because its down-regulation leads to hyperglycemia, hyperinsulinemia, and insulin resistance (81–83).
Two of the clinicopathological implications of our findings, i.e. that (i) DAGL activity is a minimal requirement for insulin secretion and (ii) prolonged exposure to heightened eCB concentrations drives insulin release, outline a link between increased circulating eCB levels (5), elevated tissue 2-AG and AEA content, and up-regulated DAGLα in β cells (4) and insulin hypersecretion in obesity (51, 84). The mechanism we identified suggests that acute CB1R activation may be critical for the adaptation of pancreatic β cells to insulin resistance. Our findings provide a molecular backbone to develop tissue selective CB1R antagonists, which, given their insulin re-sensitizing effects (71), can offer the rescue of insulin secretion to maintain adequate to tissue demands.
Acknowledgments
We thank K. Ledent, B. F. Cravatt, and P. Doherty for knock-out tissues, P. Maechler for INS-1E cells, G. Ortar for the kind gift of OMDM 188, A. C. Cuello and B. Meister for antibodies, and R. Verde for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants DA023214 (to T. H.), DA011322 (to K. Mac.), and DA021696 (to K. Mac.), Foundation for Polish Science Grants TEAM/2010–5/2 (to A. D.) and MPD/2009/4 (to A. D.), National Science Center (NCN) Grant UMO-2011/03/B/NZ4/03055 (to A. D.), Scottish Universities Life Science Alliance (to T. H.), Vetenskapsrådet (to T. H.), Hjärnfonden (to T. H.), European Commission Grant HEALTH-F2-2007-201159 (to T. H.), and the Novo Nordisk Foundation (to T. H.).
- eCB
- endocannabinoid
- FAAH
- fatty acid amide hydrolase
- ABHD6
- α/β-hydrolase domain containing 6
- ACEA
- N-(2-chloroethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide
- AEA
- anandamide
- Akt
- protein kinase B
- AM 251
- N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbox-amide
- CB1R
- type 1 cannabinoid receptor
- DAGLα/β
- sn-1-diacylglycerol lipases α and β
- ERK1/2
- extracellular signal-regulated kinase 1/2
- FA
- focal adhesion
- FAK
- focal adhesion kinase
- FAKi14
- FAK inhibitor 14
- GPR55
- orphan G protein-coupled receptor 55
- JWH133
- (6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetra-hydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran
- JZL 184
- 4-[bis(1,3-benzodioxol-5-yl)hydroxymethyl]-1-piperidine-carboxylic acid 4-nitrophenyl ester
- KRBH
- Krebs-Ringer bicarbonate HEPES buffer
- O-2050
- (6aR,10aR)-1-hydroxy-3-(1-methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo [b,d]pyran
- OMDM 188
- N-formyl-l-isoleucine-(1S)-1-[[(2S,3S)-3-hexyl-4-oxo-2-oxetanyl]methyl]dodecyl ester
- MGL
- monoacylglycerol lipase
- NAPE-PLD/N-PLD
- N-acyl phosphatidylethanolamine-specific phospholipase D
- PB
- sodium phosphate buffer
- ANOVA
- analysis of variance
- TRPV1
- vanilloid type 1 channels
- 2-AG
- 2-arachidonoylglycerol.
