

Abstract
The distribution of circulating lipoprotein particles affects the risk for cardiovascular disease (CVD) in humans. Lipoproteins are historically defined by their density, with low-density lipoproteins positively and high-density lipoproteins (HDLs) negatively associated with CVD risk in large populations. However, these broad definitions tend to obscure the remarkable heterogeneity within each class. Evidence indicates that each class is composed of physically (size, density, charge) and compositionally (protein and lipid) distinct subclasses exhibiting unique functionalities and differing effects on disease. HDLs in particular contain upward of 85 proteins of widely varying function that are differentially distributed across a broad range of particle diameters. We hypothesized that the plasma lipoproteins, particularly HDL, represent a continuum of phospholipid platforms that facilitate specific protein–protein interactions. To test this idea, we separated normal human plasma using three techniques that exploit different lipoprotein physicochemical properties (gel filtration chromatography, ionic exchange chromatography, and preparative isoelectric focusing). We then tracked the co-separation of 76 lipid-associated proteins via mass spectrometry and applied a summed correlation analysis to identify protein pairs that may co-reside on individual lipoproteins. The analysis produced 2701 pairing scores, with the top hits representing previously known protein–protein interactions as well as numerous unknown pairings. A network analysis revealed clusters of proteins with related functions, particularly lipid transport and complement regulation. The specific co-separation of protein pairs or clusters suggests the existence of stable lipoprotein subspecies that may carry out distinct functions. Further characterization of the composition and function of these subspecies may point to better targeted therapeutics aimed at CVD or other diseases.
Lipoproteins are circulating emulsions of protein and lipid that play important roles, both positive and negative, in cardiovascular disease (CVD).1 Historically defined by their density as separated by ultracentrifugation, the major lipoprotein classes include the neutral lipid ester-rich very low-density and low-density lipoproteins (VLDLs and LDLs, respectively), which function to transport triglyceride and cholesterol from the liver to the peripheral tissues. Significant epidemiological evidence, in vitro studies, animal experiments, and human clinical trials have shown that high-LDL cholesterol is a bona fide causative factor in CVD (1). In contrast, protein- and phospholipid-rich high-density lipoproteins (HDLs) are thought to mediate the reverse transport of cholesterol from the periphery to the liver for catabolism and to perform anti-oxidative and anti-inflammatory functions (reviewed in Refs. 2 and 3). A host of human epidemiology and animal studies indicate that HDLs are atheroprotective (4). However, recent clinical trials of therapeutics that generically raise HDL, at least as measured by its cholesterol levels, have failed to confer the expected CVD protections (5–7).
Although these traditional density-centric definitions have been used for nearly 40 years, accumulating evidence indicates that they are not particularly reflective of lipoprotein compositional and functional complexity. With respect to most physical traits (size, charge, lipid content, protein content, etc.), one can demonstrate significant heterogeneity within each density class. This suggests that particle subspecies exist with unique functions and effects on disease. For example, LDL can be resolved into large, buoyant and small, dense forms (8), with subjects carrying more cholesterol in the small, dense LDL exhibiting a greater CVD risk (9). HDL is particularly noted for heterogeneity, as it can be separated into numerous subfractions by density (10), diameter (11), charge (12), and major apolipoprotein content (13). Most strikingly, recent applications of soft-ionization mass spectrometry (MS) have identified upward of 85 HDL proteins with functions that go well beyond the structural apolipoproteins, lipid transport proteins, and lipid-modifying enzymes known from previous biochemical studies (14, 15). Many of these proteins imply functions as diverse as complement regulation, acute phase response, protease inhibition, and innate immunity (16). Individual HDL subspecies can apparently draw from this palette of proteins to produce distinct particles of distinct function. One well-defined HDL subfraction, termed trypanosome lytic factor, contains apolipoprotein apoA-I, haptoglobin-related protein, and apoL-I. Working together, these proteins enter the trypanosome brucei brucei and kill it via lysosomal disruption (17). There are numerous other instances of on-particle protein cooperation in HDL related to CVD (reviewed in Ref. 15). Furthermore, two-dimensional electrophoresis studies by Asztalos and colleagues (18), as well as our own work (11, 19), strongly support the concept that certain apolipoproteins segregate among different HDL particles. These observations present the intriguing possibility that the phospholipids of HDLs act as an organizing platform that facilitates the assembly of specific protein complexes (20). Such subspecies could have important functional implications in the context of CVD protection, inflammation, or even innate immune function. Furthermore, this subspeciation may explain why therapeutics that raise HDL cholesterol levels across the board have not yet shown promise with regard to CVD.
