

Background: Tapasin edits the MHC I peptide repertoire and is highly polymorphic in birds but not mammals.
Results: Two chicken MHC I alleles differ in peptide binding properties and participate in an allele-specific interaction with tapasin.
Conclusion: Tapasin-MHC alleles have co-evolved by balancing interaction characteristics against MHC peptide-binding ability.
Significance: Variations in the functional attributes of tapasin and MHC I alleles determine effective antigen presentation.
Keywords: Antigen Presentation, Evolution, Immunology, Major Histocompatibility Complex (MHC), Protein-Protein Interactions, Chicken MHC, Co-evolution, Peptide Selection, Tapasin
Abstract
MHC class I molecules display peptides at the cell surface to cytotoxic T cells. The co-factor tapasin functions to ensure that MHC I becomes loaded with high affinity peptides. In most mammals, the tapasin gene appears to have little sequence diversity and few alleles and is located distal to several classical MHC I loci, so tapasin appears to function in a universal way to assist MHC I peptide loading. In contrast, the chicken tapasin gene is tightly linked to the single dominantly expressed MHC I locus and is highly polymorphic and moderately diverse in sequence. Therefore, tapasin-assisted loading of MHC I in chickens may occur in a haplotype-specific way, via the co-evolution of chicken tapasin and MHC I. Here we demonstrate a mechanistic basis for this co-evolution, revealing differences in the ability of two chicken MHC I alleles to bind and release peptides in the presence or absence of tapasin, where, as in mammals, efficient self-loading is negatively correlated with tapasin-assisted loading. We found that a polymorphic residue in the MHC I α3 domain thought to bind tapasin influenced both tapasin function and intrinsic peptide binding properties. Differences were also evident between the MHC alleles in their interactions with tapasin. Last, we show that a mismatched combination of tapasin and MHC alleles exhibit significantly impaired MHC I maturation in vivo and that polymorphic MHC residues thought to contact tapasin influence maturation efficiency. Collectively, this supports the possibility that tapasin and BF2 proteins have co-evolved, resulting in allele-specific peptide loading in vivo.
Introduction
MHC I molecules help protect the host against infection and cancer by binding and presenting peptides to cytotoxic T cells. The peptides displayed at the cell surface are usually of intracellular origin and are loaded into MHC I molecules in the endoplasmic reticulum (ER)3 with the assistance of the proteins that constitute the peptide loading complex. The peptide loading complex assembles around the peptide transporter associated with antigen presentation (TAP) with MHC I tethered to TAP via tapasin (1), ERp57 conjugated to tapasin (2), and calreticulin interacting with both MHC I and ERp57 to form a synergistically strong network of individually weak intermolecular interactions (3).
MHC I molecules are thought to load an optimal peptide cargo in two stages, where a low affinity peptide cargo is initially bound, probably representing the most abundant peptides in the ER, and then exchanged for a higher affinity cargo (4). Tapasin enhances MHC I peptide loading in several ways: tapasin localizes and stabilizes unloaded MHC I molecules at the site of peptide import, and the interaction between tapasin and TAP stabilizes the TAP transporter, thus increasing peptide supply. Perhaps most significant is that tapasin increases not just the rate and extent of peptide loading but the discrimination that occurs between peptides for binding (5). This “editing” function ensures that MHC I becomes loaded with high affinity peptides, which allow prolonged cell surface expression (6), rather than loading more prevalent low affinity peptides (5, 7). Mammalian MHC I alleles differ in their dependence upon tapasin for high affinity peptide loading (8, 9). Whereas alleles like HLA-B*44:02 rely upon tapasin, alleles such as HLA-B*44:05 do not and efficiently self-load their peptide repertoire (5).
Two sites of interaction have been identified between tapasin and MHC I (3, 4, 10–12). The first involves the N-terminal tapasin domain, which is thought to interact with a loop underlying the short α2-1 helix of the MHC I peptide binding groove. The second involves the membrane-proximal tapasin domain, which is thought to interact with a loop in the MHC I α3 domain. The mechanism by which tapasin achieves high affinity peptide selection is poorly understood but may involve stabilization of MHC I conformations where iterative cycles of peptide binding and release occur that only high affinity peptides withstand. Variation in the dependence of MHC I alleles upon tapasin may reflect differences in the propensity for MHC I molecules to adopt conformations that facilitate peptide binding and exchange.
Tapasin is evolutionarily well conserved; however, the location of the gene within the MHC and its allelic diversity differs between mammals and non-mammals. In most mammals, the tapasin gene is in the extended class II region, far from the multiple MHC I loci (13). Few polymorphisms have been documented in tapasin or TAP, with no functional distinctions between alleles (14–16). Thus, it seems likely that in most mammals, tapasin and TAP function universally for whichever MHC I molecules are expressed.
The best characterized non-mammalian MHC is that of the chicken, which has been described as minimal and essential, with the tapasin, TAP, and MHC I loci in a small and simple region virtually never disrupted by recombination (17, 18). In contrast to most mammals, the highly polymorphic chicken TAP genes have distinct transport specificities that match the peptide motif of the single dominantly expressed MHC I (BF2) molecule (19). We have also found that chicken tapasin is highly polymorphic and moderately divergent in sequence (20).4 Thus, it seems likely that different chicken haplotypes use functionally distinct combinations of TAP and tapasin alleles, with optimal peptide loading resulting from alleles of these proteins that have evolved within stable haplotypes to share complementary functional attributes.
We sought to test this hypothesis by comparing the functional attributes of the tapasin and MHC I alleles that are expressed in the B15 and B19 haplotypes. The dominantly expressed MHC I molecules in these haplotypes, BF2*1501 and BF2*1901, are very similar in sequence (21) (Table 1 and Fig. 1) and bind very similar peptides on the cell surfaces (22, 23) but are expressed in haplotypes that encode different tapasin alleles. Thus, BF2*1501 is expressed with Tapasin*15, whereas BF2*1901 is expressed with Tapasin*12 (the tapasin allele found in both the B12 and B19 haplotypes).4 Intriguingly, two of the eight polymorphisms between BF2*1501 and BF2*1901 are at regions that are thought to bind tapasin directly. We therefore sought to determine how the peptide binding properties and the ability to bind tapasin differ between BF2*1501 and BF2*1901, the contribution that polymorphic amino acids thought to contact tapasin have on these functional attributes, and whether tapasin alleles possess different functional properties.
TABLE 1.
BF2 amino acid polymorphisms
Amino acid polymorphisms between BF2*1501 and BF2*1901 alleles are tabulated. Residues are numbered from the first residue of the mature protein and the domain in which they are located.
| Amino acid position, domain | BF2*1501 | BF2*1901 |
|---|---|---|
| 22, α1 | Tyr | Phe |
| 69, α1 | Thr | Ser |
| 79, α1 | Thr | Ile |
| 95, α2 | Leu | Trp |
| 111, α2 | Ser | Arg |
| 113, α2 | Asp | Tyr |
| 126, α2 | Asp | Gly |
| 220, α3 | Gln | Arg |
FIGURE 1.
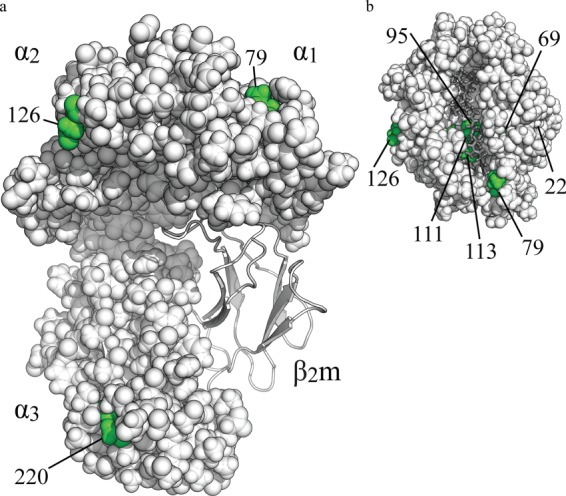
Model of BF2*1501, depicting the location of the eight amino acids that differ between BF2*1501 and BF2*1901. The BF2*1501 structure is based upon the BF2*2101 structure (25) and is shown in a space-filling format with polymorphic amino acids shown in green. β2-Microglobulin (β2m) is shown in gray in a ribbon format, and peptide is shown in dark gray. a, side view; b, view from above the peptide binding groove. Polymorphic residue 22 is buried beneath the α1 helix. The side chains of residues 95 and 111 are on separate β strands with their side chains orientated toward each other.
EXPERIMENTAL PROCEDURES
Production of BF2 and Tapasin-jun Proteins
BF2-fos Proteins
DNA encoding amino acids 1–271 of the mature BF2*1501 and BF2*1901 proteins was amplified by PCR with primers introducing 5′ NdeI and 3′ NcoI sites and cloned into pET22b plasmid (Invitrogen). The Fos leucine zipper sequence (GGSGG linker, thrombin site, and Fos leucine peptide) was amplified by PCR from HLA-B*08-fos DNA (24) using primers introducing 5′ NcoI and 3′ HindIII sites and subsequently cloned into the BF2-containing plasmid. Site-directed mutants were produced by PCR methods analogous to those recommended in the QuikChange mutagenesis kit (Stratagene).
