

Background: Mechanisms underlying SGK1 activation are incompletely understood in epithelial cells.
Results: Store-operated Ca2+ entry up-regulates SGK1, thereby modulating the lethal effects of Ca2+ overloading on mitochondrial membrane potential.
Conclusion: Ca2+-induced SGK1 activates cytoprotective signaling and modifies mitochondrial function in epithelial cells.
Significance: This work reveals a cytoprotective role for SGK1 in necrosis and has potential relevance for epithelial cell protection and cancer treatment.
Keywords: Apoptosis, Calcium Signaling, Mitochondrial Apoptosis, Necrosis (Necrotic Death), Signal Transduction, SGK1, Cytoprotection, Mitochondrial Membrane Potential
Abstract
Serum and glucocorticoid-regulated kinase 1 (SGK1) encodes a phosphatidylinositol 3-kinase-dependent serine/threonine kinase that is rapidly induced in response to cellular stressors and is an important cell survival signal. Previous studies have suggested that an increase in cytoplasmic Ca2+ concentration ([Ca2+]c) is required for increased SGK1 expression, but the subcellular source of Ca2+ regulating SGK1 transcription remains uncertain. Activation of endoplasmic reticulum stress (ERS) with thapsigargin (TG) increased SGK1 mRNA and protein expression in MDA-MB-231 cells. Intracellular Ca2+ imaging revealed that store-operated Ca2+ entry played a prominent role in SGK1 induction by TG. Neither ERS nor release of Ca2+ from the ER was sufficient to activate SGK1. Prolonged elevation of intracellular Ca2+ levels, however, triggered cell death with a much greater proportion of the cells undergoing necrosis rather than apoptosis. A relative increase in the percentage of cells undergoing necrosis was observed in cells expressing a short hairpin RNA targeted to the SGK1 gene. Necrotic cell death evoked by cytoplasmic Ca2+ overloading was associated with persistent hyperpolarization of the inner mitochondrial membrane and a modest increase in calpain activation, but did not involve detectable caspase 3 or caspase 7 activation. The effects of cytoplasmic Ca2+ overloading on mitochondrial membrane potential were significantly reduced in cells expressing SGK1 compared with SGK1-depleted cells. Our findings indicate that store-operated Ca2+ entry regulates SGK1 expression in epithelial cells and suggest that SGK1-dependent cytoprotective signaling involves effects on maintaining mitochondrial function.
Introduction
Serum and glucocorticoid-regulated kinase 1 (SGK1)4 is a transcriptionally induced serine-threonine kinase of the AGC subfamily of protein kinases that is also regulated by posttranslational modification. The SGK1 gene, an immediate early response gene, was identified from serum- or glucocorticoid-stimulated transcripts in a rat mammary epithelial cell line (1, 2). Transcription of SGK1 is also rapidly induced in non-malignant human breast epithelial cells by glucocorticoids, progesterone, or serum (3) and in mouse mammary epithelial cells following oxidative, osmotic, and ultraviolet radiation stress (4). Activation of SGK1 can be affected by many kinases, including 3-phosphoinositide-dependent protein kinase 1 mTOR and PI3K (5–7). Disruption of SGK1 activation can occur by ubiquitination (8). In contrast to other rapidly degraded protein kinases, neither the catalytic activity of SGK1 nor activation site phosphorylation is required for ubiquitin modification and degradation. Instead, SGK1 degradation requires a lysine-less six-amino acid (amino acids 19–24) hydrophobic motif (GMVAIL) within the N-terminal domain that also serves to target SGK1 to the endoplasmic reticulum (ER) and mitochondria (9). Interaction with the stress-associated E3 ligase C terminus of Hsc (heat shock cognate protein) 70-interacting protein) (CHIP) is also required for ubiquitin modification and rapid proteasomal degradation of SGK1 (10). Multiple intracellular signal transduction pathways have been implicated in the regulation of SGK1 gene expression (3, 11–14). Intracellular Ca2+ regulates SGK1 gene expression in A6 renal cells (15) and SGK1 kinase activity in CHO, CHO-insulin receptor (CHO-IR), and HepG2 cells (16). Activation of SGK1 during cell stress has been shown to be Ca2+-dependent. Hypotonic stress and Ca2+ overloading increased SGK1 mRNA and protein levels in A6 cells (15). The effects of osmotic stress were attenuated following chelation of intracellular Ca2+ with 1,2-bis (o-aminophenoxy) ethane-N,N,N′,N′-tetraacetate (BAPTA) and by N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide, an inhibitor of calmodulin. Exposure of CHO, CHO-insulin receptor, and HepG2 cells to supraphysiological levels of insulin also increased SGK1 kinase activity. The activation of SGK1 by insulin was inhibited by BAPTA and calmodulin antagonists (16). These studies indicate an important role of Ca2+ signaling pathways in SGK1 gene expression and kinase activity. However, the mechanisms underlying Ca2+-dependent activation of SGK1 remain unresolved.
SGK1 participates in the regulation of a wide range of cellular functions, including ion channel activity, Na+/H+ exchange, glucose and amino acid transport, glucose metabolism, gene transcription, hormone secretion, cell volume, proliferation, and cell death (17). SGK1 activity maintains electrolyte homeostasis in kidney epithelial cells by regulating epithelial sodium channel and ROMK1 (Kir 1.1) potassium channel expression (18), affects cardiomyocyte Na+ and K+ fluxes (19), increases Na+/H+ exchange in renal epithelial cells (20), and modifies carbohydrate metabolism (21). SGK1 also increases embryonic rat hippocampal neurite formation through direct effects on microtubule polymerization (22) and contributes to neuronal plasticity (23). SGK1 phosphorylation and inhibition of B-Raf kinase activity is important for cell cycle regulation in HEK293 cells (24).
SGK1 is part of a cytoprotective signaling network that inhibits apoptosis (17, 25, 26). Phosphorylation and inactivation of forkhead receptor-L1 (FKHR-L1 or FOXO3a) has been implicated in SGK1-dependent cell survival signaling (27, 28). SGK1 activation of IKKβ inhibits breast cancer cell apoptosis (29). In the context of tumor formation and progression, the increased expression of SGK1 associated with invasive breast cancer and myeloma cells suggests that increased SGK1 activity may confer a selective advantage to the survival and proliferation of tumorigenic cells (30, 31). Conversely, the cytoprotective effects of SGK1 signaling may enhance survival of cells following ischemia (17) and therefore could be a novel therapeutic target for treatment of stroke and myocardial infarction.
Ischemic cell death involves apoptosis and necrosis. Unlike apoptosis, necrosis was once thought to be an uncontrolled form of cell death consequent to mechanical or oxidative stress that resulted in irreversible cellular damage to the plasma membrane, bioenergetic capacity, and DNA (32). This view of necrotic death has undergone a significant revision in recent years, and it is now believed to involve dynamic interplay of multiple death signaling pathways (32–34). Accumulation of cytoplasmic and mitochondrial Ca2+ is critical in the propagation and execution phases of necrosis (33, 35, 36). Sustained elevation of intracellular Ca2+ (or Ca2+ overloading) contributes to necrotic cell death following ischemia in neurons (37, 38), hepatocytes (39), and cardiomyocytes (40). Whether SGK1 provides prosurvival signaling in necrotic cell death is not known.
Here, we used a combination of biochemical, pharmacological, and Ca2+ imaging approaches to define mechanisms underlying the Ca2+ signals that regulate SGK1 expression. In addition, we used shRNA to define cytoprotective signals requiring SGK1 expression in Ca2+-overloaded cells. Our results suggest that store-operated Ca2+ entry and subsequent calmodulin-dependent protein kinase II (CaMKII) activity regulate SGK1 induction in MDA-MB-231 cells. We also discovered that necrotic cell death produced by a sustained increase in intracellular Ca2+ was attenuated by SGK1 prosurvival signals associated with maintenance of the mitochondrial membrane potential.
EXPERIMENTAL PROCEDURES
Chemicals
Thapsigargin (TG), BAPTA, tunicamycin (TNC), cyclopiazonic acid (CPA), calmidazolium, DTT, and ionomycin were obtained from Sigma-Aldrich. KN93 and STO609 were purchased from Calbiochem.