To address this hypothesis, we began to think of lipoproteins as a continuum of phospholipid platforms that support the assembly of specific protein complexes analogous to those in cells that perform coordinated biological functions (i.e. ribosomes, centrosomes, etc.). Two common methods for characterizing protein complexes are tandem affinity purification (21) and immunoprecipitation. Both rely on the specific pull-down of a target protein (by either an introduced affinity tag or an antibody) followed by the identification of co-precipitated proteins via MS. Unfortunately, tandem affinity purification strategies are impractical in humans, and we have found that immunoprecipitation experiments with human plasma lipoproteins result in a high false-positive rate due to the low abundance of most of these proteins, particularly those in HDLs. Therefore, we took an alternative approach called co-separation analysis, a method based on the principle that stable protein complexes can be identified by tracking their co-migration as they undergo biochemical separation by multiple orthogonal approaches (22). Native proteins are analyzed in an unbiased manner without affinity tags or antibodies, and purification to homogeneity is not necessary for the identification of putative protein complexes.
Most current studies of the lipoprotein proteome utilize samples isolated via density ultracentrifugation because contaminating lipid-unassociated lipoproteins, which can be highly abundant and obscure the identification of targeted lipid-associated proteins, are thus removed prior to the analysis. In previous work, we characterized the use of a calcium silica hydrate (CSH) resin that allowed the specific isolation of phospholipid-associated proteins and their subsequent MS identification without ultracentrifugation (11). This advance enabled the use of a variety of non-density-based separation methods for the study of plasma lipoproteins. Here, we take advantage of this to analyze the proteome of human plasma lipoproteins separated via three separation techniques that exploit different physicochemical properties: (i) gel filtration chromatography (size), (ii) anion exchange chromatography (charge interaction), and (iii) isoelectric focusing. By tracking the co-migration of specific proteins across these separations (Fig. 1), we identified a host of putative protein pairings, including the previously known trypanosome lytic factor HDL fraction, for further biochemical verification and characterization.
Fig. 1.
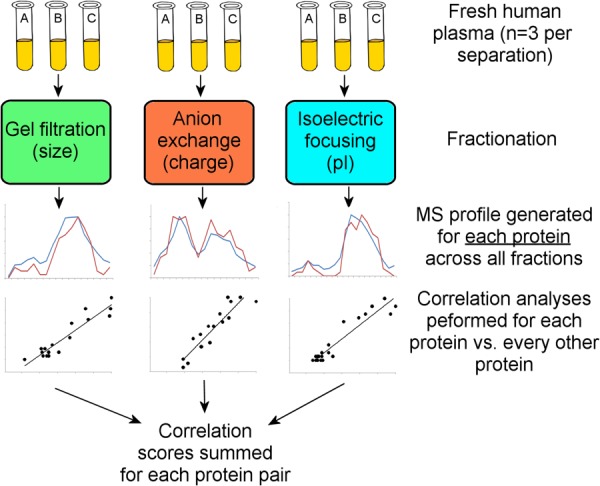
Overview of the multi-dimensional separation co-migration analysis used in this study (see “Experimental Procedures” for details).
EXPERIMENTAL PROCEDURES
Human Subjects and Plasma Collection
Venous blood was collected from three fasted (≥12 h), apparently healthy normolipidemic (total cholesterol between 125 and 200 mg/dl; HDL-C ≥ 40 mg/dl; triglycerides < 150 mg/dl) male donors (ages 21, 22, and 34) by a trained phlebotomist using BD Vacutainer® Plus citrate tubes containing buffered sodium citrate (0.105 m) as an anticoagulant. Cellular components were pelleted via centrifugation at ∼1590 × g for 15 min in a Horizon mini-E (Quest Diagnostics, Madison, NJ) at room temperature. Plasma was stored at 4 °C until subjected to biochemical separation, always within 20 min. Samples were never frozen. Human subjects provided informed consent according to an approved protocol as overseen by the University of Cincinnati Institutional Review Board.
Gel Filtration Chromatography
Fresh plasma (370 μl) was applied to three Superdex 200 10/300 GL columns (GE Healthcare) arranged in series as described previously (11). The sample was processed at a flow rate of 0.3 ml/min in standard Tris buffer (10 mm Tris, 0.15 m NaCl, 1 mm EDTA, 0.2% NaN3, pH 8.2). 17 fractions of 1.5 ml each that encompassed the usable separation range of the column system (i.e. between the void and total volumes) were collected.
Anion Exchange Chromatography
Fresh plasma (370 μl) was mixed with buffer A (50 mm Tris, 1 mm EDTA, pH 8.2) to a total volume of 2 ml and applied to a MonoQ 5/50 GL anion exchange column (GE Healthcare) at a flow rate of 1 ml/min at room temperature. A gradient of buffer B (0% to 100%, buffer A + 500 mm sodium perchlorate, pH 8.2) was used to elute plasma components from the resin over a total volume of 25 ml. 23 fractions of 1 ml each were collected. This method was adapted from Ref. 23.