BF2 Proteins
DNA encoding amino acids 1–271 of mature BF2*1501 and BF2*1901 proteins was amplified by PCR from BF2-fos DNA using primers that replaced the 3′ NcoI site with a stop codon followed by a HindIII site and cloned into pET22b plasmid.
Peptide-loaded BF2 Complexes
Peptide-loaded BF2-fos or BF2 complexes were obtained by refolding solubilized inclusion bodies of BF2-fos or BF2 heavy chains with chicken β2-microglobulin (as described (25)) and UV-labile peptide KRLIGjRY (26) (Peptide Synthetics; j represents 3-amino-3-(2-nitro) phenyl-propionic acid) as in Ref. 27.
Tapasin-jun Proteins
DNA encoding amino acids 23–397 of the primary sequence of chicken tapasin alleles (Tapasin*02, AM403065; Tapasin*12, AM403068; Tapasin*14, AM403069; Tapasin*15, AM403070; Tapasin*21, AM403072 (20)) was amplified by PCR from cDNA using primers introducing 5′ BglII and 3′ EcoRI sites and cloned into pMT/BiP (Invitrogen). The His6 tag present in the Jun leucine zipper of human tapasin-jun (24) was replaced by PCR mutagenesis with an HA epitope. The modified Jun leucine zipper sequence (GGSGG linker, thrombin site, Jun leucine peptide, HA tag, and stop codon) was amplified by PCR using primers introducing 5′ EcoRI and 3′ XbaI sites and cloned into tapasin-containing pMT/BiP plasmid. Stable polyclonal transfectants of S2 cells were obtained by co-transfecting 1 μg of tapasin-jun DNA with 50 ng of pCoHygro plasmid (Invitrogen) using Fugene 6 (Roche Applied Science), and hygromycin selection. Transfectants were adapted to Express 5 serum-free medium (Invitrogen), and tapasin-jun expression was induced with 500 μm CuSO4. Supernatants were harvested 3 days later and frozen with 10% glycerol or purified immediately using anti-HA-agarose (Sigma) and eluted either by brief exposure to glycine, pH 2.5, with immediate neutralization or by the addition of HA peptide. Tapasin-jun was purified to >95%, as ascertained by SDS-PAGE, and was dialyzed against 25 mm Tris, pH 8, 150 mm sodium chloride, 50 mm l-arginine, 50 mm l-glutamic acid, 10% glycerol.
Biotinylated Tapasin-jun Proteins
PCR mutagenesis was used to insert a BirA motif (GLNDIFEAQKIEWHE) between the HA tag and stop codon of Tapasin*12-jun and Tapasin*15-jun. To enable in vivo biotinylation, the E. coli birA gene with the KDEL ER retention motif was amplified by PCR from pDisplay birA (28) using primers introducing a 5′ BglII site followed by a Myc epitope and a 3′ EcoRI site and was then cloned into pMT/BiP. Stable polyclonal co-transfectants of S2 cells were obtained by co-transfecting 0.5 μg of tapasin-jun DNA, 0.5 μg of myc-birA DNA, and 50 ng of pCoHygro plasmid. Protein expression was induced in Express 5 serum-free medium supplemented with 10 μm biotin.
Fluorescence Polarization Experiments
The Affinity at Which KRLIGK*RY Is Bound by BF2-fos Molecules
A final concentration of 0.3 μm BF2*15fos or BF2*19fos molecules was exposed to ∼360-nm light for 20 min at 4 °C. Various concentrations of KRLIGK*RY (where K* represents 5′-TAMRA-labeled lysine) peptide were then added to aliquots of the empty BF2-fos molecules, each in a total volume of 60 μl. Fluorescence polarizations measurements were taken after being left at room temperature for ∼22 h using an Analyst AD (Molecular Devices) with 530-nm excitation and 580-nm emission filters and 561-nm dichroic mirror. Binding of KRLIGK*RY is reported in millipolarization units (mP) and is obtained from the equation, mP = 1000 × (S − G × P)/(S + G × P), where S and P are background-subtracted fluorescence count rates (S = polarized emission filter is parallel to the excitation filter; P = polarized emission filter is perpendicular to the excitation filter), and G (grating) is an instrument- and assay-dependent factor.
Association Rate Measurements
For association rate measurements (Fig. 2b), empty BF2-fos complexes were obtained by exposing purified BF2-fos complexes to ∼360-nm light for 20 min at 4 °C. 0.375 μm KRLIGK*RY was added to 0.65 μm empty BF2-fos complexes, and binding was followed. In Fig. 2, g and h, 0.026 μm KRLIGK*RY was added to 0.45 μm empty BF2-fos complexes, and KRLIGK*RY binding was followed in the presence or absence of 0.25 μm tapasin-jun proteins. In Fig. 3, a–c, 0.1 μm KRLIGK*RY was added to 0.3 μm empty BF2-fos complexes, and KRLIGK*RY binding was followed in the presence or absence of 0.25 μm Tapasin*21jun protein.
FIGURE 2.

In vitro analysis of MHC I peptide binding characteristics. a, affinity at which KRLIGK*RY is bound by BF2*15fos or BF2*19fos molecules. The BF2-fos molecules were rendered empty by exposure to UV light and then allowed to bind different concentrations of KRLIGK*RY. Fluorescence polarization measurements were taken after ∼22 h at room temperature. Binding of KRLIGK*RY is reported in mP. Unbound KRLIGK*RY is assumed to have an mP level of 50. b, binding of KRLIGK*RY to empty BF2*15fos or BF2*19fos. c, dissociation of KRLIGK*RY. Excess unlabeled peptide (Comp) or buffer (None) was added to BF2*15fos or BF2*19fos loaded with KRLIGK*RY. d and e, comparison of catalyzed dissociation. Buffer (None) or excess unlabeled peptide with (Comp+Tpn), or without tapasin-jun (Comp) was added to BF2*15fos (d) or BF2*19fos (e) loaded with KRLIGK*RY. Tapasin*12-jun was paired with BF2*19fos, and Tapasin*15jun was paired with BF2*15fos. The data shown are from a representative experiment. f, the half-life of KRLIGK*RY dissociation measured over ∼1 day. Individual results (dots) from 10–11 experiments are shown with the (mean) average depicted as a bar. Details of calculations are provided under “Experimental Procedures.” Statistically significant differences (i.e. p < 0.05) between the indicated results are bracketed. g and h, binding of KRLIGK*RY to empty BF2*15fos (g) or BF2*19fos (h) molecules in the presence or absence of tapasin-jun. The data shown are from a representative experiment. i, tapasin-jun allele enhancement of KRLIGK*RY dissociation. Dissociation assays were conducted as in d and e, using the indicated proteins. Tapasin specific activity was calculated as described under “Experimental Procedures,” with individual results (dots) and (mean) average specific activity (bars) from four experiments.
FIGURE 3.
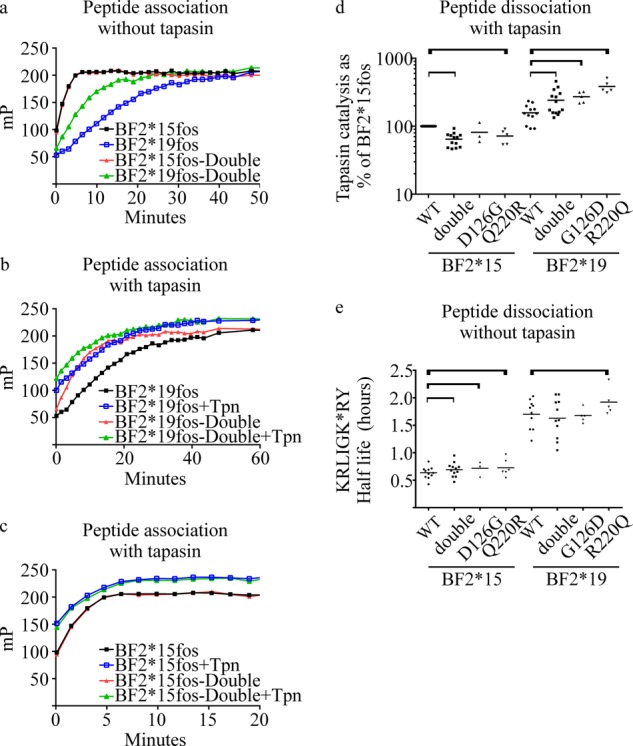
In vitro analysis of the peptide binding characteristics of mutant BF2-fos molecules. a, binding of KRLIGK*RY to empty WT or double mutants of BF2*15fos or BF2*19fos. b and c, binding of KRLIGK*RY to empty WT or double mutants of BF2*19fos (b) or BF2*15fos (c) molecules in the presence or absence of Tapasin*21jun. A representative experiment is shown. d and e, dissociation assays were conducted using WT and position 126 and 220 double or single mutants of BF2*15fos or BF2*19fos with or without tapasin-jun as in Fig. 2. Individual results (dots) and (mean) averages (bars) from 10–12 experiments are shown. Statistically significant differences (i.e. p < 0.05) between the indicated results are bracketed. d, the extent to which tapasin-jun enhanced KRLIGK*RY dissociation for each BF2-fos molecule (calculated as “tapasin bonus” under “Experimental Procedures”) was normalized to the calculated “tapasin bonus” of BF2*15fos in each experiment. The same tapasin-jun allele was used for all BF2-fos molecules within each experiment, but different tapasin-jun alleles were used in some experiments, with the majority of experiments using Tapasin*21jun (mismatched for both BF2-fos alleles), with no difference observed between tapasin-jun alleles. The experiments that included single mutants used just Tapasin*21jun for all BF2-fos molecules and fewer experiments: BF2*15fos D126G = 3, BF2*15fos Q220R = 6, BF2*19fos G126D = 5, BF2*19fos R220Q = 5. It is likely that the small magnitude of tapasin bonus that BF2*15fos experiences may confound the calculation of statistical significance from replicate experiments. e, the half-life of KRLIGK*RY dissociation for each BF2-fos molecule in the absence of tapasin measured over ∼1 day, shown as in Fig. 2f.