Cell Culture
MDA-MB-231 human breast cancer cells were obtained from the ATCC (Rockville, MD). MCF10A-Myc, human breast epithelial cells (overexpressing human c-myc), have been described previously (41). The human keratinocyte cell line (HaCaT) was a kind gift of Dr. Yu-Ying He at the University of Chicago. Cells were cultured in DMEM or DMEM-F12 (BioWhittaker) supplemented with 10% heat-inactivated FCS (Gemini BioProducts), 100 units/ml penicillin, and 100 μg/ml streptomycin.
Measurement of Intracellular Ca2+ Concentration
[Ca2+]c in MDA-MB-231 or HaCaT cells was determined as described previously (20, 35). Cells were loaded with Fura-2 by applying 1 μm Fura-2 acetoxymethyl ester (Molecular Probes, Inc.) for 15 min at 37 °C in Krebs Ringers Bicarbonate HEPES buffer (pH 7.4) supplemented with 10 mm d-glucose (KRBH10) and 0.015% (v/v) Pluronic F-127 (Molecular Probes, Inc.). Cells were visualized with a Nikon TE-2000U inverted microscope equipped for epifluorescence. Fura-2 dual-excitation ratiometric microspectrophotometry was used to quantitatively measure [Ca2+]c in single cells as described (42). Digitized images were recorded with a Cascade 650 charge-coupled device (Roper Instruments) at 10-s intervals. Experiments were performed at 37 °C, and cells were superfused constantly in a microperfusion chamber with KRBH10 containing test reagents. Background-subtracted fluorescence intensity measurements from individual cells were converted to molar values of [Ca2+] using an in vivo calibration method (43). Data are expressed as the fold change in [Ca2+]c normalized to the average basal [Ca2+]c recorded during a 60-s period prior to addition of test reagents. OriginPro data analyses (version 7.0) and graphing software (OriginLab Corp., Northampton, MA) was used for curve integration analysis of the [Ca2+] responses.
Western Immunoblotting
Lysates of MDA-MB-231, MCF10A-Myc, or HaCaT cells were prepared as described previously (8). Cells were washed one time with 1× PBS and lysed in Laemmli 2× buffer and lysates incubated at 95 °C for 5 min. Proteins in the lysates were separated on 9% SDS-PAGE gels, electrophoretically transferred to nitrocellulose membrane (Bio-Rad), visualized with Ponceau S Red dye solution, and blocked in a 5% skim milk/Tris-buffered saline and 0.1% Tween 20 solution (TBST). Nitrocellulose membranes were exposed to the following antibodies: anti-SGK1 (1:1000; Assay Designs), anti-P-SGK1 Ser-422 (1:1000; Santa Cruz Biotechnology), anti-BiP (1:1000, BD Biosciences), with anti-fodrin (1:100) Cell Signaling Technology) or anti-β-actin (1:5000; Sigma-Aldrich) as a loading control, in TBST for 1 h (for anti-BiP and anti-β-actin) or overnight (for anti-SGK1 antibodies). The blots were incubated with goat anti-rabbit HRP or anti-mouse HRP-conjugated antibodies (1:5000; Santa Cruz Biotechnology) and visualized using an ECL chemiluminescent kit per manufacturer's instructions (GE Healthcare). Densitometry was performed on the Bio-Rad GS-670 imaging densitometer, analyzed using Quantity One one-dimensional analysis software (Bio-Rad).
Measurements of Gene Expression
Cells were washed in 1× PBS, and total RNA was isolated using the RNeasy mini kit (Qiagen). Total RNA (1 μg) was then used for cDNA synthesis using a TaqMan reverse transcription kit (Applied Biosystems) in a GeneAmp PCR system 9700 (Applied Biosystems). Reverse-transcribed RNA was combined with SYBR PCR Master Mix Green (Applied Biosystems) and primer pairs for SGK1 or the GAPDH gene. Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was performed using the ABI Prism 7300 (Applied Biosystems), and the data were analyzed as described previously (44).
Flow Cytometry
MDA-MB-231 cells stably expressing scrambled sequence (ss) control or SGK1 shRNA cells were treated with ionomycin or DMSO vehicle in 2.5% FCS media for 2 and 8 h. Flow cytometry was used to determine the extent and mechanism of cell death of these cells by measuring phosphatidylserine externalization by annexin A5 (conjugated to FITC and propidium iodide (PI) DNA staining. Both vital stains were applied according to the manufacturer's instructions (FITC-annexin-5 apoptosis detection kit I, BD Biosciences). The Amnis ImageStream® multispectral imaging flow cytometer was used to analyze the different stages of cell death based on staining and morphology. Fluorescence emission from the stained cells was detected in the 488-nm and 785-nm channels for annexin A5 and PI, respectively. Data analysis (n > 2,500 cells) was performed using IDEAS software. Live cells (annexin A5−/PI−), early apoptotic (annexin A5+/PI−), late apoptotic (annexin A5+/PI+), or necrotic (annexin A5−/PI+) were distinguished based on staining. Apoptotic and necrotic cell death were further distinguished based on morphology as described (45).
Mitochondrial Membrane Potential Assay
Dynamic changes in mitochondrial membrane potential (ΔΨm) were measured using a modification of an assay described previously (46). Rhodamine 123 (Rh123), a fluorescent lipophilic cationic dye that specifically partitions into negatively charged mitochondrial membranes was loaded into cells by incubation with 10 μg/ml Rh123 in KRBH10 for 20 min at 37 °C. As ΔΨm increases (hyperpolarization), Rh123 accumulates in the mitochondrial membrane that consequently caused self-quenching of the Rh123 fluorescence. Depolarization of ΔΨm, on the other hand, causes redistribution of Rh123 from mitochondria into the cytosol, which relieves intermolecular quenching and generates an increase of Rh123 fluorescence. Cells were placed into a microperifusion chamber mounted on an inverted microscope equipped for epifluorescence as described above for the Ca2+ imaging studies. Fluorescence emission (530 nm) from Rh123-loaded cells was excited at 490 nm. Fluorescence intensity measurements from single cells were recorded at 10-s intervals. Data were normalized to base-line Rh123 fluorescence intensity 60-s prior to application of test reagents and analyzed using curve integration (Origin, version 7.0).
RESULTS
SERCA (Sarco/endoplasmic Reticulum Ca2+-ATPase) Inhibition Results in Increased Expression of SGK1 Protein
SGK1 is transcriptionally induced, and SGK1 enzymatic activity is stimulated by oxidative and osmotic stress (17). To test the hypothesis that SGK1 expression could be induced by ERS, we measured SGK1 protein levels in MDA-MB-231 breast cancer cells following exposure to three well characterized activators of ERS. TG (500 nm), TNC (10 μg/ml), and DTT (5 mm) were applied for 1, 2, 4, and 8 h. TG rapidly increased the expression of SGK1 (Fig. 1A). Increased SGK1 was evident as early as 1 h and continued increasing 2–8 h after administration of TG. Neither TNC nor DTT significantly increased SGK1 expression (Fig. 1A). Induction of ERS in MDA-MB-231 cells was confirmed by measuring for the presence the GRP78 (78-kDa glucose-regulated protein), also known as immunoglobulin heavy-chain binding protein (BiP), a member of the 70-kDa heat shock protein family and a protein chaperone within the ER lumen. Consistent with stimulation of ERS, TG, TNC, and DTT treatment increased BiP protein expression 8 h after treatment (Fig. 1B). Induction of SGK1 protein expression was remarkably sensitive to low concentrations of TG: a 4-h exposure to 10 nm TG increased SGK1 (Fig. 1C).
FIGURE 1.

Thapsigargin treatment, but not other ER stressors, induces SGK1 protein expression in MDA-MB-231 human breast cancer cells. A, MDA-MB-231 cells treated with TG (500 nm), tunicamycin (10 μg/ml), or dithiothreitol (5 mm), vehicle (DMSO or H2O) for the indicated times and examined by Western blot. β-Actin was used as a loading control. B, MDA-MB-231 cells were treated with TG (500 nm), tunicamycin (10 μg/ml), or dithiothreitol (5 mm) for a longer time period from 4 to 32 h, and protein lysates were examined for total SGK1 and BiP protein expression. C, MDA-MB-231 cells were treated with various concentration of TG (from 0.1 to 500 nm) for 4 h. D, MDA-MB-231 cells were treated with 500 nm TG for 2 h, and P-SGK1 Ser-422 and total SGK1 were measured. E, immortalized keratinocytes (HaCaT) treated with thapsigargin for the indicated times, and P-SGK1 Ser-422 and total SGK1 were detected by Western blot. All data are representative of at least three independent experiments. The asterisks represent a background band seen with the total SGK1 antibody; β-actin was detected as a loading control.