Isoelectric Focusing
Fresh plasma samples were separated via in-solution preparative isolectric focusing using a Rofor apparatus (Bio-Rad, Hercules, CA). Plasma (3 ml) was mixed with 56 ml of water and 3 ml of Bio-lyte 5/7 ampholyte solution (Bio-Rad, Hercules, CA). The mixture was loaded into the apparatus and run according to manufacturer's instructions at a constant 15 W until voltage stabilized (approximately 4 h). 20 fractions of 1.5 ml each were collected using the supplied sample recovery manifold.
Fraction Lipid Analysis
Fractions collected via each separation technique were analyzed for phospholipid (PL) (enzymatic kit from Wako Diagnostics, Richmond, VA), cholesterol (CH) (enzymatic kit from Pointe Scientific, Canton, MI), and total protein content by Bradford assay (Thermo Scientific, Rockford, IL). For the isoelectric focusing (IEF) analysis, a Markwell modified Lowry assay (24) was used to track the protein, as the ampholytes appeared to interfere with the Bradford assay.
Isolation of Lipid-associated Proteins
Fractions collected via each technique were applied to a CSH resin (marketed as Lipid Removal Agent, Supelco, Sigma-Aldrich, St. Louis, MO) to isolate only those proteins associated with PL (11). In a centrifuge tube, 45 μg of CSH (from 100 mg/ml suspension stock solution in 50 mm ammonium bicarbonate) per 1 μg of PL in 400 μl of fraction were mixed gently for 30 min at room temperature. The CSH was then pelleted via centrifugation (∼2200 × g for 2 min) in a minicentrifuge, and the supernatant containing lipid-free plasma proteins was removed. The CSH was then washed twice with 50 mm ammonium bicarbonate.
MS Analysis of Fractions
The lipoprotein particles were subjected to trypsin digestion while still bound to the CSH. 1.5 μg of sequencing grade trypsin (Promega, Madison, WI) in 25 μl of 50 mm ammonium bicarbonate was added to each CSH pellet and incubated at 37 °C overnight on a rotating plate. To collect the digested peptides, the CSH was washed twice with 125 μl of 50 mm ammonium bicarbonate. Peptides were first reduced with dithiothreitol (200 mm; 30 min at 37 °C) and then carbamidomethylated with iodoacetamide (800 mm; 30 min at room temperature). Peptide solutions were then lyophilized to dryness and stored at −20 °C. For MS, dried peptides were reconstituted in 15 μl of 0.1% formic acid in water. An Agilent 1100 series Autosampler/HPLC was used to draw 0.5 μl of sample and inject it onto a C18 reverse-phase column (GRACE; 150 mm × 0.5 mm) where an acetonitrile concentration gradient (5%–30% in water with 0.1% formic acid) was used to elute peptides for online electrospray ionization MS/MS by a QStar XL mass spectrometer (Applied Biosystems, Grand Island, NY). Column cleaning was performed automatically with two cycles of a 5%–85% acetonitrile gradient lasting 15 min each between runs.
MS Data Analysis
To identify the protein composition of particles contained in the various fractions, peak lists (in the form of Mascot generic files) generated from the analysis of each fraction (Analyst QS v1.1) were scanned against the UniProtKB/Swiss-Prot Protein Knowledgebase (release 57.0, 03/2009), containing 428,636 entries, using both the Mascot (version 2.1) and X!Tandem (version 2007.01.01.1) search engines. Search criteria included human taxonomy, no fixed modifications, variable modifications of Met oxidation and carbamidomethylation, peptide tolerance and MS/MS tolerance set at ±0.15 Da, and up to three missed tryptic cleavage sites. Scaffold software (version Scaffold 3.6.4, Proteome Software) was used to validate MS/MS-based peptide and protein identifications. Peptide identification required a value of 90% probability (using data from both Mascot and X!Tandem) using the Peptide Prophet algorithm (25). Positive protein identification also required a value of 90% probability from the Protein Prophet algorithm (26). Also, a minimum of two peptides were required unless the protein in question was found with single peptide hits in multiple consecutive fractions that were consistent across all subjects. The annotated Scaffold files containing MS/MS spectra and peptide identifications for all subjects and separation techniques are available as online supplements. They can be viewed using a free viewer available at the Proteome Software website. Because equal volumes of sample were applied to the MS analysis, as opposed to equal protein contents, the relative amount of a given protein present in a given fraction should have been proportional to the number of spectral counts (i.e. the number of unweighted MS/MS spectra assigned to a particular protein) in each fraction. In no case were conclusions about the relative abundance of two different proteins drawn on the basis of peptide counting. We previously demonstrated that this approach provides a semi-quantitative abundance readout across the fractions that match well with patterns derived from immunological analyses (19).