Dissociation Rate Measurements
For dissociation rate measurements, 0.5 μm empty BF2-fos molecules were allowed to bind 0.25 μm KRLIGK*RY, and then dissociation was followed after the addition of a 500× molar excess of KRLIGKRY with or without 0.25 μm tapasin-jun proteins. All experiments were conducted at room temperature in duplicate and used PBS supplemented with 0.5 mg/ml bovine γ-globulin (Sigma).
Dissociation data from ∼24 h was processed in GraphPad Prism using one-phase exponential decay non-linear regression. The rate of peptide dissociation was taken as the calculated half-life of dissociation. Tapasin specific activity was calculated in four steps: 1) to calculate “tapasin rate enhancement,” the half-life of uncatalyzed dissociation was divided by the half-life of tapasin-catalyzed dissociation; 2) the “tapasin bonus” was the tapasin rate enhancement minus 1 (to account for the uncatalyzed reaction being divided by uncatalyzed reaction producing a tapasin rate enhancement of 1); 3) “tapasin catalysis,” the number of units of tapasin catalysis, was calculated assuming that one unit of tapasin allows the catalyzed half-life to be reached in half the time of uncatalyzed dissociation; and 4) specific activity was calculated as the number of units of tapasin activity divided by the molar concentration of tapasin-jun. Paired two-tailed t tests (GraphPad Prism) were performed to ascertain whether differences between groups of results were statistically significant, with a p value of 0.05 denoting statistically significant differences.
Surface Plasmon Resonance (SPR) Assays
Binding studies were performed at 25 °C using a Biacore T100, streptavidin chips, and HBS EP+ buffer (GE Healthcare) at a 30 μl/min flow rate. Biotinylated Tapasin*12-jun and Tapasin*15-jun proteins were immobilized on different flow cells to densities that varied between experiments in the range of ∼200–1000 RU. Monomeric BF2 complexes were repurified by size exclusion chromatography in HBS or HBS EP+ buffer 1 day before SPR and stored at 4 °C. Repurified BF2 complexes were UV-exposed for 20 min at 4 °C immediately before SPR. Kinetic rate constants were calculated from five sequential injections (2 min each or in one experiment 2.5 min) of BF2 proteins within the range of 0.37–6 μm. Regeneration of the chip was performed with a 5–10-min injection of 50 μm KRLIGKRY peptide in HBS EP+ buffer supplemented with 0.5 m NaCl to remove tapasin-bound peptide-empty BF2 molecules. Sensorgrams were corrected for bulk refractive index changes and nonspecific binding using a blank flow cell. All data were double referenced using responses from blank injections with running buffer. There was no evidence in any experiment that the binding of BF2 to tapasin was mass transport-limited. Data were processed using BiaEvaluation software (GE Healthcare) using “heterologous ligand” interaction model fitting, which fit the data better than “one-to-one” interaction models. We assume that this is because the tapasin-jun preparations contained a small proportion of protein that bound BF2 protein nonspecifically. The kinetic rates reported were therefore corrected for this nonspecific binding as described in the legend for Fig. 4. Unpaired two-tailed t tests (GraphPad Prism) were performed to ascertain whether differences between groups of results were statistically significant, with a p value of 0.05 denoting statistically significant differences.
FIGURE 4.
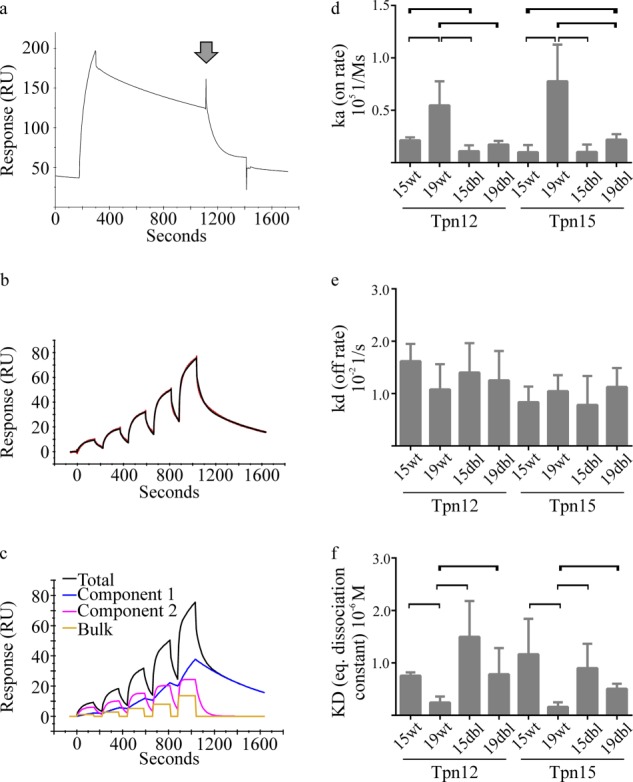
Surface plasmon resonance assays. a, sensorgram of 20 μm empty BF2*1501 binding to Tapasin*15jun (with a binding level of 2000 RU) during a 2-min injection, slow dissociation following the injection, and faster dissociation induced by injection of 50 μm KRLIGKRY peptide coincident with the arrowhead. All data are reference flow cell-corrected. b, sensorgram showing binding of empty BF2*1501-Double (concentrations between 0.375 and 6 μm) to Tapasin*12jun (with a binding level of 1035 RU) as a red line, with the fit of the “heterologous ligand” interaction model shown as a black line. c, the “heterologous ligand” interaction model fitted to the sensorgrams (including the example shown in b) was usually attributed to one slow, high affinity component (component 1), and a faster but lower affinity component (component 2), along with a “bulk effect” contribution resulting from mismatching of the refractive indices of the running buffer and sample. We anticipate that the faster but lower affinity component (component 2) represents the specific binding of monomeric BF2 proteins to tapasin, because this component appears most similar to binding profiles that were modeled by 1:1 interaction models (data not shown) and with the expected affinity of the tapasin-MHC interaction. The slow, higher affinity component (component 1) might represent a proportion of aggregated, denatured, or inactive tapasin protein that might bind MHC proteins nonspecifically and dissociate slowly. The proportion of BF2 proteins that could be loaded with peptide was consistent with the measured total protein concentration and was comparable between the different BF2 proteins examined within each experiment (data not shown). d–f, the interaction characteristics of empty WT or double mutants (labeled dbl) of BF2*1501 or BF2*1901 binding to Tapasin*12jun or Tapasin*15jun. The kinetic attributes measured were the association rate constant (ka; d) and the dissociation rate constant (kd; e). The equilibrium dissociation constant (KD; f) calculated from the dissociation rate constant divided by the association rate constant is presented as in d and e. The data in d–f were combined from three independent experiments with a variable number of replicates of each protein combination performed within each experiment (between 1 and 4 repeats). The data constitute those that were modeled satisfactorily by the heterologous ligand interaction model, where the fast, low affinity interaction characteristics are reported, and are presented as a bar chart with the S.D. value indicated by error bars. Statistically significant differences (i.e. p < 0.05) between the indicated results are bracketed.
Cellular Mismatching Experiments
BF2-myc DNA Constructs
The Myc epitope with 5′ EcoRI and 3′ stop codon followed by an XbaI site was created by annealing oligonucleotides and cloned into pcDNA3.1+ plasmid (Invitrogen). DNA encoding full-length BF2*1501 or BF2*1901 proteins was amplified by PCR with primers introducing a 5′ HindIII site and replacing the stop codon with an EcoRI site and was cloned into the myc-containing pcDNA3.1+ plasmid. Transfections of TG15 cells (reticuloendotheliosis virus-transformed lymphocytes from a homozygous B15 chicken (19)) were performed using a Nucleofector (Amaxa). Stable transfectants were cloned from single cells under G418 selection.