REFERENCES
- 1. Nolan C. J., Madiraju M. S., Delghingaro-Augusto V., Peyot M. L., Prentki M. (2006) Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 55, S16-S23 [DOI] [PubMed] [Google Scholar]
- 2. Yaney G. C., Corkey B. E. (2003) Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 46, 1297–1312 [DOI] [PubMed] [Google Scholar]
- 3. Bermúdez-Silva F. J., Suárez J., Baixeras E., Cobo N., Bautista D., Cuesta-Muñoz A. L., Fuentes E., Juan-Pico P., Castro M. J., Milman G., Mechoulam R., Nadal A., Rodríguez de Fonseca F. (2008) Presence of functional cannabinoid receptors in human endocrine pancreas. Diabetologia 51, 476–487 [DOI] [PubMed] [Google Scholar]
- 4. Starowicz K. M., Cristino L., Matias I., Capasso R., Racioppi A., Izzo A. A., Di Marzo V. (2008) Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with a high-fat diet. Obesity 16, 553–565 [DOI] [PubMed] [Google Scholar]
- 5. Matias I., Gonthier M. P., Orlando P., Martiadis V., De Petrocellis L., Cervino C., Petrosino S., Hoareau L., Festy F., Pasquali R., Roche R., Maj M., Pagotto U., Monteleone P., Di Marzo V. (2006) Regulation, function, and dysregulation of endocannabinoids in models of adipose and β-pancreatic cells and in obesity and hyperglycemia. J. Clin. Endocrinol. Metab 91, 3171–3180 [DOI] [PubMed] [Google Scholar]
- 6. Nakata M., Yada T. (2008) Cannabinoids inhibit insulin secretion and cytosolic Ca2+ oscillation in islet beta-cells via CB1 receptors. Regul. Pept. 145, 49–53 [DOI] [PubMed] [Google Scholar]
- 7. Getty-Kaushik L., Richard A.M., Deeney J. T., Krawczyk S., Shirihai O., Corkey B. E. (2009) The CB1 antagonist rimonabant decreases insulin hypersecretion in rat pancreatic islets. Obesity 17, 1856–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Petrocellis L., Marini P., Matias I., Moriello A. S., Starowicz K., Cristino L., Nigam S., Di Marzo V. (2007) Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma beta-cells. Exp. Cell Res. 313, 2993–3004 [DOI] [PubMed] [Google Scholar]
- 9. Cravatt B. F., Giang D. K., Mayfield S. P., Boger D. L., Lerner R. A., Gilula N. B. (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83–87 [DOI] [PubMed] [Google Scholar]
- 10. Dinh T. P., Carpenter D., Leslie F. M., Freund T. F., Katona I., Sensi S. L., Kathuria S., Piomelli D. (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. U.S.A. 99, 10819–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Devane W. A., Hanus L., Breuer A., Pertwee R. G., Stevenson L. A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258, 1946–1949 [DOI] [PubMed] [Google Scholar]
- 12. Sugiura T., Kondo S., Sukagawa A., Nakane S., Shinoda A., Itoh K., Yamashita A., Waku K. (1995) 2-Arachidonoylglycerol. A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 215, 89–97 [DOI] [PubMed] [Google Scholar]
- 13. Matsuda L. A., Lolait S. J., Brownstein M. J., Young A. C., Bonner T. I. (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346, 561–564 [DOI] [PubMed] [Google Scholar]
- 14. Munro S., Thomas K. L., Abu-Shaar M. (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365, 61–65 [DOI] [PubMed] [Google Scholar]
- 15. Begg M., Pacher P., Bátkai S., Osei-Hyiaman D., Offertáler L., Mo F. M., Liu J., Kunos G. (2005) Evidence for novel cannabinoid receptors. Pharmacol. Ther. 106, 133–145 [DOI] [PubMed] [Google Scholar]
- 16. Ryberg E., Larsson N., Sjögren S., Hjorth S., Hermansson N. O., Leonova J., Elebring T., Nilsson K., Drmota T., Greasley P. J. (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 152, 1092–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melck D., Bisogno T., De Petrocellis L., Chuang H., Julius D., Bifulco M., Di Marzo V. (1999) Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs). Vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Commun. 262, 275–284 [DOI] [PubMed] [Google Scholar]
- 18. Zygmunt P. M., Petersson J., Andersson D. A., Chuang H., Sørgård M., Di Marzo V., Julius D., Högestätt E. D. (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452–457 [DOI] [PubMed] [Google Scholar]