Correlation Analysis
The strategy for our correlation analysis is outlined in Fig. 1. Protein distribution profiles for each separation technique were generated using the MS peptide-count data. Within the distribution data, we used Pearson correlation coefficients to assess the similarity between the abundance profiles for all combinations of protein pairs (Eq. 1), where Xi and Yi are the abundance values of proteins X and Y in the ith fraction. Higher scores indicate protein pairs that were more consistently collected in the same fraction with each individual separation technique. The correlation scores from each of the three separation techniques for each pair of proteins were summed to obtain a combined correlation score. This score was used to determine the likelihood of particle co-habitance by the protein pair.
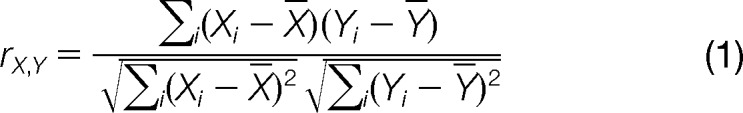 |
RESULTS
Each plasma separation technique was empirically optimized to spread the PL-containing components of plasma across as many fractions as practical. By subjecting each fraction to the CSH PL-binding resin, we limited the proteomic analysis to only those proteins that were associated with PLs. This includes all lipoproteins traditionally defined as HDL, LDL, and VLDL and even may include PL-containing entities such as microparticles (27). For each technique, PL, CH (total), and total protein were determined by means of colorimetric assay across all fractions, and the relative abundance of individual proteins, as determined by the number of spectral counts in equal volumes from each fraction, was tracked by MS.
Gel Filtration Chromatography
The gel filtration chromatography separation used here has been described previously (11). Fig. 2A displays the distribution profiles of protein, PL, and CH across the fractions. Fractions 13–19 were rich in lipid and poor in protein. Previous characterizations of these fractions indicate that this peak predominantly contains VLDLs and LDLs (i.e. apoB-containing lipoproteins). Because the separation characteristics of the column system were optimized for the HDL size range, LDLs and VLDLs were not resolved. Fractions 19–25 were PL and protein rich, with lower levels of CH. The abundance of apoA-I in this peak suggested that it contained most of the traditionally defined HDLs. However, we caution that although lipoprotein size and density are generally related, one cannot make direct comparisons between particles separated by gel filtration and traditional density-centric definitions of human lipoproteins. Fractions 25–29 were lipid poor and highly protein rich. SDS-PAGE analysis of these fractions prior to CSH treatment showed that they were dominated by serum albumin and other small, soluble lipid-free proteins that make up the bulk of circulating protein.
Fig. 2.
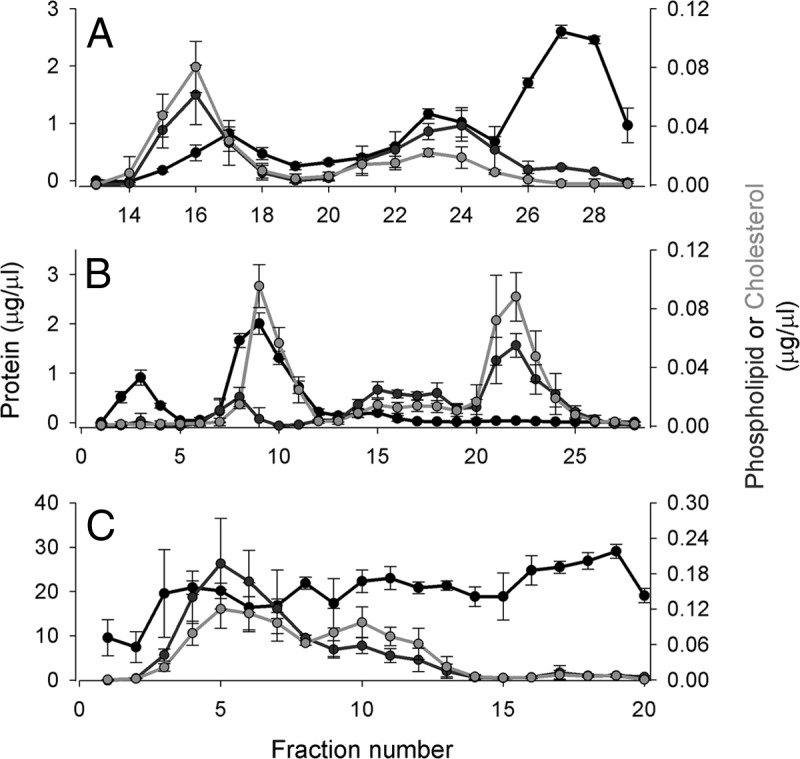
Phospholipid, total cholesterol, and total protein distribution across fractions from each separation technique. A, gel filtration chromatography on three Superdex 200 columns arranged in series (1.5-ml fractions). Fractions 1–12 are not shown as they represent the void volume of the column where no lipid or protein signal was detected. B, anion exchange chromatography on a MonoQ column (1.5-ml fractions). C, preparative isoelectric focusing on a Rotofor apparatus (Bio-Rad) using ampholytes from pH 5–7 (1.5-ml fractions). Fraction 1 represents the anode (+, or low-pH) end of the gradient, and fraction 20 is the cathode (−, or high-pH). For each separation, the total protein determined by colorimetric assay (see “Experimental Procedures”) is in black, the total cholesterol is in green, and choline-containing phospholipids are in red. The concentration listed reflects that in the fraction, not in plasma. The data show three independent separations on three normal plasma donors, and the error bars represent ±1 sample standard deviation.