35S Pulse-Chases, Myc Immunoprecipitation, and Endoglycosidase H Digestions
5 × 106 cells were incubated at 41 °C in a 5% CO2 incubator for 45–60 min in cysteine- and methionine-free RPMI supplemented with 10% dialyzed FCS. Cells were labeled by the addition of 100–200 μCi of 35S Promix (Amersham Biosciences) for 20–30 min. The chase was initiated by 10-fold dilution in prewarmed, CO2-equilibrated RPMI supplemented with 10% FCS, 2 mm cysteine, and 2 mm methionine or by brief centrifugation and resuspension in the above medium. Aliquots were taken as indicated and lysed in 100 μl of ice-cold radioimmune precipitation assay buffer (Sigma) supplemented with 10 mm iodoacetamide and 4 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (Roche Applied Science). Lysates were centrifuged at 16,000 × g at 4 °C for 15 min to remove nuclei and other subcellular structures and were then precleared with 50 μl of 50% Sepharose 4B and rotated at 4 °C for at least 1 h. Precleared samples were mixed with 50 μl of washed myc-agarose (Sigma) for at least 1 h at 4 °C before extensive washing in lysis buffer. Proteins were eluted in 100 mm sodium acetate, pH 5.4, 0.02% SDS, 100 mm β-mercaptoethanol, 4 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 10 mm iodoacetamide; heated at 85 °C for 5 min; divided as indicated; and digested or mock-digested with 5 milliunits of endoglycosidase H (Roche Applied Science) at 37 °C overnight. Samples were separated by SDS-PAGE, fixed, soaked in Amplify (Amersham Biosciences), and exposed to phosphor screens.
RESULTS
BF2*1501 and BF2*1901 Differ in Their Peptide Binding Properties in the Presence or Absence of Tapasin
The ability of different mammalian MHC I molecules to load an optimal peptide cargo independently of tapasin varies. Alleles that are able to self-load an optimal peptide cargo, such as HLA-B*44:05 and B*27:05, are expressed at the surface of tapasin-deficient cells and can present antigens effectively, whereas those alleles that are inefficient self-loaders, such as HLA-B*44:02, require tapasin in order to assemble with stable peptides and present them at the cell surface. Nevertheless, all MHC I molecules benefit to some degree from tapasin, such that those alleles that can load peptides efficiently receive a small tapasin bonus, whereas those alleles that self-load inefficiently receive a large tapasin bonus (5). To determine whether such a relationship also exists in chickens, where MHC I and tapasin appear to have co-evolved, we compared the peptide binding characteristics of BF2*1501 and BF2*1901 in the presence or absence of tapasin in vitro. We adapted the assay of Chen and Bouvier (24), which brings together in vitro the luminal domains of MHC I and tapasin using leucine zipper sequences comprising a C-terminal Fos sequence on MHC and a C-terminal Jun sequence on tapasin. BF2-fos proteins were refolded with a UV-labile peptide in order to allow generation of “empty,” peptide-receptive MHC I after UV exposure (26). To enable the comparison of the peptide binding properties of the two BF2-fos alleles, fluorescence polarization assays were used to measure the binding to MHC of a fluorophore-labeled peptide, KRLIGK*RY (where K* denotes TAMRA-labeled lysine). KRLIGK*RY is derived from the protein hsp70 and has been identified among the peptides eluted from MHC I molecules immunopurified from B15 and B19 cellular lysates (22, 23). We found KRLIGK*RY to bind both BF2 alleles with nearly identical affinity (Fig. 2a).
When we measured association (Fig. 2b) and dissociation rates (Fig. 2c), we found that BF2*15fos bound KRLIGK*RY significantly faster than BF2*19fos and that KRLIGK*RY dissociated from BF2*15fos significantly faster than from BF2*19fos, with a half-time of around 0.6 versus 1.7 h (Fig. 2, c and f). This difference in KRLIGK*RY dissociation characteristics was not due to competing KRLIGKRY peptide being bound by the two BF2-fos alleles with different affinities, because binding of KRLIGK*RY to BF2*15fos and BF2*19fos was similarly inhibited by varying concentrations of KRLIGKRY (data not shown). Dissociation of KRLIGK*RY was only apparent when excess specific peptide was added and not when an equal volume of buffer (labeled none in Fig. 2c) or excess irrelevant peptide was added (data not shown). In the absence of excess specific competing peptide, polarization values remained high, suggesting that MHC and KRLIGK*RY were in equilibrium of binding and dissociation, with excess unlabeled KRLIGKRY preventing rebinding of free KRLIGK*RY to empty MHC. Further, there appeared to be minimal loss of peptide-binding sites throughout these assays, suggesting that there was little dissociation of β2-microglobulin from the MHC heavy chain. We therefore reasoned that measuring KRLIGK*RY dissociation when specific competing peptides were added provided an indirect measurement of peptide exchange. We then determined whether tapasin might enhance peptide dissociation. Thus, BF2*15fos was paired with Tapasin*15jun and BF2*19fos with Tapasin*12jun, and dissociation of preloaded KRLIGK*RY was compared (Fig. 2, d and e). We found that tapasin increased the rate (Fig. 2f) of KRLIGK*RY dissociation from both BF2-fos alleles and that this occurred to a greater degree for BF2*19fos than for BF2*15fos.
Thus, BF2*15fos and BF2*19fos differ in a number of attributes; empty BF2*15fos molecules bind KRLIGK*RY faster than BF2*19fos, and KRLIGK*RY dissociates more readily from BF2*15fos than from BF2*19fos. Conversely, tapasin enhances KRLIGK*RY dissociation to a much greater extent for BF2*19fos. Thus, as with mammalian MHC I alleles, chicken MHC I alleles with inferior abilities to load peptides and to undergo peptide dissociation benefit from the greatest tapasin bonus, and vice versa.
Tapasin Alleles Function Universally in Vitro
A possible explanation for the difference in the magnitude of tapasin function we observed between BF2*19fos and BF2*15fos in peptide dissociation assays (Fig. 2, d–f) could have been due to differences in tapasin allele function. To investigate this further, we compared KRLIGK*RY binding to peptide-empty BF2-fos molecules, which were paired with either Tapasin*12jun or Tapasin*15jun (Fig. 2, g and h). In these experiments, BF2*15fos and BF2*19fos were UV-exposed, while KRLIGK*RY was mixed with either Tapasin*12jun or Tapasin*15jun or with buffer. The empty MHC molecules were then added to KRLIGK*RY peptide mixed with buffer or with tapasin, and fluorescence polarization measurements were taken immediately. Binding of KRLIGK*RY to BF2*1501 either in the presence or absence of tapasin reached its maximum value within the first few min, comparable with the setup time of the experiment (Fig. 2g). In contrast, binding of KRLIGK*RY to BF2*1901 was significantly slower, and more of the binding reaction was measured, although the earliest part of the binding reaction occurred before the first fluorescence polarization measurement was taken (Fig. 2h). We found for both BF2-fos alleles that tapasin increased the number of molecules that could be loaded with peptide but that there was no apparent difference between the two tapasin-jun alleles in the enhancement of KRLIGK*RY binding, although the earliest stages of peptide binding were not observed.
We also compared the catalysis of KRLIGK*RY dissociation of matched tapasin-jun and BF2-fos alleles to that of four mismatched tapasin-jun and BF2-fos pairs (Fig. 2i). Only slight differences in function were found between tapasin-jun alleles, which were not statistically significant. Thus, all tapasin alleles enhance dissociation of KRLIGK*RY from BF2*19fos to a greater extent than for BF2*15fos, with the minor differences in tapasin activity that were observed being qualitatively similar for both BF2 alleles. There was therefore no evidence that matched tapasin and BF2 alleles function differently from mismatched alleles in vitro when the proteins are artificially appended.
Tapasin “Contact” Residues 126 and 220 Modulate the Ability of Tapasin to Enhance Peptide Dissociation in Vitro
Two of the eight polymorphic differences between BF2*1501 and BF2*1901 reside at positions 126 (Asp in BF2*1501, Gly in BF2*1901) and 220 (Gln in BF2*1501, Arg in BF2*1901). In mammalian MHC I proteins, the equivalent non-polymorphic positions (129 and 224) are localized in or close to regions that have been shown by site-directed mutagenesis to be important for tapasin binding and function. To determine the extent to which these two residues might underpin the allelic differences in peptide binding characteristics between BF2*1501 and BF2*1901, we exchanged them between the two BF2-fos alleles, thereby generating the BF2*15fos D126G/Q220R mutant and the BF2*19fos G126D/R220Q mutant, referred to hereafter as BF2*15fos-Double and BF2*19fos-Double, respectively.
We first sought to determine whether the exchange of positions 126 and 220 might influence the rate at which the empty MHC molecules bind KRLIGK*RY in the absence of tapasin (Fig. 3a). We found that empty BF2*19fos-Double molecules bound KRLIGK*RY faster than BF2*19fos molecules. In comparison, we found no apparent difference in the rate that KRLIGK*RY was bound by BF2*15fos and BF2*15fos-Double molecules, although the initial stages of the peptide binding reactions were not observed.