Posttranslational modification of SGK1 is also an important regulator of SGK1 activity. Phosphorylation of SGK1 at residues Thr-256 and Ser-422 regulates its activation and downstream prosurvival signaling (6, 47). Phosphorylation of the Ser-422 site of SGK1 was detected 2 h after TG treatment of MDA-MB-231 cells (Fig. 1D). We found the same effects of TG on SGK1 total protein expression and SGK1 Ser-422 phosphorylation in keratinocyte HaCaT cells (Fig. 1E). These results are consistent with previous observations suggesting that SGK1 Ser-422 phosphorylation and subsequent activation is associated with peak steady-state levels of SGK1 (7).
Thapsigargin Treatment Results in Increased SGK1 mRNA Expression
Because SGK1 activation is initiated by transcriptional up-regulation, we measured the effects of DTT, TNC, and TG on SGK1 mRNA expression levels. One hour after application of 500 nm TG, SGK1 mRNA steady-state levels in MDA-MB-231 cells were significantly increased relative to control cells (Fig. 2). TG-induced SGK1 mRNA was significantly greater than levels evoked by TNC or DTT (p < 0.05) (Fig. 2A, left panel). SGK1 steady-state mRNA levels were also significantly increased in the MCF10A-Myc breast epithelial cell line 4 h following application of 500 nm TG (Fig. 2B, right panel).
FIGURE 2.
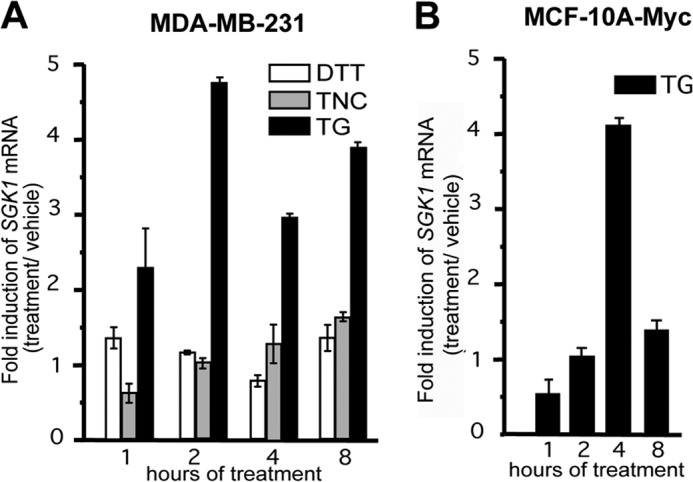
Thapsigargin induces SGK1 mRNA in mammary epithelial cells. A, MDA-MB-231 cells were treated with DTT, TNC, TG, or vehicle, and qRT-PCR was used to detect SGK1 steady-state mRNA levels. B, MCF10A-Myc cells were treated with TG (500 nm), and qRT-PCR was used to detect the SGK1 mRNA levels. The fold change of SGK1 mRNA was normalized to GAPDH mRNA, and all data are representative of three independent experiments.
Our findings indicated that TG, TNC, and DTT trigger ERS in MDA-MB-231 cells. However, ERS alone was not sufficient to increase SGK1 expression because TNC and DTT did not affect SGK1 protein induction. This led us to examine other signaling pathways activated by TG that might underlie SGK1 activation. TG has long been known to be a specific inhibitor of SERCA. ER Ca2+ homeostasis is critically dependent on SERCA activity. Irreversible inhibition of SERCA by TG causes depletion of ER Ca2+ stores and an elevation of [Ca2+]c. This raised the possibility that inhibition of SERCA and changes in [Ca2+]c, rather than ERS itself, contributed to the observed rapid induction of SGK1 by TG. To test this hypothesis, we determined the effect of CPA, a SERCA inhibitor that is structurally distinct from TG, on SGK1 expression. CPA (50–150 μm) rapidly and transiently increased expression of SGK1 (Fig. 3, top panel). The initial CPA-induced increase in SGK1 was observed within 1–2 h. Maximum levels occurred within 4 h and then declined after 8 h. CPA effects were reversible as soon as 1 h following washout (Fig. 3, bottom panel).
FIGURE 3.
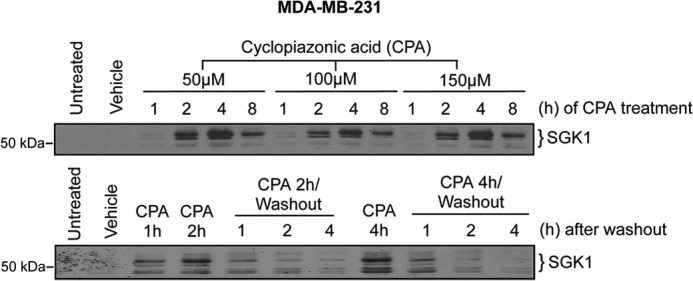
Increased SGK1 expression is dependent on SERCA pump inhibition. Western analysis of SGK1 expression following treatment with the reversible SERCA pump inhibitor CPA. Top panel, time course Western blot analysis of SGK1 following treatment with CPA (50–150 μm). Bottom panel, SGK1 expression in cells treated with CPA (50 μm) for 2 or 4h followed by a PBS washout and subsequent recovery in DMEM lacking CPA for an additional 1- 4-h.
SGK1 Expression Is Regulated by Ca2+ Influx from the Extracellular Milieu
In many cell types, inhibition of SERCA increases [Ca2+]c. We used Fura2, a fluorescent Ca2+ indicator and microspectrophotometry to measure the effects of TG on [Ca2+]c in MDA-MB-231 cells. Base-line [Ca2+]c was 59 ± 5 nm (n = 129 cells). Application of 100 nm TG rapidly increased [Ca2+]c within 90 s; after 15 min, maximum levels were increased 20- ± 4-fold (n = 29 cells) over base-line values (Fig. 4A). The increase in [Ca2+]c during a 15-min exposure to TG was decreased by 85% in cells in which the cytoplasm had been loaded with BAPTA, a Ca2+ chelator (Fig. 4A). The area under the [Ca2+]c response curve in BAPTA-loaded cells was 1,400 ± 465 compared with 9,147 ± 1,310 in control cells. Intracellular accumulation of BAPTA did not affect base-line [Ca2+]c (62 ± 6 nm; n = 34 cells). Consistent with a role of increased [Ca2+]c in the induction of SGK1, we found that chelation of intracellular [Ca2+]c with BAPTA significantly decreased TG-induced SGK1 mRNA (Fig. 4B) and protein (Fig. 4C) accumulation. SGK1 protein expression was also increased following a 4-h exposure of cells to ionomycin (1–2.5 μm), a Ca2+ ionophore (Fig. 4D). Taken together, these findings suggest that an increase in [Ca2+]c is sufficient for induction of SGK1.
FIGURE 4.

The calcium chelator, BAPTA, inhibits thapsigargin-induced SGK1 expression. MDA-MB-231 cells were pretreated with either 20 μm BAPTA for 2 h followed by TG or vehicle. A, intracellular Ca2+ was measured over time using Fura-2-AM following TG treatment (100 nm) of MDA-MB-231 cells pretreated with BAPTA (n = 41) or vehicle control (n = 47). B, MDA-MB-231 cells treated with BAPTA alone, pretreated with BAPTA, and then treated with TG or TG alone for 4 h. SGK1 mRNA levels were measured by qRT-PCR. The fold changes in SGK1 mRNA levels in treated samples were normalized to GAPDH expression and then compared with vehicle. C, Western blot analysis of SGK1. D, MDA-MB-231 cells treated with ionomycin or DMSO vehicle for SGK1 expression by Western blotting. Data representative of three independent experiments are presented.