Anion Exchange Chromatography
Lipid and protein distribution patterns from plasma separated via anion exchange chromatography are shown in Fig. 2B. There was an initial protein peak across fractions 1–5 that contained no lipid, likely representing lipid-free plasma proteins with either positive or neutral charge that were not retained on the column. Fractions 7–11 were rich in CH and protein. Fractions 13–19 were enriched in PL, while fractions 20–27 were rich in both lipids but poor in protein. Although it is not possible to assign specific peaks to specific lipoprotein density classes, it is clear that the separation profile differed dramatically from the gel filtration analysis.
Preparative IEF
Despite the theoretical relationship between anion exchange (ionic interaction) and IEF (isoelectric point) separations, Fig. 2C shows that yet another completely different plasma separation profile was obtained via IEF. The protein profile was more homogeneous across the pH gradient than in the other separations. The PL and CH were distributed across fractions 2–13.
Thus, when plasma was separated by means of three different physicochemical techniques, the distribution profiles of the protein and lipid components varied dramatically—a key requirement for the successful co-migration analysis of putative protein assemblies.
Proteomics
Each isolated fraction was bound to CSH resin to isolate PL-associated species, fully trypsinized, and then analyzed via MS. Because the same volume of each fraction was used in the analysis, the relative peptide counts for each identified protein allow for a semi-quantitative estimate of the abundance of a given protein in each fraction relative to the others (11). Overall, we identified 159 PL-associated proteins in this study across all three separation techniques and all fractions. Fig. 3 shows a Venn diagram indicating the identifications made from the analysis of each separation technique. 140 proteins were observed with anion exchange, and fewer were observed with gel filtration (106) and IEF (93). However, 76 were identified in all three analyses. This extensive degree of overlap among the separation methodologies boded well for an effective correlation analysis.
Fig. 3.
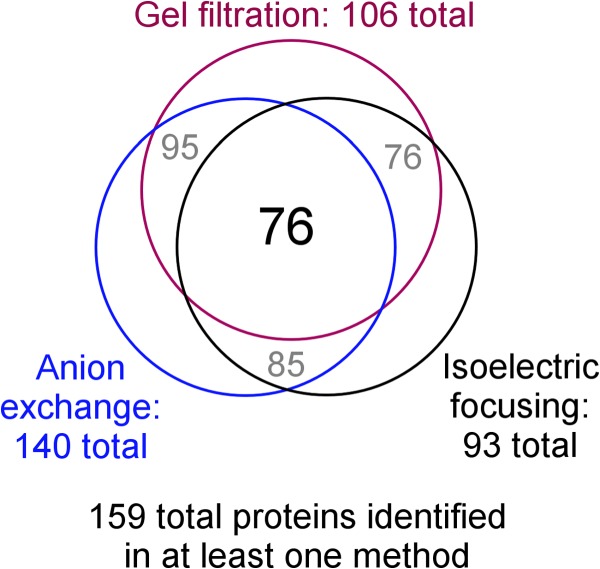
Venn diagram showing the total number of proteins identified via MS in each separation technique and the overlap between them. The criteria for protein identification are laid out under “Experimental Procedures.”