Next, these peptide binding experiments were conducted in the presence of Tapasin*21jun, an allele that is mismatched for both BF2-fos alleles and which functioned identically to other tapasin alleles in the enhancement of KRLIGK*RY dissociation (Fig. 2i). We found that in either the presence or absence of Tapasin*21jun, BF2*19fos-Double bound KRLIGK*RY faster than BF2*19fos, with the earlier stages of the BF2*19fos-Double binding reaction occurring before the earliest fluorescence polarization measurement (Fig. 3b). As before (Fig. 3a), peptide binding to empty BF2*15fos or BF2*15fos-Double molecules was too fast to observe the earlier stages of the binding reaction, but there was no apparent difference in the binding of KRLIGK*RY to empty BF2*15fos or BF2*15fos-Double molecules in the presence of tapasin during the stages of the peptide binding reaction that were observed (Fig. 3c). As before, tapasin increased the number of BF2 molecules that were loaded with peptide. Collectively, this shows that empty WT BF2-fos molecules differ in the rates at which peptides are bound, that positions 126 and 220 can indirectly influence MHC-intrinsic peptide loading rates (i.e. unassisted by tapasin), and that tapasin increases the efficiency of MHC peptide loading.
We next conducted peptide dissociation experiments using WT or double mutant BF2-fos molecules in the presence or absence of tapasin (Fig. 3, d and e). We found that the double mutation had a significant effect on the magnitude that tapasin enhanced KRLIGK*RY dissociation for both BF2fos-Double mutants, reducing it for BF2*15fos-Double and enhancing it for BF2*19fos-Double (Fig. 3d). In contrast, we found that the double mutation did not drastically change the rate (Fig. 3e) at which KRLIGK*RY dissociated from either BF2-fos allele in the absence of tapasin. Thus, BF2*15fos and BF2*15fos-Double were more similar to each other than to either BF2*19fos molecule, and vice versa. These results suggest that polymorphisms at positions 126 and 220, which have presumably arisen as a result of co-evolution with polymorphic tapasin proteins, can influence the peptide-editing function of the tapasin-MHC I heterodimer.
We also created single position mutants, where either position 126 or position 220 were exchanged between BF2-fos alleles and measured the ability of tapasin to catalyze KRLIGK*RY dissociation. We found that, like BF2*19-Double, both single position BF2*19fos mutants experienced greater tapasin enhancement of KRLIGK*RY dissociation than did BF2*19fos (Fig. 3d). Similar to BF2*15fos-Double, we found that the BF2*15fos position 220 mutant experienced a statistically significant decrease in tapasin enhancement of KRLIGK*RY dissociation in comparison with the WT molecule. Likewise, the BF2*15fos position 126 mutant experienced decreased tapasin enhancement of KRLIGK*RY dissociation in comparison with the WT molecule, although this difference was not statistically significant when experiment-to-experiment variation was taken into account.
The effect that position 126 has on tapasin function might be anticipated given the proximity of position 126 to the peptide binding groove. However, the localization of position 220 and the influence that exchange of this residue had on tapasin function was intriguing. When considered alongside the finding that in the absence of tapasin KRLIGK*RY dissociated significantly more slowly from the 220 mutants of both BF2-fos alleles in comparison with WT molecules (Fig. 3e), we suggest there is intramolecular communication between the α3 domain and the peptide binding groove and that by interacting with BF2 position 220, tapasin exerts an allosteric influence on the peptide binding domain.
BF2*1501 and BF2*1901 Bind Tapasin at Different Rates
We sought to determine whether the difference in the magnitude of tapasin function experienced by BF2*1501 and BF2*1901 when tapasin and BF2 proteins were artificially tethered by Fos-Jun leucine zippers might coincide with differences in binding interactions between MHC I and tapasin alleles. We reasoned that the Fos-Jun pairing is likely to mimic the anchoring of tapasin and BF2 proteins to the same plane of the ER membrane that is normally achieved by their transmembrane domains, whereas the Gly-Ser linkers that separate the luminal domains from the leucine zipper sequences allow the proteins sufficient flexibility to interact in a physiologically relevant fashion.
The binding of MHC I and tapasin has not been directly quantitated, although binding of mammalian MHC I and tapasin (or tapasin-ERp57) proteins has been demonstrated (7, 29, 30). Numerous indirect observations, such as MHC I co-immunoprecipitation studies with conformation-specific antibodies, suggest that the preferred ligand for tapasin is “peptide-receptive” and not “peptide-occupied” MHC I. In order to directly quantitate the interaction between BF2 and tapasin proteins, we conducted SPR experiments with immobilized Tapasin*12jun or Tapasin*15jun and soluble BF2 proteins, lacking any leucine zipper sequence, in the fluid phase. The BF2 proteins were refolded with a UV-conditional ligand and were rendered empty by UV exposure immediately prior to exposing them to tapasin. We found UV exposure to be an absolute requirement for binding of BF2*1501 or BF2*1901 to tapasin and found that injection of a high affinity peptide into the flow cell caused rapid dissociation of tapasin-bound MHC (Fig. 4a). We calculated the half-life of the interaction between peptide-bound BF2*1501 molecules and Tapasin*15 to be ∼7.5 times faster than the dissociation rate of peptide-empty BF2*1501 molecules. This provides further support to the hypothesis that the preferred substrate for tapasin is peptide-receptive MHC I molecules.
After correcting for nonspecific binding of MHC proteins (as described under “Experimental Procedures” and in the legend to Fig. 4, b and c), we compared the rates at which empty BF2*1501 and BF2*1901 bound to and dissociated from Tapasin*12jun or Tapasin*15jun (Fig. 4, d and e, first, second, fifth, and sixth columns). The combined results of three experiments performed at different times with different protein preparations show that BF2*1901 bound tapasin significantly faster than BF2*1501 (Figs. 4d and 5, a and b) but that there was no clear difference in the rates that empty BF2*1501 and BF2*1901 dissociated from tapasin (Fig. 4e and 5, a and b). Reassuringly, the rates measured for BF2 binding to tapasin, which are in the range of 104 to 105 m−1 s−1, appear in keeping with estimates for the transport rate of peptides by TAP (31).
FIGURE 5.
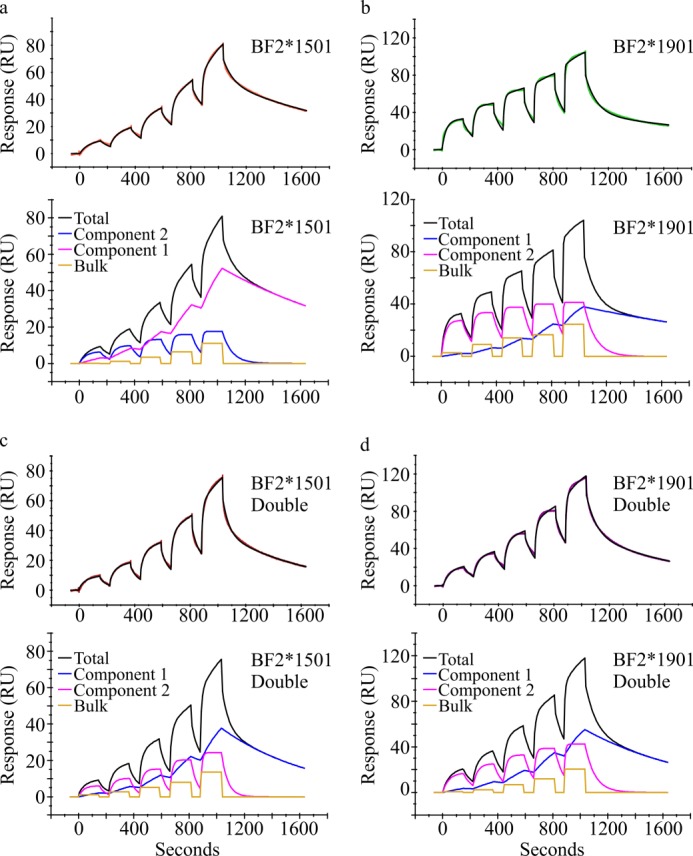
Comparison of the modeling of the wild-type and double mutant sensorgrams via the heterologous ligand interaction model. a, BF2*1501; b, BF2*1901; c, BF2*1501-Double; d, BF2*1901-Double. The top panels show the binding of the indicated protein (concentrations between 0.375 and 6 μm) to Tapasin*12jun (with a binding level of 1035 RU) as a colored line, with the fit of the “heterologous ligand” interaction model shown as a black line. The bottom panels show how the total binding was attributed between the two components and the bulk effect. The binding of component 2 is taken to represent the specific binding of monomeric BF2 proteins to tapasin as discussed in the legend to Fig. 4 and under “Results.”
The equilibrium dissociation constants calculated from the measured on and off rates (Fig. 4f) show that BF2*1901 bound tapasin with higher affinity than BF2*1501. The differences that these BF2 alleles exhibit in on rates (ka; Fig. 4d) and equilibrium dissociation constants (KD; Fig. 4f) were evident when these BF2 proteins bound to either Tapasin*12jun or Tapasin*15jun. Thus, although there was no clear evidence that matched combinations of MHC-tapasin alleles interact differently from mismatched alleles in vitro, the finding that BF2*1501 and BF2*1901 bound either tapasin allele with a similar pattern of interaction kinetics lends further support to the existence of an allelic difference in the tapasin binding characteristics of these BF2 alleles in vitro.