- 19. Bermudez-Silva F. J., Sanchez-Vera I., Suárez J., Serrano A., Fuentes E., Juan-Pico P., Nadal A., Rodríguez de Fonseca F. (2007) Role of cannabinoid CB2 receptors in glucose homeostasis in rats. Eur. J. Pharmacol. 565, 207–211 [DOI] [PubMed] [Google Scholar]
- 20. Li C., Jones P. M., Persaud S. J. (2010) Cannabinoid receptors are coupled to stimulation of insulin secretion from mouse MIN6 beta-cells. Cell Physiol. Biochem. 26, 187–196 [DOI] [PubMed] [Google Scholar]
- 21. Vilches-Flores A., Delgado-Buenrostro N. L., Navarrete-Vázquez G., Villalobos-Molina R. (2010) CB1 cannabinoid receptor expression is regulated by glucose and feeding in rat pancreatic islets. Regul. Pept. 163, 81–87 [DOI] [PubMed] [Google Scholar]
- 22. Henquin J. C., Ishiyama N., Nenquin M., Ravier M. A., Jonas J. C. (2002) Signals and pools underlying biphasic insulin secretion. Diabetes 51, S60-S67 [DOI] [PubMed] [Google Scholar]
- 23. Thurmond D. C., Gonelle-Gispert C., Furukawa M., Halban P. A., Pessin J. E. (2003) Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 17, 732–742 [DOI] [PubMed] [Google Scholar]
- 24. Orci L., Gabbay K. H., Malaisse W. J. (1972) Pancreatic beta-cell web. Its possible role in insulin secretion. Science 175, 1128–1130 [DOI] [PubMed] [Google Scholar]
- 25. Rondas D., Tomas A., Halban P. A. (2011) Focal adhesion remodeling is crucial for glucose-stimulated insulin secretion and involves activation of focal adhesion kinase and paxillin. Diabetes 60, 1146–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rondas D., Tomas A., Soto-Ribeiro M., Wehrle-Haller B., Halban P.A. (2012) Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem. 287, 2423–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schober M., Raghavan S., Nikolova M., Polak L., Pasolli H. A., Beggs H. E., Reichardt L. F., Fuchs E. (2007) Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J. Cell Biol. 176, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arous C., Rondas D., Halban P. A. (2013) Non-muscle myosin IIA is involved in focal adhesion and actin remodelling controlling glucose-stimulated insulin secretion. Diabetologia 56, 792–802 [DOI] [PubMed] [Google Scholar]
- 29. Jewell J.L., Luo W., Oh E., Wang Z., Thurmond D. C. (2008) Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 283, 10716–10726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henquin J. C., Mourad N. I., Nenquin M. (2012) Disruption and stabilization of beta-cell actin microfilaments differently influence insulin secretion triggered by intracellular Ca2+ mobilization or store-operated Ca2+ entry. FEBS Lett. 586, 89–95 [DOI] [PubMed] [Google Scholar]
- 31. Derkinderen P., Toutant M., Burgaya F., Le Bert M., Siciliano J. C., de Franciscis V., Gelman M., Girault J. A. (1996) Regulation of a neuronal form of focal adhesion kinase by anandamide. Science 273, 1719–1722 [DOI] [PubMed] [Google Scholar]
- 32. Laezza C., Pisanti S., Malfitano A. M., Bifulco M. (2008) The anandamide analog, Met-F-AEA, controls human breast cancer cell migration via the RHOA/RHO kinase signaling pathway. Endocr. Relat. Cancer 15, 965–974 [DOI] [PubMed] [Google Scholar]
- 33. Obara Y., Ueno S., Yanagihata Y., Nakahata N. (2011) Lysophosphatidylinositol causes neurite retraction via GPR55, G13 and RhoA in PC12 cells. PLoS One 6, e24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nithipatikom K., Gomez-Granados A. D., Tang A. T., Pfeiffer A. W., Williams C. L., Campbell W. B. (2012) Cannabinoid receptor type 1 (CB1) activation inhibits small GTPase RhoA activity and regulates motility of prostate carcinoma cells. Endocrinology 153, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ren X. D., Kiosses W. B., Sieg D. J., Otey C. A., Schlaepfer D. D., Schwartz M. A. (2000) Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113, 3673–3678 [DOI] [PubMed] [Google Scholar]