We next determined the source of Ca2+ that triggered increased SGK1 expression following TG exposure. TG-induced elevation of [Ca2+]c is caused by a Ca2+ leak into the cytoplasm following SERCA inhibition and Ca2+ influx through plasma membrane store-operated Ca2+ channels. The increase in [Ca2+]c following a 15-min application of 100 nm TG was reduced 93% in cells incubated in a Ca2+-free solution (Fig. 5A). The average ± S.E. area under the [Ca2+]c response curve was 931 ± 57 (n = 29 cells) in a Ca2+-free buffer compared with 13,826 ± 1,687 in an extracellular solution containing 2.5 mm Ca2+ (n = 42 cells). In Ca2+-free solutions, 135 ± 9 s after administration of TG, [Ca2+]c increased transiently to a maximum amplitude 2.7 ± 0.2 times the base-line resting level (Fig. 5A). These results suggest that influx of extracellular Ca2+ via store-operated Ca2+ entry is the major source of the rise in [Ca2+]c evoked by TG. Parallel experiments demonstrated that Ca2+ influx was required for induction of SGK1. In the absence of extracellular Ca2+, TG treatment did not induce detectable levels of SGK1 protein (Fig. 5B). Our findings suggest that influx of extracellular Ca2+ through store-operated Ca2+ channels underlies Ca2+-mediated activation of SGK1 in MDA-MB-231 cells.
FIGURE 5.
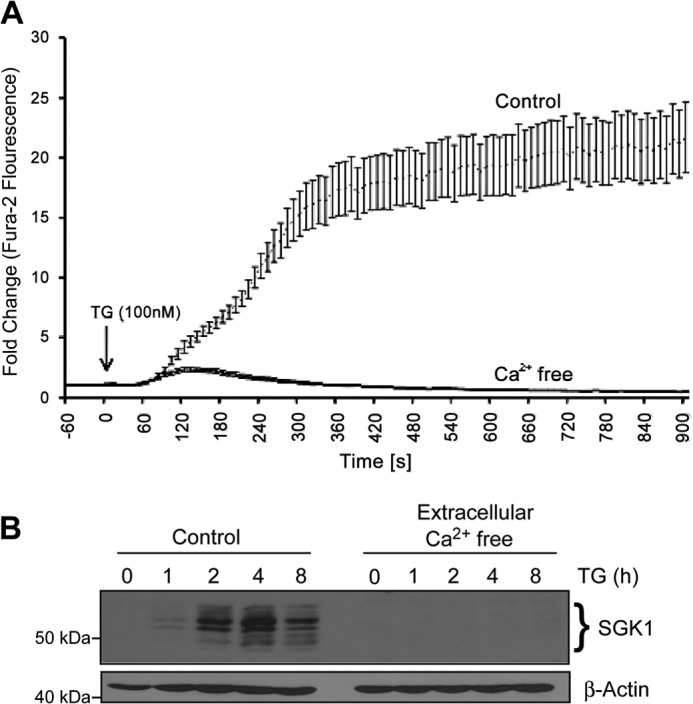
SGK1 induction by TG requires extracellular calcium. A, MDA-MB-231 cells were maintained in Ca2+-free DMEM under low serum conditions (2.5% FCS) and pretreated with 20 μm BAPTA or DMSO vehicle control for 2 h, loaded with Fura-2AM, and subsequently stimulated with TG (100 nm). To monitor intracellular Ca2+ release, Fura-2 fluorescence was measured over time. B, SGK1 protein expression was examined by Western blot analysis of total cell lysates. β-Actin was used as a loading control.
Calmodulin-mediated Signaling Is Required for Ca2+-mediated SGK1 Induction
Calmodulin and CaMKII are important downstream effectors of intracellular Ca2+ signal transduction and have been shown to play a role in the induction of SGK1 mRNA expression in A6 renal cells following hypotonic shock (11). We used pharmacological agents to probe the role of calmodulin, CaMKII, and CaM-kinase kinase (CaMKK) in TG-induced SGK1 expression in MDA-MB-231 cells. Application of calmidazolium, a well characterized inhibitor of calmodulin, inhibited TG-stimulated SGK1 protein induction by 98% (Fig. 6A). Short term SGK1 mRNA expression in MDA-MB-231 cells treated with TG was also inhibited by KN93 and STO609, inhibitors of CaMKII and CaMKK, respectively (Fig. 6B); both reagents significantly decreased the effect of TG on SGK1 induction within 1 h. However, although inhibition of TG-induced SGK1 mRNA by STO609 was transient, in the presence of a combination of KN93 and STO609, the effect of TG on SGK1 mRNA induction was significantly decreased at all time points studied (Fig. 6B). TG-induced SGK1 protein levels were reduced by 86% in cells pretreated with KN93 (Fig. 6C). SGK1 protein levels were actually higher in STO609-pretreated cells. These data suggest that CaMKII, and not CaMKK, is required for mediating TG-induced SGK1 expression.
FIGURE 6.
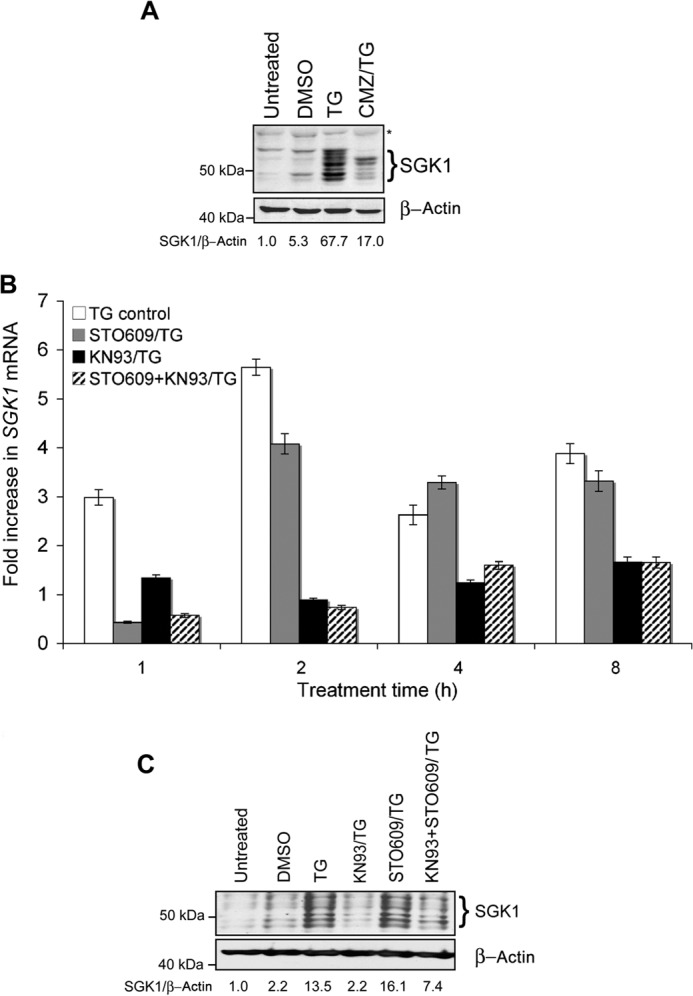
The calmodulin/CamKII pathway is required for calcium-mediated SGK1 induction. A, MDA-MB-231 cells were treated with TG alone or pretreated with calmizidium chloride (CMZ) prior to TG (CMZ/TG) and total SGK1 detected by Western blot. B, total RNA was isolated from cells treated with TG alone, or pretreated with either KN93, STO609, or both. qRT-PCR was performed to detect steady-state SGK1 mRNA levels and then normalized to GAPDH levels. C, SGK1 expression following pretreatment with either TG alone; co-treatment with KN93 and TG; co-treatment with STO609 and TG; co-treatment with KN93, STO609, and TG; DMSO vehicle control; or untreated control. Data shown are representative of three independent experiments.
SGK1 Expression Attenuates Cell Death Induced by Ca2+ Overloading
We next explored the role of [Ca2+]c-induced SGK1 expression in maintaining cell viability. Prolonged elevation of [Ca2+]c or calcium overloading is a pathway for initiating cellular demise. We reasoned that Ca2+-dependent induction of SGK1 expression might contribute to mitigating Ca2+-induced cell death. We selected MDA-MB-231 cells stably expressing SGK1 shRNA (7) and exhibiting no detectable SGK1 despite either ionomycin (Fig. 7A, right panel) or TG (data not shown) treatment. In contrast, cells stably expressing a control scrambled sequence (ss) shRNA, SGK1 protein levels increased 1–4 h following the application of 2.5 μm ionomycin (Fig. 7A, left panel).
FIGURE 7.