Exchanging Positions 126 and 220 between BF2 Alleles Changes the Interaction between Tapasin and BF2*1901 but Not for BF2*1501
To further characterize the allele-specific interaction of tapasin and BF2, we measured the effect of exchanging residues 126 and 220 between BF2*1501 and BF2*1901 on the binding to tapasin (Fig. 4, d–f). In particular, we wanted to know whether the effect on the magnitude of tapasin function we observed in vitro for the double mutants (Fig. 3d) correlated with effects on binding.
We first compared the tapasin binding kinetics of the BF2*1901-Double mutant to the wild-type BF2 molecules (Figs. 4 (d and e) and 5 (b and d)). Interestingly, we found that BF2*1901-Double bound to either tapasin allele with a significantly slower on rate than was apparent for BF2*1901, being more similar to the on rate measured for BF2*1501. Again there was no difference in off rates. The decreased tapasin-binding rate resulted in BF2*1901-Double binding tapasin with lower affinity than BF2*1901, being more similar to the affinity with which BF2*1501 bound tapasin (Fig. 4f). In contrast, we found that the tapasin interaction kinetics were not as dramatically affected by the double mutation for BF2*1501 (Figs. 4 (d and e) and 5 (a and c)). The on and off rates measured for BF2*15-Double binding to either tapasin allele were most similar to those found for BF2*1501, although we found that BF2*15-Double bound Tapasin*12 with a significantly slower on rate than BF2*1501. In agreement with the small change in Tapasin*12 binding rate and the unchanged dissociation rates, we found that the equilibrium dissociation constants calculated for BF2*1501-Double binding to either tapasin allele were not statistically different from those calculated for BF2*1501 (Fig. 4f).
Thus, as in the peptide-loading experiments (Fig. 3, a–c), the exchange of positions 126 and 220 affected the tapasin binding properties of the BF2*1901 molecule to a greater extent than for BF2*1501. It is noteworthy that despite the apparently detrimental alterations to the tapasin interaction properties brought about by the double mutation to BF2*1901, BF2*1901-Double mutants experienced greater tapasin enhancement of peptide dissociation than BF2*1901 (Fig. 3d). Similarly, although the tapasin binding properties of BF2*1501-Double molecules were similar to those of BF2*1501, BF2*1501-Double molecules experienced less tapasin enhancement of peptide dissociation than BF2*1501 (Fig. 3d). This suggests that tapasin binding properties alone do not define the magnitude of tapasin function and that the identity of the amino acids at positions 126 and 220 significantly influences the extent of tapasin function.
In Vivo Maturation of BF2*1901 Is Impaired When Expressed in B15 Cells but Is Restored by Exchanging Positions 126 and 220
Our in vitro analysis showed that BF2*1501 and BF2*1901 differ (a) in their ability to self-load peptide cargo, (b) as substrates for tapasin-catalyzed peptide dissociation, and (c) in the kinetics and affinity of their interaction with tapasin in vitro. BF2 positions 126 and 220 influenced (a) the magnitude that tapasin enhanced peptide dissociation from both BF2 alleles, (b) the interaction of BF2*1901 with tapasin and, (c) the ability of BF2*1901 to load peptide cargo without tapasin. We therefore sought to determine the impact that mismatching tapasin and BF2 alleles has on BF2 peptide loading in vivo, and in particular we wondered whether exchanging positions 126 and 220 has a greater impact on BF2*1901 peptide loading than on that of BF2*1501, as suggested by the in vitro fluorescence polarization and SPR assays. Thus, we assessed the transport through the secretory pathway of newly synthesized myc-tagged BF2*1501 and BF2*1901 molecules or their respective double mutants expressed in B15 cells that endogenously express Tapasin*15 and BF2*1501 molecules. Previous work has shown that the trafficking of stable MHC I molecules through the cis-Golgi network indicates that they are loaded with high affinity peptides resulting from peptide editing (5).
We first compared the maturation of BF2*15myc, BF2*19myc, and BF2*19myc-Double (Fig. 6a). We found that whereas BF2*15myc matured efficiently, BF2*19myc matured very inefficiently. In comparison with BF2*19myc, we found that BF2*19myc-Double showed significantly improved trafficking through the secretory pathway, approaching that seen for BF2*15myc. The improved maturation of BF2*19myc-Double is probably a consequence of an improved ability to self-load peptide cargo (Fig. 3a) and the greater catalysis of peptide dissociation that tapasin mediates (Fig. 3d). The impaired maturation of BF2*19myc molecules expressed in B15 cells suggests that BF2*19myc molecules were unable to load an optimal peptide cargo efficiently, presumably being unable to bind to or receive benefit from tapasin in vivo. Instead, it seems that BF2*19myc molecules were forced to rely on their inefficient ability to load an optimal peptide cargo (Fig. 2).
FIGURE 6.

Comparison of maturation of BF2*15myc, BF2*19myc, BF2*19myc-Double (a), or BF2*15myc-Double (b) in TG15 cells. Only the bands corresponding to BF2myc molecules are shown from a representative experiment. EHS, molecular weight of endoglycosidase H-sensitive, or immature, BF2 molecules; EHR, molecular weight of endoglycosidase H-resistant, or mature, BF2 molecules. The maturation of BF2*15myc and BF2*19myc molecules has been compared in more than eight experiments using two different clones, and their maturation has been compared with that of BF2*19myc-Double in at least two experiments using two different clones and with that of BF2*15myc-Double in two experiments. Comparable results were observed in each experiment. The graphs depict the rate and extent of the acquisition of resistance of the BF2myc molecules to endoglycosidase H digestion averaged from two experiments.
We next compared the maturation of BF2*15myc, BF2*19myc, and BF2*15myc-Double (Fig. 6b). We found that BF2*15myc-Double displayed slightly impaired maturation (Fig. 6b) in comparison with BF2*15myc. This is consistent with our observations that BF2*1501-Double has similar self-loading and tapasin binding properties to BF2*1501 but that tapasin enhanced peptide dissociation from BF2*15fos-Double to a lesser extent than from BF2*15fos.
Collectively, our analysis of the maturation of BF2myc molecules expressed in B15 cells shows that the maturation of mismatched BF2 molecules can be severely compromised. We found a greater effect of mismatching alleles in vivo than in vitro, suggesting that allelic differences in tapasin binding and/or function are most apparent in a physiological environment. Importantly, we found that BF2 positions 126 and 220 have a significant influence on BF2 maturation. This may be attributable to the effect these residues have both on the ability of tapasin to enhance peptide dissociation and on the intrinsic peptide binding properties of BF2*1901 molecules. We suggest that polymorphisms at these positions have evolved to “fine tune” the function of the tapasin-MHC I heterodimer and can also influence the intrinsic peptide binding properties of the BF2 allele.
DISCUSSION
The effect of tapasin on MHC I peptide loading and editing has been intensively researched (1, 5–7, 24). Studies of mammalian MHC I molecules show that some MHC I alleles are more effective at loading an optimal peptide cargo independently of tapasin, and thus there is diversity in the extent that tapasin enhances peptide loading. Our comparison of BF2*1501 and BF2*1901 showed that this is also the case in the chicken MHC, where BF2*1501 is more effective than BF2*1901 at loading peptides and undergoing peptide dissociation without tapasin, and consequently BF2*1501 benefits less from the action of tapasin. Thus, BF2*1501 and BF2*1901 bear certain similarities in their loading properties to HLA-B*44:05 and B*44:02, which may define the extremes of tapasin dependence in mammals. This suggests that efficient self-loading and significant tapasin enhancement of peptide loading are fundamentally opposing characteristics of MHC I function. There may have been several evolutionary pressures that led to MHC I alleles varying in their dependence on tapasin to achieve optimal peptide loading. For example, MHC alleles with the ability to self-load efficiently may have arisen in response to viral subversion of tapasin (32, 33). However, it is also clear that pathogen sequence diversification has driven increased MHC I polymorphism (34). Therefore, it is possible that tapasin has facilitated diversification of the MHC I peptide binding groove by allowing “new” MHC alleles to function even when the amino acid changes that allow them to present new epitopes might destabilize the peptide-receptive protein and render the new MHC allele unable to self-load. Tapasin may therefore allow the spread of such MHC alleles to confer survival advantage. Thus, although BF2*1501 and BF2*1901 are quite similar in sequence and in peptide-binding specificity, our findings suggest that these alleles have evolved to use different peptide loading mechanisms; whereas BF2*1901 appears to have evolved to benefit from the loading enhancement that tapasin affords, BF2*1501 appears to have evolved to self-load efficiently.