- 36. Merglen A., Theander S., Rubi B., Chaffard G., Wollheim C. B., Maechler P. (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145, 667–678 [DOI] [PubMed] [Google Scholar]
- 37. Ortar G., Bisogno T., Ligresti A., Morera E., Nalli M., Di Marzo V. (2008) Tetrahydrolipstatin analogues as modulators of endocannabinoid 2-arachidonoylglycerol metabolism. J. Med. Chem. 51, 6970–6979 [DOI] [PubMed] [Google Scholar]
- 38. Ledent C., Valverde O., Cossu G., Petitet F., Aubert J. F., Beslot F., Böhme G. A., Imperato A., Pedrazzini T., Roques B. P., Vassart G., Fratta W., Parmentier M. (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283, 401–404 [DOI] [PubMed] [Google Scholar]
- 39. Szot G. L., Koudria P., Bluestone J. A. (2007) Murine pancreatic islet isolation. J. Vis. Exp. 255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshida T., Fukaya M., Uchigashima M., Miura E., Kamiya H., Kano M., Watanabe M. (2006) Localization of diacylglycerol lipase-α around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J. Neurosci. 26, 4740–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Slipetz D. M., O'Neill G. P., Favreau L., Dufresne C., Gallant M., Gareau Y., Guay D., Labelle M., Metters K. M. (1995) Activation of the human peripheral cannabinoid receptor results in inhibition of adenylyl cyclase. Mol. Pharmacol. 48, 352–361 [PubMed] [Google Scholar]
- 42. Schlosburg J. E., Blankman J. L., Long J. Z., Nomura D. K., Pan B., Kinsey S. G., Nguyen P. T., Ramesh D., Booker L., Burston J. J., Thomas E. A., Selley D. E., Sim-Selley L. J., Liu Q. S., Lichtman A. H., Cravatt B. F. (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 13, 1113–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gao Y., Vasilyev D. V., Goncalves M. B., Howell F. V., Hobbs C., Reisenberg M., Shen R., Zhang M. Y., Strassle B.W., Lu P., Mark L., Piesla M. J., Deng K., Kouranova E. V., Ring R. H., Whiteside G. T., Bates B., Walsh F. S., Williams G., Pangalos M. N., Samad T. A., Doherty P. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 30, 2017–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mulder J., Zilberter M., Pasquaré S. J., Alpár A., Schulte G., Ferreira S. G., Köfalvi A., Martín-Moreno A. M., Keimpema E., Tanila H., Watanabe M., Mackie K., Hortobágyi T., de Ceballos M. L., Harkany T. (2011) Molecular reorganization of endocannabinoid signalling in Alzheimer's disease. Brain 134, 1041–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kano M., Ohno-Shosaku T., Hashimotodani Y., Uchigashima M., Watanabe M. (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 89, 309–380 [DOI] [PubMed] [Google Scholar]
- 46. Cristino L., Starowicz K., De Petrocellis L., Morishita J., Ueda N., Guglielmotti V., Di Marzo V. (2008) Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience 151, 955–968 [DOI] [PubMed] [Google Scholar]
- 47. Muccioli G. G., Labar G., Lambert D. M. (2008) CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. Chembiochem. 9, 2704–2710 [DOI] [PubMed] [Google Scholar]
- 48. De Marchi N., De Petrocellis L., Orlando P., Daniele F., Fezza F., Di Marzo V. (2003) Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis. 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rasmussen I., Pedersen L. H., Byg L., Suzuki K., Sumimoto H., Vilhardt F. (2010) Effects of F/G-actin ratio and actin turn-over rate on NADPH oxidase activity in microglia. BMC. Immunol. 11, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vilches-Flores A., Hauge-Evans A. C., Jones P. M., Persaud S. J. (2013) Chronic activation of cannabinoid receptors in vitro does not compromise mouse islet function. Clin. Sci. 124, 467–478 [DOI] [PubMed] [Google Scholar]