SGK1 induction contributes to protection from apoptosis and necrosis. A, MDA-MB-231 cells stably expressing SGK1 shRNA or a control “scrambled sequence” shRNA were treated with ionomycin (2.5 μm) for the indicated time. B, SGK1 or control shRNA-expressing MDA-MB-231 cells were treated with 2.5 μm ionomycin for 8 h in the presence of a medium containing 2.5% serum, stained with annexin A5 (A5) and PI, and analyzed by flow cytometry. C, the different stages of cell death were delineated based on whether they were healthy (A5−/PI−), early apoptotic (A5+/PI−), late apoptotic (A5+/PI+), or necrotic (A5−/PI+). Inset shows representative microscopic images of these categories from the ImageStream flow cytometer. B.F, bright field. A5, annexin A5. For both B and C, the graph represents the average cell death of triplicate control or SGK1 shRNA samples treated with ionomycin or vehicle. The error bars represent the S.E. *, p < 0.05 compared with vehicle treated control or SGK1 shRNA. **, p < 0.05 compared with ionomycin treated control shRNA. The two-tailed Student's t test was used to determine significance.
Diminished SGK1 expression in the SGK1 shRNA cells was also associated with increased cell death following Ca2+ overload. Flow cytometric detection of annexin A5 and PI staining was used to characterize the type of cell death induced by exposure to ionomycin for 2 and 8 h. Cells undergoing early stages of apoptosis stain positive for annexin A5 by binding to phosphatidylserine on the cell surface. Late apoptotic and necrotic cells lose membrane integrity and lack an ability to exclude PI, a fluorescent cationic DNA binding molecule. A 2-h exposure to ionomycin did not evoke statistically significant increased cell death in SGK1 shRNA cells versus control (data not shown). Application of ionomycin for 8 h, however, induced cell death in 31 and 50% of control and SGK1 shRNA cells, respectively (Fig. 7B). Brightfield imaging of the early apoptotic cells (annexin A5+/PI−) indicated that they retained a relatively normal cellular morphology, but late apoptotic cells (annexin A5+/PI+) were irregularly shaped. Necrotic cells (annexin A5−/PI+) maintained normal cell morphology. We estimated the type of cell death based on staining of annexin A5 and PI (Fig. 7C). At 8 h, ionomycin did not significantly induce cell death characteristic of early apoptosis (annexin A5+/PI−). Nevertheless, the percentage of cells undergoing late apoptosis was significantly increased in the SGK1 shRNA cells (1.1% following vehicle treatment compared with 8.6% after ionomycin exposure). These findings suggest that ionomycin-induced apoptosis plays a modest role in MDA-MB-231 cell death consequent to Ca2+ overloading.
In contrast to the relatively low percentage of early and late apoptotic cells following Ca2+ overloading, necrotic cell death appeared to be the predominant form of cellular demise evoked by ionomycin. After an 8-h exposure to ionomycin, necrotic cell death (reflected by annexin A5−/PI+ staining cells) was observed in 23 and 40% of the control ss shRNA and SGK1 shRNA cells, respectively. These data suggest that SGK1 plays an important cytoprotective role in Ca2+-induced necrotic cell death.
SGK1 Expression Attenuates the Effects of Cytoplasmic Ca2+ Overloading on Mitochondrial Membrane Potential
Our findings suggest that SGK1 prosurvival signals affect the biochemical and molecular events that regulate necrotic cell death. Caspases, a family of proteolytic enzymes, are key executioners of cell death (48) and can be activated by cytoplasmic Ca2+ overloading (49). We used flow cytometry and Western analysis to measure the effects of cytoplasmic Ca2+ overloading on caspase activation. A 24-h application of ionomycin did not activate caspase 3 or caspase 7 in either control or SGK1-depleted cells as detected by either flow cytometry or Western analysis (data not shown). This suggested that cytoplasmic Ca2+ overloading likely triggered MDA-MB-231 cell death by a caspase-independent mechanism.
Caspase-independent cell death has been associated with sustained elevation of intracellular Ca2+ following application of ionomycin (50). Multiple cellular proteins can contribute to Ca2+-mediated cell death, including proteases, Ca2+-dependent kinases and phosphatases, and endonucleases (48). Calpains, cysteine proteases that are activated by an increase in intracellular Ca2+, play an important role in neuronal cell death (33, 51, 52). To determine whether calpain activation was involved in MDA-MB-231 cell death induced by sustained elevation of cytoplasmic Ca2+, we next measured relative cleavage of α-fodrin, a calpain substrate, by Western blot following ionomycin treatment. Modest fodrin cleavage was observed in control and SGK1 shRNA cells following ionomycin treatment.5 Using densitometric assessment of immunoreactive cleaved fodrin normalized to actin, the extent of α-fodrin cleavage was only slightly (1.4-fold) greater in SGK1 shRNA expressing cells compared with ss shRNA control cells.5 This modest increase in fodrin cleavage in SGK1-depleted cells suggests that SGK1 expression does not primarily inhibit Ca2+-mediated necrosis through blockade of calpain activation.
Changes in mitochondrial membrane potential (ΔΨm) accompany the initiation of apoptosis (35), whereas persistent hyperpolarization of mitochondria is associated with necrotic cell death (53–56). We measured time-dependent changes in Δψm using rhodamine 123, a fluorescent potentiometric indicator. Under resting conditions, base-line Δψm in the control and SGK1 shRNA cells was not significantly different (p = 0.42, n = 246 and 122 control and SGK1 shRNA cells, respectively). A 60-min application of 2.5 μm ionomycin produced two patterns of Δψm response in the control and SGK1 shRNA cells (Fig. 8). In 62% of control cells (44/71 cells) expressing a scrambled sequence (ss) shRNA, Ca2+ overloading evoked a rapid and transient depolarization of Δψm. The maximum level of mitochondrial depolarization in the control cells occurred within 10 min and was sustained over the next 25 min. Subsequently, Δψm hyperpolarized (Fig. 8). This pattern of response was observed in only 14% of the SGK1-depleted cells (12/76 cells). In 86% of the SGK1-shRNA cells (64/76 cells), Δψm rapidly hyperpolarized in the presence of the Ca2+ ionophore. After 20 min, Δψm in the SGK1-depleted cells reached a maximum level of hyperpolarization that remained until the end of the experiment (Fig. 8). In contrast, the persistent Δψm hyperpolarization that occurred during Ca2+ overloading was detected in only 38% of the control cells (27/71 cells). These results suggest that a primary mechanism whereby SGK1 induction decreases Ca2+-dependent necrosis through preventing persistent hyperpolarization of the mitochondrial membrane.
FIGURE 8.

SGK1 prevents Ca2+ overloading-induced persistent hyperpolarization of mitochondria. Time course of Δψm response in MDA-MB-231 cells after application of ionomycin (2.5 μm). Cells were loaded with the cationic fluorescent dye, rhodamine 123 (Rh123). Data are expressed as normalized Rh123 fluorescence (F/F0), where F is the Rh123 measured at each time point and F0 is the average Rh123 fluorescence measured in the cells during the 60-s period prior to the addition of ionomycin (relative Rh123FLI). As mitochondria hyperpolarize relative to base-line levels, Rh123 accumulates within the mitochondria. Intermolecular crowding of Rh123 causes fluorescence quenching and a decrease in fluorescence emission. Conversely, as mitochondria depolarize, Rh123 is released from the mitochondria into the cytosol, which relieves quenching and increases Rh123 fluorescence. Note that ionomycin evokes persistent hyperpolarization of mitochondria in MDA-MB-231 cells that do not express SGK1 (SGK1 shRNA). In control cells expressing a scrambled sequence shRNA that does not target any known gene (darker gray line), the Ca2+ ionophore causes transient depolarization of Δψm that repolarizes to suprabasal levels. Results depict mean ± S.E. of 71 control and 76 SGK1 shRNA cells.
DISCUSSION
In this study, we found that only a subset of chemicals causing ERS are capable of inducing SGK1 expression in human epithelial cell lines. The mechanism of SGK1 induction in this subset is specific to a Ca2+-dependent signal transduction mechanism. The application of those chemical inducers of ERS that inhibit ER Ca2+-ATPases both activated transcription of the SGK1 gene and greatly increased steady-state SGK1 protein levels. SGK1 induction did not occur in cells loaded with a Ca2+ chelator or maintained in a Ca2+-free extracellular solution but was observed in cells treated with ionomycin, a Ca2+ ionophore. Our findings are in accordance with results in CHO cells (16) and Xenopus laevis A6 renal epithelial cells (15) suggesting a role for increased intracellular Ca2+ in inducing SGK1 expression. However, we show for the first time that an increase in [Ca2+]c stemming from Ca2+ entry through store-operated Ca2+ channels up-regulates SGK1 expression and that SGK1 up-regulation attenuates mitochondrial membrane hyperpolarization and necrotic cell death induced by Ca2+ overloading.