The protein distributions across the gel filtration fractions are shown as a heat map in Fig. 4. Each identified protein is listed with its relative abundance for each fraction (expressed in relation to the fraction with the highest peptide count for that particular protein). The proteins were arranged in order from largest to smallest diameter based on the peak fraction. The identified proteins were distributed in distinct patterns across the size gradient. Some, such as apoA-I and apoA-II, could be found in almost every fraction, whereas others, such as apoM, were localized to distinct peaks. Still others such as vitronectin resolved into two separate peaks. ApoB appeared primarily in larger fractions 14–18, consistent with the presence of VLDL and LDL in the cholesterol peak in these same fractions apparent in Fig. 2A. Although apoA-I appeared in all fractions, it was most concentrated in fractions 19–27, indicating that much of the traditionally defined HDL was in this region.
Fig. 4.

Heat map of protein distribution patterns as determined via gel filtration chromatography. Fraction numbers are indicated across the top. The proteins identified with this method (n = 3 donors) are listed on the left. Equal volumes of each fraction were applied to the CSH resin, trypsinized, and then isolated via MS. The spectral counts (i.e. the number of unique peptides identified for each protein) were determined for each protein in each fraction. The fraction that contained the highest peptide count for a given protein was normalized to 1, and all other fractions for that protein were scaled accordingly. The highest relative values are colored red and gradate to yellow for the lowest values (blue indicates that no peptides were found). The proteins were ordered from largest to smallest based on the peak fraction. No determination can be made about the relative abundance of different proteins from this analysis. All proteins indentified via this method are shown.
Fig. 5 shows the corresponding heat map for the anion exchange separation. Again, the identified proteins resolved in a range of different patterns. ApoB appeared as two peaks at fractions 8–10, with the majority in fractions 18–25. ApoA-I was again found in all PL-containing fractions, but it appeared to be distributed in two general peaks centered on fractions 9 and 15. Fig. 6 shows the corresponding analysis from preparative IEF. Here, apoB was found as a rather tight band at fractions 4–10, with relatively smaller amounts found elsewhere. ApoA-I was again found in all PL-containing fractions but was concentrated more or less in a single peak centered on fraction 12. To make a crude comparison to a typical agarose gel separation (28, 29), it appeared that fractions 6–8 represented the beta migration band, whereas fractions 12–14 formed the alpha band.
Fig. 5.

Heat map of protein distribution patterns as determined via anion exchange chromatography. The figure is laid out exactly as for Fig. 4 except that the proteins were ordered from least to highest ionic character based on the peak fraction. To conserve space, only those proteins that appeared in at least one other method are shown (105 of 140 total).
Fig. 6.

Heat map of protein distribution patterns as determined via preparative isoelectric focusing. The figure is laid out exactly as for Fig. 4 except that the proteins are ordered from lowest to highest isoelectric point (pI) based on the peak fraction. All proteins identified with this method are shown.
Correlation Analysis
As indicated above, the separation correlation analysis aims to identify pairs of proteins that reside on the same phospholipid particle. Using the hypothetical example of two proteins (A and B) that strictly and exclusively reside on the same lipoprotein particle, one predicts that the abundance profiles of proteins A and B would overlap in all three separations. However, proteins C and D, which do not reside on the same particle, may co-migrate by chance in one of the techniques but will fail to co-migrate across all three separations. The analysis used a correlation scoring system to identify pairs of proteins whose distribution profiles in a given separation were similar or even partially overlapping. Proteins consistently identified in the same fractions were given higher correlation scores, with a value of 1 representing perfect correlation across all fractions. The individual correlation scores for a given pair of proteins for each separation technique were then combined to produce an overall score reflecting the likelihood that those proteins will reside together on a single particle. Thus, a perfect correlation across all separations would result in a score of 3, whereas poorly correlated protein pairs would exhibit scores closer to 0 or less.
In total, scores were assigned to 2701 protein pairs with a scoring range from 2.87 to −1.47. Table I lists the top 50 ranked pairs in this analysis representing protein pairs scoring in the top 2%. The complete list is in supplemental Table S1. The top-scoring pairs contained the various combinations of the fibrinogen α, β, and γ-chain with a score of >2.8, indicating almost complete co-migration across the separations. The normalized elution profiles for the α and β subunits are shown in Fig. 7A, confirming the tight correlations. The next highest scoring pair was apoA-I and apoA-II (Fig. 7B). Of interest, the trypanosome lytic factor described in the Introduction was identified as a co-migration between apoL-I and haptoglobin-related protein that scored within the top 3% of protein pairs at #79 (also shown in Table I). The co-migration of these two proteins is shown in Fig. 7C. The remaining protein pairs listed in Table I and the top of supplemental Table S1 represent potentially novel protein interactions among PL-associated plasma proteins, the majority of which have not yet been reported to our knowledge.