Interestingly, we found that in the absence of tapasin, the rate at which KRLIGK*RY dissociated from the position 220 mutants of both BF2 alleles was significantly diminished in comparison with the WT molecules. However, we did not find such alterations for the double position 126 and 220 mutants. This suggests that a functional relationship exists between position 220 and the peptide-binding domain and position 126 in particular, which helps to define intrinsic peptide binding properties. It seems likely that β2-microglobulin may also participate in this intramolecular communication (35). Also supporting the concept that peptide binding properties are collectively defined by the whole MHC molecule is the finding that exchanging positions 126 and 220 improved the rate at which empty BF2*19fos-Double molecules bound peptide without tapasin.
Our in vitro analysis of the double mutants suggests that BF2 positions 126 and 220 exert significant influence on the ability of tapasin to function. We found that tapasin enhancement of KRLIGK*RY dissociation was greatest for both BF2-fos alleles when the residues derived from BF2*1501 were present (i.e. BF2*15fos and BF2*19fos-Double) and that both positions 126 and 220 independently contribute to influence the extent of tapasin function. The finding that tapasin function was influenced by BF2 position 220 strongly suggests that the membrane-proximal interaction between tapasin and BF2 allows tapasin to exert allosteric control of the peptide binding groove in order to enhance peptide dissociation. We suggest that the membrane-proximal MHC I-tapasin interaction allows the MHC I peptide binding domain to become more ordered and more readily adopt conformations conducive to enhancing peptide dissociation. This finding is consistent with previous observations, including the ablation of tapasin-jun function for the HLA-B*08fos E222K mutant (24), which collectively suggest that even when artificially tethered, the membrane-proximal tapasin-MHC interaction is of key significance. Interestingly, there is a prominent cluster of polymorphisms in the immunoglobulin domain of chicken tapasin that may influence this membrane-proximal interaction in an allele-specific manner (20).4
Our analysis of empty BF2 molecules binding KRLIGK*RY showed that tapasin increased the number of peptide-loaded BF2 molecules, consistent with functions attributed to tapasin previously (5, 7, 29, 36, 37). We did not find any evidence that tapasin catalyzed the rate at which peptides were bound by empty BF2 molecules, although we were unable to observe the earlier stages of the peptide binding reactions where catalysis may be most evident. It was, however, clear that tapasin allowed peptide binding reactions to reach equilibrium earlier. Combined with the finding that dissociation of KRLIGK*RY from BF2 molecules was catalyzed by tapasin, we suggest that tapasin-mediated peptide editing may result from tapasin minimizing the time that MHC molecules are unreceptive to peptide loading combined with increasing peptide dissociation rates, allowing many peptides to be sampled, consistent with the proposed “two-step” loading process (4).
Direct binding assays confirmed that peptide-deficient MHC I molecules are the preferred substrates of tapasin. Binding affinities between empty BF2 molecules and tapasin varied between 0.1 and 1.5 × 106 m, depending on the combination of alleles. Thus, the tapasin-MHC I interaction is relatively weak, consistent with suggestions from previous studies and the requirement for leucine zipper sequences to be used in vitro (24). We found BF2*1501 and BF2*1901 differ in their interactions with tapasin, with empty BF2*1901 molecules binding tapasin faster than BF2*1501, forming higher affinity interactions than empty BF2*1501 molecules. If the peptides that become bound by MHC I molecules are of high affinity, peptide-loaded MHC I molecules are poor substrates for tapasin, and such molecules will quickly dissociate from the peptide loading complex. However, if peptides are bound and then rapidly released, empty MHC I molecules either remain bound by tapasin or rebind tapasin to start another cycle. Thus, MHC I alleles like BF2*1901 that bind tapasin with fast interaction kinetics might experience more peptide sampling cycles, allowing greater potential for tapasin to catalyze peptide exchange.
Our analysis of the maturation of myc-tagged BF2 molecules expressed in B15 cells showed that BF2 maturation can be compromised by the mismatching of tapasin and BF2 alleles. The impaired maturation of BF2*19myc molecules was surprising, given our in vitro results, suggesting that tapasin alleles function in an allele-dependent way in vivo and/or that BF2-tapasin alleles interact in an allele-dependent manner in vivo. It is tempting to speculate that allelic differences in tapasin binding or function may only become apparent when tapasin is conjugated to ERp57, although chicken tapasin lacks a residue equivalent to cysteine 95, to which ERp57 conjugates in mammals (17). Importantly, we found that exchange of positions 126 and 220 between BF2 alleles influenced maturation efficiency. This in vivo mismatching of tapasin and BF2 alleles and the influence of BF2 positions 126 and 220 on maturation efficiency provide the best support for the notion that chicken tapasin and BF2 proteins have co-evolved, resulting in allele-specific peptide loading.
We suggest that in different MHC haplotypes, the tapasin and BF2 alleles have co-evolved to balance a number of functional attributes in different ways. Thus, in haplotypes like B19, where the MHC alleles have poor self-loading ability and whose amino acids at positions 126 and 220 confer poor tapasin functionality in comparison with their BF2*1501 counterparts, these BF2 alleles appear to have evolved to rely upon tapasin for efficient peptide loading; thus, these MHC I alleles bind tapasin quickly and with high affinity. This therefore allows plentiful opportunity for tapasin to load and edit the peptide cargo. In contrast, in other MHC haplotypes, such as B15, where the BF2 alleles are intrinsically more efficient at loading an optimal peptide cargo and whose amino acids at BF2 positions 126 and 220 confer greater tapasin functionality, there is likely to have been less pressure for the tapasin and BF2 interaction to evolve to interact rapidly and with high affinity, and there is less scope for tapasin to enhance peptide loading efficiency.
In conclusion, the BF2 alleles of the B15 and B19 haplotypes have different intrinsic peptide binding properties. These BF2 alleles also differ in their interactions with tapasin and in the extent that they benefit from tapasin. Our comparison of WT BF2 molecules and position 126 and 220 mutants shows that these polymorphisms are relevant for tapasin function and can profoundly influence binding to tapasin. We found BF2 position 220, located in the MHC I α3 domain, to be a key determinant of both intrinsic peptide binding efficiency and the ability of tapasin to function, suggesting that tapasin exerts allosteric control on MHC I peptide binding properties through this site. We found that when tapasin and BF2 alleles were mismatched in vivo that MHC I maturation could be severely impaired but that exchange of polymorphic tapasin contacts at positions 126 and 220 between the BF2 alleles alleviated this impairment. Collectively, we believe that this evidence supports the notion that efficient peptide loading of chicken MHC I requires tapasin and BF2 alleles from the same haplotype, as previously proposed (17, 23), which have co-evolved to balance tapasin-BF2 interaction characteristics against MHC I peptide binding properties.
Acknowledgments
We thank Dr. Patrick Duriez and Leon Douglas of the Cancer Research UK Core Protein Production Facility for assistance with protein purification, Drs. Ian Mockridge and Ruth French for SPR expertise, Professor Joern Werner for helpful advice, Professor Marlene Bouvier for providing HLA-B*08-fos and human tapasin-jun, Dr. Mark Howarth for the gift of pDisplay birA, and Dr. Nick van Hateren for help producing figures and other invaluable help and advice. We thank Dr. Denise Boulanger for critical reading of the manuscript.
This work was supported by Cancer Research UK Program Grant C7056A (to T. E.).
A. van Hateren, C. Tregaskes, R. Carter, A. P. Williams, J. P. Jacob, T. Elliott, and J. Kaufman, manuscript in preparation.
- ER
- endoplasmic reticulum
- TAP
- transporter associated with antigen presentation
- SPR
- surface plasmon resonance
- mP
- millipolarization units
- RU
- response units
- TAMRA
- tetramethylrhodamine.