Acute induction of ERS results from leakage of intraluminal ER Ca2+ following, for example, SERCA inhibition. Subsequently, altered protein folding and post-translational modification of nascent polypeptides triggers the unfolded protein response (UPR). This signaling cascade facilitates restoration of normal cellular function and viability following ERS (57, 58). The transient activation of the UPR maintains cellular homeostasis in the presence of acute stress by relieving ERS through reducing translation, up-regulating the expression of genes encoding ER chaperones, and decreasing misfolded or mutant proteins through ER-associated degradation (59, 60). Additional cellular signals that may participate in the recovery from ERS have not been completely defined; the stress-responsive SGK1 gene presented an excellent candidate gene for participating in the recovery from ERS, especially in light of the role of SGK1 in regulating cation flux (18) and its mitochondrial and ER subcellular localization (9).
We tested the hypothesis that ERS would increase SGK1 expression. Because ERS can be caused by different experimental conditions, we determined the effects of three chemical reagents that induce ERS by distinct mechanisms. As expected, TG, TNC, and DTT rapidly increased BiP levels, an indicator of ERS and the UPR. However, induction of ERS per se was insufficient to up-regulate SGK1 transcription. TG, a sesquiterpene lactone compound derived from the root of the Thapsia garganica plant and CPA, a mycotoxin produced by Penicillium cyclopium, stimulated robust expression of SGK1. Neither TNC, a product of Streptomyces lysosuperficus that inhibits GlcNAc phosphotransferase and post-translational modification of glycoproteins, nor DTT (or Cleland's reagent) which interferes with the protein folding within the ER, induced SGK1. This suggests that signals downstream of the UPR do not in themselves contribute to SGK1 induction.
SERCA transports Ca2+ from the cytosol into the ER lumen and is the central regulator of ER Ca2+ homeostasis. TG and CPA specifically inhibit SERCA, and in many cell types, increase [Ca2+]c by a combination of release of Ca2+ sequestered within the ER lumen and influx of extracellular Ca2+ through store-operated Ca2+ channels (61, 62). Application of TG to MDA-MB-231 cells resulted in the rapid and irreversible increase in [Ca2+]c. However, in the absence of Ca2+ in the extracellular medium, an experimental condition that excludes any contribution of store-operated Ca2+ channels to the influx of Ca2+, we observed a modest increase in Ca2+ levels 2 min following TG treatment. This increase in [Ca2+]c likely stems from ER store leakage but did not affect SGK1 expression compared with the influx of extracellular Ca2+, suggesting that the main mechanism of SGK1 induction following SERCA inhibition is extracellular Ca2+ rather than the release of Ca2+ from ER stores. This observation is consistent with findings in other cell systems, demonstrating control of gene expression specifically by Ca2+ entry through plasmalemmal Ca2+ channels (63).
SERCA inhibition causes a rapid depletion of ER Ca2+ stores (64). Persistent depletion of ER Ca2+ stores has been associated with induction of ERS and apoptosis (65, 66). We investigated the role of ER Ca2+ depletion in SGK1 induction using BAPTA. BAPTA, a chelator of Ca2+, blocked the TG-induced increase in SGK1 expression and [Ca2+]c. This suggests that the rise in [Ca2+]c was necessary and sufficient for up-regulation of SGK1 expression following SERCA inhibition. Our findings also suggest that depletion of ER Ca2+ stores alone is not sufficient to up-regulate SGK1 transcription.
Signal transduction by intracellular Ca2+ is mediated through a wide range of effector proteins that undergo Ca2+-dependent conformational changes and consequently activate distal Ca2+-dependent physiological processes. For example, calmodulin (CaM), a ubiquitous Ca2+-binding protein, has no intrinsic enzymatic activity but integrates Ca2+ signaling by transducing changes in [Ca2+] to downstream enzymes such as Ca2+/CaM-activated protein kinases, CaMKK, CaMKI, CaMKII, and CaMKIV. Previous studies have demonstrated that phosphorylation and activation of SGK1 following stimulation of CHO-IR cells is regulated by CaMKK in a PI3K-independent manner (16). In addition, calmodulin-dependent signaling was implicated in up-regulation of SGK1 mRNA expression in A6 cells following exposure to hypotonic stress conditions (15). Interestingly, our data suggest that calmodulin/CaMKII activity is necessary for Ca2+-dependent induction of SGK1 mRNA and protein expression in MDA-MB-231 human breast epithelial cells exposed to TG. Taken together with our Ca2+ flux measurements, the aggregate data suggest that an influx of Ca2+ through store-operated Ca2+ channels is coupled with activation of CaMKK and CaMKII signaling cascades that in turn up-regulate SGK1 gene expression, leading to increased SGK1 steady-state protein levels and activity.
Because SGK1 expression plays a role in mitigating cell death following exposure to many stressors, including chemotherapy and growth factor withdrawal, we speculated that increased SGK1 expression might promote cell survival following Ca2+ influx. Indeed, we found that ionomycin treatment induced a higher percentage of MDA-MB-231 cells undergoing late apoptotic and necrotic cell death in cells in which SGK1 expression was diminished by an SGK1-specific shRNA. Ionomycin-induced necrosis was partially inhibited in SGK1-expressing cells after 8 h of ionomycin treatment. Necrosis and apoptosis are independent biological processes and apoptosis is not (always) a requisite step for necrosis. We do not yet understand the precise mechanisms linking Ca2+ overload, SGK1 induction, cell survival, and preservation of mitochondrial membrane potential. Although it has long been appreciated that overloading mitochondria with Ca2+ leads to apoptotic and necrotic cell death, whether SGK1 plays a specific role in inhibiting accumulation of Ca2+ in mitochondria and/or attenuating loss of mitochondrial membrane potential during Ca2+ influx remains uncertain. Additional studies will be necessary to define the interrelationship between SGK1 expression, maintenance of mitochondrial membrane potential, and protection from Ca2+ overload-induced cell death.
In summary, we have demonstrated that contrary to our initial hypothesis that all ERS and/or UPR signals would result in increased expression of the stress-associated protein SGK1, SGK1 induction appears to respond specifically to increased [Ca2+]c. Among ER stressors, only compounds also causing increased epithelial cell [Ca2+]c stimulated a rapid and robust induction of SGK1 mRNA and protein levels followed by phosphorylation at the SGK1 active site Ser422. Furthermore, our findings suggest that Ca2+-dependent activation of SGK1 transcription is caused by opening of store-operated Ca2+ channels in the plasma membrane and the influx of extracellular Ca2+ from the extracellular milieu rather than from the release of Ca2+ from the intraluminal ER. Moreover, we demonstrate that Ca2+-dependent SGK1 induction and activation contribute to attenuating changes in mitochondrial membrane polarity and are independent of caspase activation. Taken together, these observations suggest that SGK1 could function as a central responder to calcium-mediated mitochondrial membrane dysfunction and subsequent cell death. Future studies will address the hypothesis that SGK1 induction and/or increased activity may specifically regulate cation flux within the mitochondria, thereby attenuating cell death.
Acknowledgments
We thank Dr. Gopal Thinakaran for advice on antibodies and measurement of ERS at the outset of this project. We also thank Dr. Ryan Duggan from the University of Chicago Flow Cytometry Facility for expertise on flow cytometry experiments.
This work was supported by National Institutes of Health Grants R01 CA089208 and R21 CA149472 (to S. D. C.), R01 DK074966 and R01 DK092616 (to M. W. R.), as well as the University of Chicago's Breast Cancer SPORE National Institutes of Health Grant P50 CA125183 and Comprehensive Cancer Center Support Grant for Core Facilities, National Institutes of Health Grant P30 CA014599.
D. R. Brickley, A. S. Agyeman, R. F. Kopp, B. A. Hall, M. C. Harbeck, L. Belova, P. A. Volden, W. Wu, M. W. Roe, and S. D. Conzen, unpublished observations.