Table I. Top 50 phospholipid-associated protein pairings as determined by composite correlation score (from 2701 scored pairs ranging from 2.87 to −1.47).
| Rank | Protein A | Protein B | Correlation score |
|||
|---|---|---|---|---|---|---|
| Gel filtration | Anion exchange | IEF | Comp.a | |||
| 1 | Fibrinogen β chain | Fibrinogen γ chain | 0.95 | 0.97 | 0.95 | 2.87 |
| 2 | Fibrinogen α chain | Fibrinogen β chain | 0.98 | 0.98 | 0.90 | 2.86 |
| 3 | ApoA-I | ApoA-II | 0.96 | 0.94 | 0.95 | 2.85 |
| 4 | Fibrinogen α chain | Fibrinogen γ chain | 0.94 | 0.97 | 0.92 | 2.83 |
| 5 | IgG h. chain V-III region BRO | IgG κ chain C region | 0.92 | 0.91 | 0.90 | 2.73 |
| 6 | Complement factor B | Histidine-rich glycoprotein | 0.91 | 0.95 | 0.81 | 2.67 |
| 7 | ApoA-IV | Kallistatin | 0.79 | 0.94 | 0.93 | 2.67 |
| 8 | IgGγ-1 chain C region | IgG κ chain C region | 0.94 | 0.77 | 0.92 | 2.62 |
| 9 | Complement C8 α | Complement C8 β | 0.99 | 0.88 | 0.74 | 2.61 |
| 10 | Insulin-like growth factor-binding protein 1 | Inter-α-trypsin inhibitor 4 | 0.87 | 0.99 | 0.76 | 2.61 |
| 11 | Insulin-like growth factor-binding protein 1 | Extracellular matrix protein 1 | 0.79 | 0.89 | 0.92 | 2.59 |
| 12 | IgG κ chain C region | IgG λ-1 chain C region | 0.97 | 0.87 | 0.74 | 2.58 |
| 13 | Complement factor I | Hemopexin | 0.92 | 0.92 | 0.73 | 2.56 |
| 14 | Inter-α-trypsin inhibitor 1 | Inter-α-trypsin inhibitor 2 | 0.94 | 0.99 | 0.59 | 2.51 |
| 15 | Complement factor B | n-acetylmuramoyl-l-ala. amid. | 0.95 | 0.96 | 0.59 | 2.50 |
| 16 | IgG γ-1 chain C region | IgG λ-1 chain C region | 0.91 | 0.75 | 0.84 | 2.50 |
| 17 | IgG heavy chain V-III reg. BRO | IgG γ-1 chain C region | 0.82 | 0.83 | 0.79 | 2.44 |
| 18 | Complement factor C4-B | Complement C5 | 0.65 | 0.84 | 0.93 | 2.43 |
| 19 | IgG κ chain C region | IgG λ chain V-I region HA | 0.87 | 0.76 | 0.80 | 2.43 |
| 20 | IgG heavy chain V-III reg. BRO | IgG λ-1 chain C region | 0.89 | 0.80 | 0.72 | 2.41 |
| 21 | IgG γ-1 chain C region | IgG λ chain V-I region HA | 0.84 | 0.86 | 0.70 | 2.41 |
| 22 | IgG heavy chain V-III reg. BRO | IgG λ chain V-I region HA | 0.85 | 0.76 | 0.76 | 2.37 |
| 23 | Complement C1s | Complement factor C4-B | 0.95 | 0.49 | 0.92 | 2.35 |
| 24 | α-1-antichymotrypsin | ApoJ | 0.87 | 0.58 | 0.87 | 2.32 |
| 25 | Complement factor B | Coagulation factor XII | 0.85 | 0.93 | 0.51 | 2.29 |
| 26 | Carboxypeptidase B2 | Kallistatin | 0.83 | 0.92 | 0.53 | 2.27 |
| 27 | Extracellular matrix protein 1 | Inter-α-trypsin inhibitor 4 | 0.71 | 0.91 | 0.65 | 2.27 |
| 28 | Histidine-rich glycoprotein | n-acetylmuramoyl-l-ala. amid. | 0.91 | 0.93 | 0.40 | 2.25 |
| 29 | IgG λ-1 chain C region | Ig λ chain V-I region HA | 0.79 | 0.78 | 0.65 | 2.21 |
| 30 | Antithrombin-III | Pigment epithelial-derived factor | 1.00 | 0.85 | 0.34 | 2.18 |
| 31 | α-1-antichymotrypsin | Kininogen-1 | 0.90 | 0.40 | 0.88 | 2.18 |
| 32 | ApoA-II | Extracellular matrix protein 1 | 0.48 | 0.83 | 0.87 | 2.18 |
| 33 | α-1B-glycoprotein | α-2-HS-glycoprotein | 0.84 | 0.50 | 0.82 | 2.16 |
| 34 | ApoH | Plasminogen | 0.83 | 0.90 | 0.42 | 2.15 |
| 35 | ApoA-I | Extracellular matrix protein 1 | 0.51 | 0.78 | 0.84 | 2.13 |
| 36 | Complement C9 | Heparin cofactor 2 | 0.39 | 0.80 | 0.91 | 2.09 |
| 37 | ApoJ | Inter-α-trypsin inhibitor 4 | 0.84 | 0.71 | 0.55 | 2.09 |
| 38 | ApoA-I | ApoM | 0.73 | 0.53 | 0.82 | 2.08 |
| 39 | Complement C1s | Complement C5 | 0.44 | 0.74 | 0.90 | 2.08 |
| 40 | α-1B-glycoprotein | Coagulation factor XII | 0.44 | 0.99 | 0.64 | 2.07 |
| 41 | Antithrombin-III | ApoA-IV | 0.30 | 0.92 | 0.83 | 2.05 |
| 42 | ApoA-II | ApoM | 0.72 | 0.44 | 0.87 | 2.04 |
| 43 | ApoC-I | Complement C7 | 0.67 | 0.64 | 0.72 | 2.03 |