- 51. Matias I., Gatta-Cherifi B., Tabarin A., Clark S., Leste-Lasserre T., Marsicano G., Piazza P. V., Cota D. (2012) Endocannabinoids measurement in human saliva as potential biomarker of obesity. PLoS One 7, e42399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simon G. M., Cravatt B. F. (2008) Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acylethanolamine precursors in mouse brain. J. Biol. Chem. 283, 9341–9349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bisogno T., Howell F., Williams G., Minassi A., Cascio M. G., Ligresti A., Matias I., Schiano-Moriello A., Paul P., Williams E. J., Gangadharan U., Hobbs C., Di Marzo V., Doherty P. (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 163, 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berghuis P., Rajnicek A. M., Morozov Y. M., Ross R. A., Mulder J., Urbán G. M., Monory K., Marsicano G., Matteoli M., Canty A., Irving A. J., Katona I., Yanagawa Y., Rakic P., Lutz B., Mackie K., Harkany T. (2007) Hardwiring the brain. Endocannabinoids shape neuronal connectivity. Science 316, 1212–1216 [DOI] [PubMed] [Google Scholar]
- 55. Long J. Z., Li W., Booker L., Burston J. J., Kinsey S. G., Schlosburg J. E., Pavón F. J., Serrano A. M., Selley D. E., Parsons L. H., Lichtman A. H., Cravatt B. F. (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 5, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hsieh C., Brown S., Derleth C., Mackie K. (1999) Internalization and recycling of the CB1 cannabinoid receptor. J. Neurochem. 73, 493–501 [DOI] [PubMed] [Google Scholar]
- 57. Kim W., Lao Q., Shin Y. K., Carlson O. D., Lee E. K., Gorospe M., Kulkarni R. N., Egan J. M. (2012) Cannabinoids induce pancreatic beta-cell death by directly inhibiting insulin receptor activation. Sci. Signal. 5, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keimpema E., Alpár A., Howell F., Malenczyk K., Hobbs C., Hurd Y. L., Watanabe M., Sakimura K., Kano M., Doherty P., Harkany T. (2013) Diacylglycerol lipase α manipulation reveals developmental roles for intercellular endocannabinoid signaling. Sci. Rep. 3, 2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Juan-Picó P., Fuentes E., Bermúdez-Silva F. J., Javier Díaz-Molina F., Ripoll C., Rodríguez de Fonseca F., Nadal A. (2006) Cannabinoid receptors regulate Ca2+ signals and insulin secretion in pancreatic beta-cell. Cell Calcium 39, 155–162 [DOI] [PubMed] [Google Scholar]
- 60. Di Marzo V., De Petrocellis L. (2012) Why do cannabinoid receptors have more than one endogenous ligand? Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 3216–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sato Y., Nenquin M., Henquin J. C. (1998) Relative contribution of Ca2+-dependent and Ca2+-independent mechanisms to the regulation of insulin secretion by glucose. FEBS Lett. 421, 115–119 [DOI] [PubMed] [Google Scholar]
- 62. Zhang W., Efanov A., Yang S. N., Fried G., Kolare S., Brown H., Zaitsev S., Berggren P. O., Meister B. (2000) Munc-18 associates with syntaxin and serves as a negative regulator of exocytosis in the pancreatic beta-cell. J. Biol. Chem. 275, 41521–41527 [DOI] [PubMed] [Google Scholar]
- 63. Nomura D. K., Long J. Z., Niessen S., Hoover H. S., Ng S. W., Cravatt B. F. (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Derkinderen P., Valjent E., Toutant M., Corvol J. C., Enslen H., Ledent C., Trzaskos J., Caboche J., Girault J. A. (2003) Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J. Neurosci. 23, 2371–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gómez del Pulgar T., Velasco G., Guzmán M. (2000) The CB1 cannabinoid receptor is coupled to the activation of protein kinase B/Akt. Biochem. J. 347, 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cai E. P., Casimir M., Schroer S. A., Luk C. T., Shi S. Y., Choi D., Dai X. Q., Hajmrle C., Spigelman A. F., Zhu D., Gaisano H. Y., MacDonald P. E., Woo M. (2012) In vivo role of focal adhesion kinase in regulating pancreatic beta-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes 61, 1708–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Golubovskaya V. M., Nyberg C., Zheng M., Kweh F., Magis A., Ostrov D., Cance W. G. (2008) A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 51, 7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Matias I., Di Marzo V. (2007) Endocannabinoids and the control of energy balance. Trends Endocrinol. Metab. 18, 27–37 [DOI] [PubMed] [Google Scholar]