- SGK1
- serum and glucocorticoid-regulated kinase 1
- BAPTA
- 1,2-bis (o-aminophenoxy) ethane-N,N,N′,N′-tetraacetate acetoxy-methylester
- CaMKK
- calmodulin-dependent protein kinase kinase
- CaMKII
- Ca2+/camodulin-dependent protein kinase II
- CPA
- cyclopiazonic acid
- ERS
- endoplasmic reticulum stress
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- ss
- scrambled sequence
- TG
- thapsigargin
- TNC
- tunicamycin
- qRT-PCR
- quantitative RT-PCR
- DMSO
- dimethyl sulfoxide
- PI
- propidium iodide
- Rh123
- rhodamine 123
- UPR
- unfolded protein response.
REFERENCES
- 1. Webster M. K., Goya L., Ge Y., Maiyar A. C., Firestone G. L. (1993) Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol. Cell Biol. 13, 2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Webster M. K., Goya L., Firestone G. L. (1993) Immediate-early transcriptional regulation and rapid mRNA turnover of a putative serine/threonine protein kinase. J. Biol. Chem. 268, 11482–11485 [PubMed] [Google Scholar]
- 3. Mikosz C. A., Brickley D. R., Sharkey M. S., Moran T. W., Conzen S. D. (2001) Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J. Biol. Chem. 276, 16649–16654 [DOI] [PubMed] [Google Scholar]
- 4. Leong M. L., Maiyar A. C., Kim B., O'Keeffe B. A., Firestone G. L. (2003) Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J. Biol. Chem. 278, 5871–5882 [DOI] [PubMed] [Google Scholar]
- 5. Loffing J., Flores S. Y., Staub O. (2006) Sgk kinases and their role in epithelial transport. Annu. Rev. Physiol. 68, 461–490 [DOI] [PubMed] [Google Scholar]
- 6. Park J., Leong M. L., Buse P., Maiyar A. C., Firestone G. L., Hemmings B. A. (1999) Serum and glucocorticoid-inducible kinase (SGK1) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 18, 3024–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belova L., Brickley D. R., Ky B., Sharma S. K., Conzen S. D. (2008) Hsp90 regulates the phosphorylation and activity of serum- and glucocorticoid-regulated kinase-1. J. Biol. Chem. 283, 18821–18831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brickley D. R., Mikosz C. A., Hagan C. R., Conzen S. D. (2002) Ubiquitin modification of serum and glucocorticoid-induced protein kinase-1 (SGK-1). J. Biol. Chem. 277, 43064–43070 [DOI] [PubMed] [Google Scholar]
- 9. Bogusz A. M., Brickley D. R., Pew T., Conzen S. D. (2006) A novel N-terminal hydrophobic motif mediates constitutive degradation of serum- and glucocorticoid-induced kinase-1 by the ubiquitin-proteosome pathway. FEBS J. 273, 2913–2928 [DOI] [PubMed] [Google Scholar]
- 10. Belova L., Sharma S., Brickley D. R., Nicolarsen J. R., Patterson C., Conzen S. D. (2006) Ubiquitin-proteosome degradation of serum- and glucocorticoid-regulated kinase-1 (SGK-1) is mediated by the chaperone-dependent E3 ligase CHIP. Biochem. J. 400, 235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sheng H., Sun T., Cong B., He P., Zhang Y., Yan J., Lu C., Ni X. (2008) Corticotropin-releasing hormone stimulates SGK-1 kinase expression in cultured hippocampal neurons via CRH-R1. Am. J. Physiol. Endocrinol. Metab. 295, E938–946 [DOI] [PubMed] [Google Scholar]
- 12. Fejes-Tóth G., Náray-Fejes-Tóth A. (2007) Early aldosterone-regulated genes in cardiomyocytes: clues to cardiac remodeling? Endocrinology 148, 1502–1510 [DOI] [PubMed] [Google Scholar]
- 13. Náray-Fejes-Tóth A., Fejes-Tóth G. (2000) The sgk, an aldosterone-induced gene in mineralocorticoid target cells, regulates the epithelial sodium channel. Kidney Int. 57, 1290–1294 [DOI] [PubMed] [Google Scholar]
- 14. Náray-Fejes-Tóth A., Fejes-Tóth G., Volk K. A., Stokes J. B. (2000) SGK is a primary glucocorticoid-induced gene in the human. J. Steroid Biochem. Mol. Biol. 75, 51–56 [DOI] [PubMed] [Google Scholar]
- 15. Taruno A., Niisato N., Marunaka Y. (2008) Intracellular calcium plays a role as the second messenger of hypotonic stress in gene regulation of SGK1 and ENaC in renal epithelial A6 cells. Am. J. Physiol. Renal Physiol. 294, F177–186 [DOI] [PubMed] [Google Scholar]
- 16. Imai S., Okayama N., Shimizu M., Itoh M. (2003) Increased intracellular calcium activates serum and glucocorticoid-inducible kinase 1 (SGK1) through a calmodulin-calcium calmodulin dependent kinase kinase pathway in Chinese hamster ovary cells. Life Sci. 72, 2199–2209 [DOI] [PubMed] [Google Scholar]
- 17. Lang F., Böhmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (2006) (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151–1178 [DOI] [PubMed] [Google Scholar]
- 18. Chen S. Y., Bhargava A., Mastroberardino L., Meijer O. C., Wang J., Buse P., Firestone G. L., Verrey F., Pearce D. (1999) Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. U.S.A. 96, 2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aoyama T., Matsui T., Novikov M., Park J., Hemmings B., Rosenzweig A. (2005) Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation 111, 1652–1659 [DOI] [PubMed] [Google Scholar]
- 20. Yun C. C., Chen Y., Lang F. (2002) Glucocorticoid activation of Na+/H+ exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J. Biol. Chem. 277, 7676–7683 [DOI] [PubMed] [Google Scholar]
- 21. Boini K. M., Hennige A. M., Huang D. Y., Friedrich B., Palmada M., Boehmer C., Grahammer F., Artunc F., Ullrich S., Avram D., Osswald H., Wulff P., Kuhl D., Vallon V., Häring H. U., Lang F. (2006) Serum- and glucocorticoid-inducible kinase 1 mediates salt sensitivity of glucose tolerance. Diabetes 55, 2059–2066 [DOI] [PubMed] [Google Scholar]
- 22. Yang Y. C., Lin C. H., Lee E. H. (2006) Serum- and glucocorticoid-inducible kinase 1 (SGK1) increases neurite formation through microtubule depolymerization by SGK1 and by SGK1 phosphorylation of tau. Mol. Cell Biol. 26, 8357–8370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tai D. J., Su C. C., Ma Y. L., Lee E. H. (2009) SGK1 phosphorylation of IκB kinase α and p300 up-regulates NF-κB activity and increases N-methyl-d-aspartate receptor NR2A and NR2B expression. J. Biol. Chem. 284, 4073–4089 [DOI] [PubMed] [Google Scholar]
- 24. Zhang B. H., Tang E. D., Zhu T., Greenberg M. E., Vojtek A. B., Guan K. L. (2001) Serum- and glucocorticoid-inducible kinase SGK phosphorylates and negatively regulates B-Raf. J. Biol. Chem. 276, 31620–31626 [DOI] [PubMed] [Google Scholar]
- 25. Rusai K., Wagner B., Roos M., Schmaderer C., Strobl M., Boini K. M., Grenz A., Kuhl D., Heemann U., Lang F., Lutz J. (2009) The serum and glucocorticoid-regulated kinase 1 in hypoxic renal injury. Cell Physiol. Biochem. 24, 577–584 [DOI] [PubMed] [Google Scholar]
- 26. Catela C., Kratsios P., Hede M., Lang F., Rosenthal N. (2010) Serum and glucocorticoid-inducible kinase 1 (SGK1) is necessary for vascular remodeling during angiogenesis. Dev. Dyn. 239, 2149–2160 [DOI] [PubMed] [Google Scholar]
- 27. Brunet A., Park J., Tran H., Hu L. S., Hemmings B. A., Greenberg M. E. (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell Biol. 21, 952–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richards J. S., Sharma S. C., Falender A. E., Lo Y. H. (2002) Expression of FKHR, FKHRL1, and AFX genes in the rodent ovary: evidence for regulation by IGF-1, estrogen, and the gonadotropins. Mol. Endocrinol. 16, 580–599 [DOI] [PubMed] [Google Scholar]
- 29. Zhang L., Cui R., Cheng X., Du J. (2005) Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating IκB kinase. Cancer Res. 65, 457–464 [PubMed] [Google Scholar]