REFERENCES
- 1. Sadasivan B., Lehner P. J., Ortmann B., Spies T., Cresswell P. (1996) Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity 5, 103–114 [DOI] [PubMed] [Google Scholar]
- 2. Dick T. P., Bangia N., Peaper D. R., Cresswell P. (2002) Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity 16, 87–98 [DOI] [PubMed] [Google Scholar]
- 3. Van Hateren A., James E., Bailey A., Phillips A., Dalchau N., Elliott T. (2010) The cell biology of major histocompatibility complex class I assembly. Towards a molecular understanding. Tissue Antigens 76, 259–275 [DOI] [PubMed] [Google Scholar]
- 4. Lewis J. W., Elliott T. (1998) Evidence for successive peptide binding and quality control stages during MHC class I assembly. Curr. Biol. 8, 717–720 [DOI] [PubMed] [Google Scholar]
- 5. Williams A. P., Peh C. A., Purcell A. W., McCluskey J., Elliott T. (2002) Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 16, 509–520 [DOI] [PubMed] [Google Scholar]
- 6. Howarth M., Williams A., Tolstrup A. B., Elliott T. (2004) Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc. Natl. Acad. Sci. U.S.A. 101, 11737–11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wearsch P. A., Cresswell P. (2007) Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat. Immunol. 8, 873–881 [DOI] [PubMed] [Google Scholar]
- 8. Greenwood R., Shimizu Y., Sekhon G. S., DeMars R. (1994) Novel allele-specific, post-translational reduction in HLA class I surface expression in a mutant human B cell line. J. Immunol. 153, 5525–5536 [PubMed] [Google Scholar]
- 9. Peh C. A., Burrows S. R., Barnden M., Khanna R., Cresswell P., Moss D. J., McCluskey J. (1998) HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity 8, 531–542 [DOI] [PubMed] [Google Scholar]
- 10. Dong G., Wearsch P. A., Peaper D. R., Cresswell P., Reinisch K. M. (2009) Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity 30, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu Y. Y., Turnquist H. R., Myers N. B., Balendiran G. K., Hansen T. H., Solheim J. C. (1999) An extensive region of an MHC class I α2 domain loop influences interaction with the assembly complex. J. Immunol. 163, 4427–4433 [PubMed] [Google Scholar]
- 12. Suh W. K., Derby M. A., Cohen-Doyle M. F., Schoenhals G. J., Früh K., Berzofsky J. A., Williams D. B. (1999) Interaction of murine MHC class I molecules with tapasin and TAP enhances peptide loading and involves the heavy chain α3 domain. J. Immunol. 162, 1530–1540 [PubMed] [Google Scholar]
- 13. The MHC sequencing consortium (1999) Complete sequence and gene map of a human major histocompatibility complex. Nature 401, 921–923 [DOI] [PubMed] [Google Scholar]
- 14. Copeman J., Bangia N., Cross J. C., Cresswell P. (1998) Elucidation of the genetic basis of the antigen presentation defects in the mutant cell line. 220 reveals polymorphism and alternative splicing of the tapasin gene. Eur. J. Immunol. 28, 3783–3791 [DOI] [PubMed] [Google Scholar]
- 15. Furukawa H., Kashiwase K., Yabe T., Ishikawa Y., Akaza T., Tadokoro K., Tohma S., Inoue T., Tokunaga K., Yamamoto K., Juji T. (1998) Polymorphism of TAPASIN and its linkage disequilibria with HLA class II genes in the Japanese population. Tissue Antigens 52, 279–281 [DOI] [PubMed] [Google Scholar]
- 16. Herberg J. A., Sgouros J., Jones T., Copeman J., Humphray S. J., Sheer D., Cresswell P., Beck S., Trowsdale J. (1998) Genomic analysis of the Tapasin gene, located close to the TAP loci in the MHC. Eur. J. Immunol. 28, 459–467 [DOI] [PubMed] [Google Scholar]
- 17. Kaufman J., Milne S., Göbel T. W., Walker B. A., Jacob J. P., Auffray C., Zoorob R., Beck S. (1999) The chicken B locus is a minimal essential major histocompatibility complex. Nature 401, 923–925 [DOI] [PubMed] [Google Scholar]
- 18. Wong G. K., Liu B., Wang J., Zhang Y., Yang X., Zhang Z., Meng Q., Zhou J., Li D., Zhang J., Ni P., Li S., Ran L., Li H., Li R., Zheng H., Lin W., Li G., Wang X., Zhao W., Li J., Ye C., Dai M., Ruan J., Zhou Y., Li Y., He X., Huang X., Tong W., Chen J., Ye J., Chen C., Wei N., Dong L., Lan F., Sun Y., Yang Z., Yu Y., Huang Y., He D., Xi Y., Wei D., Qi Q., Li W., Shi J., Wang M., Xie F., Zhang X., Wang P., Zhao Y., Li N., Yang N., Dong W., Hu S., Zeng C., Zheng W., Hao B., Hillier L. W., Yang S. P., Warren W. C., Wilson R. K., Brandstrom M., Ellegren H., Crooijmans R. P., van der Poel J. J., Bovenhuis H., Groenen M. A., Ovcharenko I., Gordon L., Stubbs L., Lucas S., Glavina T., Aerts A., Kaiser P., Rothwell L., Young J. R., Rogers S., Walker B. A., van Hateren A., Kaufman J., Bumstead N., Lamont S. J., Zhou H., Hocking P. M., Morrice D., de Koning D. J., Law A., Bartley N., Burt D. W., Hunt H., Cheng H. H., Gunnarsson U., Wahlberg P., Andersson L., Kindlund E., Tammi M. T., Andersson B., Webber C., Ponting C. P., Overton I. M., Boardman P. E., Tang H., Hubbard S. J., Wilson S. A., Yu J., Yang H. (2004) A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature 432, 717–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker B. A., Hunt L. G., Sowa A. K., Skjødt K., Göbel T. W., Lehner P. J., Kaufman J. (2011) The dominantly expressed class I molecule of the chicken MHC is explained by coevolution with the polymorphic peptide transporter (TAP) genes. Proc. Natl. Acad. Sci. U.S.A. 108, 8396–8401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Hateren A. (2006) Function of chicken tapasin in MHC class I antigen presentation. Ph.D. thesis, University of Southampton, Southampton, UK [Google Scholar]
- 21. Shaw I., Powell T. J., Marston D. A., Baker K., van Hateren A., Riegert P., Wiles M. V., Milne S., Beck S., Kaufman J. (2007) Different evolutionary histories of the two classical class I genes BF1 and BF2 illustrate drift and selection within the stable MHC haplotypes of chickens. J. Immunol. 178, 5744–5752 [DOI] [PubMed] [Google Scholar]
- 22. Wallny H. J., Avila D., Hunt L. G., Powell T. J., Riegert P., Salomonsen J., Skjødt K., Vainio O., Vilbois F., Wiles M. V., Kaufman J. (2006) Peptide motifs of the single dominantly expressed class I molecule explain the striking MHC-determined response to Rous sarcoma virus in chickens. Proc. Natl. Acad. Sci. U.S.A. 103, 1434–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaufman J., Völk H., Wallny H. J. (1995) A “minimal essential Mhc” and an “unrecognized Mhc.” Two extremes in selection for polymorphism. Immunol. Rev. 143, 63–88 [DOI] [PubMed] [Google Scholar]
- 24. Chen M., Bouvier M. (2007) Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J. 26, 1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koch M., Camp S., Collen T., Avila D., Salomonsen J., Wallny H. J., van Hateren A., Hunt L., Jacob J. P., Johnston F., Marston D. A., Shaw I., Dunbar P. R., Cerundolo V., Jones E. Y., Kaufman J. (2007) Structures of an MHC class I molecule from B21 chickens illustrate promiscuous peptide binding. Immunity 27, 885–899 [DOI] [PubMed] [Google Scholar]
- 26. Rodenko B., Toebes M., Hadrup S. R., van Esch W. J., Molenaar A. M., Schumacher T. N., Ovaa H. (2006) Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 1, 1120–1132 [DOI] [PubMed] [Google Scholar]
- 27. Garboczi D. N., Hung D. T., Wiley D. C. (1992) HLA-A2-peptide complexes. Refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. U.S.A. 89, 3429–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Howarth M., Ting A. Y. (2008) Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat. Protoc. 3, 534–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schoenhals G. J., Krishna R. M., Grandea A. G., 3rd, Spies T., Peterson P. A., Yang Y., Früh K. (1999) Retention of empty MHC class I molecules by tapasin is essential to reconstitute antigen presentation in invertebrate cells. EMBO J. 18, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rizvi S. M., Raghavan M. (2006) Direct peptide-regulatable interactions between MHC class I molecules and tapasin. Proc. Natl. Acad. Sci. U.S.A. 103, 18220–18225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yewdell J. W., Reits E., Neefjes J. (2003) Making sense of mass destruction. Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 3, 952–961 [DOI] [PubMed] [Google Scholar]
- 32. Bennett E. M., Bennink J. R., Yewdell J. W., Brodsky F. M. (1999) Cutting edge. Adenovirus E19 has two mechanisms for affecting class I MHC expression. J. Immunol. 162, 5049–5052 [PubMed] [Google Scholar]
- 33. Lybarger L., Wang X., Harris M. R., Virgin H. W., 4th, Hansen T. H. (2003) Virus subversion of the MHC class I peptide-loading complex. Immunity 18, 121–130 [DOI] [PubMed] [Google Scholar]
- 34. Apanius V., Penn D., Slev P. R., Ruff L. R., Potts W. K. (1997) The nature of selection on the major histocompatibility complex. Crit. Rev. Immunol. 17, 179–224 [DOI] [PubMed] [Google Scholar]
- 35. Hee C. S., Beerbaum M., Loll B., Ballaschk M., Schmieder P., Uchanska-Ziegler B., Ziegler A. (2013) Dynamics of free versus complexed β2-microglobulin and the evolution of interfaces in MHC class I molecules. Immunogenetics 65, 157–172 [DOI] [PubMed] [Google Scholar]
- 36. Rizvi S. M., Raghavan M. (2010) Mechanisms of function of tapasin, a critical major histocompatibility complex class I assembly factor. Traffic 11, 332–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zarling A. L., Luckey C. J., Marto J. A., White F. M., Brame C. J., Evans A. M., Lehner P. J., Cresswell P., Shabanowitz J., Hunt D. F., Engelhard V. H. (2003) Tapasin is a facilitator, not an editor, of class I MHC peptide binding. J. Immunol. 171, 5287–5295 [DOI] [PubMed] [Google Scholar]