- 69. Silvestri C., Di Marzo V. (2013) The endocannabinoid system in energy homeostasis and the ethiology of metabolic disorders. Cell Metab. 17, 475–490 [DOI] [PubMed] [Google Scholar]
- 70. Di Marzo V. (2008) The endocannabinoid system in obesity and type 2 diabetes. Diabetologia 51, 1356–1367 [DOI] [PubMed] [Google Scholar]
- 71. Kunos G., Osei-Hyiaman D., Liu J., Godlewski G., Bátkai S. (2008) Endocannabinoids and the control of energy homeostasis. J. Biol. Chem. 283, 33021–33025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. De Petrocellis L., Melck D., Palmisano A., Bisogno T., Laezza C., Bifulco M., Di Marzo V. (1998) The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 95, 8375–8380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Keimpema E., Tortoriello G., Alpár A., Capsoni S., Arisi I., Calvigioni D., Hu S. S., Cattaneo A., Doherty P., Mackie K., Harkany T. (2013) Nerve growth factor scales endocannabinoid signaling by regulating monoacylglycerol lipase turnover in developing cholinergic neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 1935–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lauckner J. E., Hille B., Mackie K. (2005) The cannabinoid agonist WIN55,212–2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 19144–19149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mombouli J. V., Schaeffer G., Holzmann S., Kostner G. M., Graier W. F. (1999) Anandamide-induced mobilization of cytosolic Ca2+ in endothelial cells. Br. J. Pharmacol. 126, 1593–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dalton G. D., Peterson L. J., Howlett A. C. (2013) CB1 cannabinoid receptors promote maximal FAK catalytic activity by stimulating cooperative signaling between receptor tyrosine kinases and integrins in neuronal cells. Cell Signal. 25, 1665–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhou D., Song Z. H. (2002) CB1 cannabinoid receptor-mediated tyrosine phosphorylation of focal adhesion kinase-related non-kinase. FEBS Lett. 525, 164–168 [DOI] [PubMed] [Google Scholar]
- 78. Daval M., Gurlo T., Costes S., Huang C. J., Butler P. C. (2011) Cyclin-dependent kinase 5 promotes pancreatic beta-cell survival via Fak-Akt signaling pathways. Diabetes 60, 1186–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Saleem S., Li J., Yee S. P., Fellows G. F., Goodyer C. G., Wang R. (2009) β1-Integrin/FAK/ERK signalling pathway is essential for human fetal islet cell differentiation and survival. J. Pathol. 219, 182–192 [DOI] [PubMed] [Google Scholar]
- 80. Higuchi M., Kihara R., Okazaki T., Aoki I., Suetsugu S., Gotoh Y. (2013) Akt1 promotes focal adhesion disassembly and cell motility through phosphorylation of FAK in growth factor-stimulated cells. J. Cell Sci. 126, 745–755 [DOI] [PubMed] [Google Scholar]
- 81. Cheung A. T., Wang J., Ree D., Kolls J. K., Bryer-Ash M. (2000) Tumor necrosis factor-α induces hepatic insulin resistance in obese Zucker (fa/fa) rats via interaction of leukocyte antigen-related tyrosine phosphatase with focal adhesion kinase. Diabetes 49, 810–819 [DOI] [PubMed] [Google Scholar]
- 82. Huang D., Khoe M., Ilic D., Bryer-Ash M. (2006) Reduced expression of focal adhesion kinase disrupts insulin action in skeletal muscle cells. Endocrinology 147, 3333–3343 [DOI] [PubMed] [Google Scholar]
- 83. Bisht B., Srinivasan K., Dey C. S. (2008) In vivo inhibition of focal adhesion kinase causes insulin resistance. J. Physiol. 586, 3825–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Matias I., Di Marzo V. (2006) Endocannabinoid synthesis and degradation, and their regulation in the framework of energy balance. J. Endocrinol. Invest. 29, 15–26 [PubMed] [Google Scholar]