- 30. Sahoo S., Brickley D. R., Kocherginsky M., Conzen S. D. (2005) Coordinate expression of the PI3-kinase downstream effectors serum and glucocorticoid-inducible kinase (SGK-1) and Akt-1 in human breast cancer. Eur. J. Cancer 41, 2754–2759 [DOI] [PubMed] [Google Scholar]
- 31. Fagerli U. M., Ullrich K., Stühmer T., Holien T., Köchert K., Holt R. U., Bruland O., Chatterjee M., Nogai H., Lenz G., Shaughnessy J. D., Jr., Mathas S., Sundan A., Bargou R. C., Dörken B., Børset M., Janz M. (2011) Serum/glucocorticoid-regulated kinase (SGK1) is a prominent target gene of the transcriptional response to cytokines in multiple myeloma and supports the growth of myeloma cells. Oncogene 30, 3198–3206 [DOI] [PubMed] [Google Scholar]
- 32. Vanlangenakker N., Vanden Berghe T., Krysko D. V., Festjens N., Vandenabeele P. (2008) Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 8, 207–220 [DOI] [PubMed] [Google Scholar]
- 33. Festjens N., Vanden Berghe T., Vandenabeele P. (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 1757, 1371–1387 [DOI] [PubMed] [Google Scholar]
- 34. McCall K. (2010) Genetic control of necrosis-another type of programmed cell death. Curr. Opin. Cell Biol. 22, 882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orrenius S., Zhivotovsky B., Nicotera P. (2003) Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565 [DOI] [PubMed] [Google Scholar]
- 36. Verkhratsky A. (2007) Calcium and cell death. Subcell. Biochem. 45, 465–480 [DOI] [PubMed] [Google Scholar]
- 37. Szydlowska K., Tymianski M. (2010) Calcium, ischemia and excitotoxicity. Cell Calcium 47, 122–129 [DOI] [PubMed] [Google Scholar]
- 38. Gouriou Y., Demaurex N., Bijlenga P., De Marchi U. (2011) Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie 93, 2060–2067 [DOI] [PubMed] [Google Scholar]
- 39. Chang W. J., Chehab M., Kink S., Toledo-Pereyra L. H. (2010) Intracellular calcium signaling pathways during liver ischemia and reperfusion. J. Invest. Surg. 23, 228–238 [DOI] [PubMed] [Google Scholar]
- 40. Bell J. R., Mellor K. M., Wollermann A. C., Delbridge L. M. (2011) Cardiac ischaemic stress: cardiomyocyte Ca2+, sex and sex steroids. Clin. Exp. Pharmacol. Physiol. 38, 717–723 [DOI] [PubMed] [Google Scholar]
- 41. Moran T. J., Gray S., Mikosz C. A., Conzen S. D. (2000) The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 60, 867–872 [PubMed] [Google Scholar]
- 42. Landa L. R., Jr., Harbeck M., Kaihara K., Chepurny O., Kitiphongspattana K., Graf O., Nikolaev V. O., Lohse M. J., Holz G. G., Roe M. W. (2005) Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 β-cell line. J. Biol. Chem. 280, 31294–31302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harbeck M. C., Chepurny O., Nikolaev V. O., Lohse M. J., Holz G. G., Roe M. W. (2006) Simultaneous optical measurements of cytosolic Ca2+ and cAMP in single cells. Sci. STKE 2006, pl6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pang D., Kocherginsky M., Krausz T., Kim S. Y., Conzen S. D. (2006) Dexamethasone decreases xenograft response to paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol. Ther. 5, 933–940 [DOI] [PubMed] [Google Scholar]
- 45. George T. C., Basiji D. A., Hall B. E., Lynch D. H., Ortyn W. E., Perry D. J., Seo M. J., Zimmerman C. A., Morrissey P. J. (2004) Distinguishing modes of cell death using the ImageStream multispectral imaging flow cytometer. Cytometry A. 59, 237–245 [DOI] [PubMed] [Google Scholar]
- 46. Zhou Y. P., Pena J. C., Roe M. W., Mittal A., Levisetti M., Baldwin A. C., Pugh W., Ostrega D., Ahmed N., Bindokas V. P., Philipson L. H., Hanahan D., Thompson C. B., Polonsky K. S. (2000) Overexpression of Bcl-xL in β-cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am. J. Physiol. Endocrinol. Metab. 278, E340-E351 [DOI] [PubMed] [Google Scholar]
- 47. Kobayashi T., Cohen P. (1999) Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 339, 319–328 [PMC free article] [PubMed] [Google Scholar]
- 48. Orrenius S., Nicotera P., Zhivotovsky B. (2011) Cell death mechanisms and their implications in toxicology. Toxicol. Sci. 119, 3–19 [DOI] [PubMed] [Google Scholar]
- 49. Broughton B. R., Reutens D. C., Sobey C. G. (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40, e331-e339 [DOI] [PubMed] [Google Scholar]
- 50. Pradelli L. A., Bénéteau M., Ricci J. E. (2010) Mitochondrial control of caspase-dependent and -independent cell death. Cell Mol. Life Sci. 67, 1589–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamashima T. (2012) Hsp70.1 and related lysosomal factors for necrotic neuronal death. J. Neurochem. 120, 477–494 [DOI] [PubMed] [Google Scholar]
- 52. Perez-Pinzon M. A., Stetler R. A., Fiskum G. (2012) Novel mitochondrial targets for neuroprotection. J. Cereb. Blood Flow Metab. 32, 1362–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skulachev V. P. (2006) Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 11, 473–485 [DOI] [PubMed] [Google Scholar]
- 54. Frosali S., Leonini A., Ettorre A., Di Maio G., Nuti S., Tavarini S., Di Simplicio P., Di Stefano A., (2009) Role of intracellular calcium and S-glutathionylation in cell death induced by a mixture of isothiazolinones in HL60 cells. Biochim. Biophys. Acta 1793, 572–583 [DOI] [PubMed] [Google Scholar]
- 55. Choi K., Kim J., Kim G. W., Choi C. (2009) Oxidative stress-induced necrotic cell death via mitochondria-dependent burst of reactive oxygen species. Curr. Neurovasc. Res. 6, 213–222 [DOI] [PubMed] [Google Scholar]
- 56. Perl A., Fernandez D. R., Telarico T., Doherty E., Francis L., Phillips P. E. (2009) T-cell and B-cell signaling biomarkers and treatment targets in lupus. Curr. Opin. Rheumatol. 21, 454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu J., Kaufman R. J. (2006) From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13, 374–384 [DOI] [PubMed] [Google Scholar]
- 58. Schröder M. (2008) Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 65, 862–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Paschen W., Yatsiv I., Shoham S., Shohami E. (2004) Brain trauma induces X-box protein 1 processing indicative of activation of the endoplasmic reticulum unfolded protein response. J. Neurochem. 88, 983–992 [DOI] [PubMed] [Google Scholar]
- 60. Yoshida H. (2007) ER stress and diseases. FEBS J. 274, 630–658 [DOI] [PubMed] [Google Scholar]
- 61. Treiman M., Caspersen C., Christensen S. B. (1998) A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol. Sci. 19, 131–135 [DOI] [PubMed] [Google Scholar]
- 62. Putney J. W., Jr. (2007) New molecular players in capacitative Ca2+ entry. J. Cell Sci. 120, 1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nilius B., Prenen J., Vennekens R., Hoenderop J. G., Bindels R. J., Droogmans G. (2001) Modulation of the epithelial calcium channel, ECaC, by intracellular Ca2+. Cell Calcium 29, 417–428 [DOI] [PubMed] [Google Scholar]
- 64. Thastrup O., Dawson A. P., Scharff O., Foder B., Cullen P. J., Drøbak B. K., Bjerrum P. J., Christensen S. B., Hanley M. R. (1989) Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions 27, 17–23 [DOI] [PubMed] [Google Scholar]
- 65. Mekahli D., Bultynck G., Parys J. B., De Smedt H., Missiaen L. (2011) Endoplasmic reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3, pii:a004317, 10.1101/cshperspect.a004317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhou Y. P., Teng D., Dralyuk F., Ostrega D., Roe M. W., Philipson L., Polonsky K. S. (1998) Apoptosis in insulin-secreting cells. Evidence for the role of intracellular Ca2+ stores and arachidonic acid metabolism. J. Clin. Invest. 101, 1623–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]