| 44 | Complement factor B | Hemopexin | 0.45 | 0.92 | 0.64 | 2.01 |
| 45 | α-1B-glycoprotein | n-acetylmuramoyl-l-ala. amid. | 0.90 | 0.89 | 0.21 | 2.01 |
| 46 | α-2-macroglobulin | Ig μ chain C region | 0.72 | 0.57 | 0.70 | 1.99 |
| 47 | Coagulation factor XII | Histidine-rich glycoprotein | 0.66 | 0.84 | 0.48 | 1.98 |
| 48 | Plasminogen | Serotransferrin | 0.98 | 0.73 | 0.27 | 1.98 |
| 49 | ApoJ | Haptoglobin | 0.77 | 0.28 | 0.93 | 1.98 |
| 50 | ApoA-IV | Carboxypeptidase B2 | 0.46 | 0.88 | 0.63 | 1.97 |
| 79 | ApoL-I | Haptoglobin-related protein | 0.72 | 0.70 | 0.39 | 1.81 |
a Composite score (i.e. sum of individual separation correlation scores).
Fig. 7.

Examples of co-migration patterns across the three separation techniques. The normalized peptide counts (same numbers shown in the heat maps in Figs. 4–6) have been plotted across the fractions for each separation technique for select protein pairs determined to have a high co-separation score. A, fibrinogen α-chain (blue) and fibrinogen β-chain (red). B, ApoA-I (blue) and apoA-II (red). C, ApoL-I (blue) and haptoglobin-related protein (red) (i.e. previously known components of trypanosome lytic factor (see text)). Data are averages from three donors. Error bars were not included for clarity.
DISCUSSION
Dong et al. (22) elegantly demonstrated the utility of an orthogonal separation analysis aimed at identifying protein–protein interactions. They first separated an E. Coli cell lysate via gel filtration chromatography. The fractions were pooled at regular intervals, concentrated, and then applied to an anion exchange column to separate proteins according to ionic character. Some of these fractions were then analyzed with MS to track the protein elution patterns. A total of 103 nonribosomal proteins were detected. Of these, 35 were constituents of 13 known protein complexes that had been previously identified with tandem affinity purification approaches. 95% of these were found to co-elute with known complex component partners. A similar idea was used to isolate subpopulations of centrosomal proteins separated by sucrose gradient ultracentrifugation (30). These studies demonstrate the utility of co-separation approaches for identifying unknown protein-interaction networks.
In this study, we took a parallel, rather than sequential, approach and added a third separation technique, isoelectric focusing, to further reduce the incidence of coincidental co-migration of proteins. We considered using density gradient ultracentrifugation in our analysis but eventually ruled it out because in preliminary experiments (a) we detected only about half as many lipid-associated proteins as detected with the other methods and (b) we consistently noted that the high salt concentrations and g-forces necessary for the separation altered the proteomic composition of the fractions, particularly with regard to some complement components, apoE, and the apoCs. This is consistent with previous reports of alterations of HDL composition by certain ultracentrifugation methods (31, 32). The use of the CSH allowed us to take advantage of multiple, non-density-based separation techniques, and this study represents the first proteomic descriptions of human plasma lipoproteins separated by means of anion-exchange chromatography and preparative IEF. The widely varying protein and lipid profiles resulting from each technique allowed us to track the co-migration of protein pairs across the separations and identify protein pairs that may exist on specific lipoprotein subparticles.
The highest scoring protein pairs involved fibrinogen, which is composed of two sets of three polypeptide subunits (α, β, and γ) held together by disulfide linkages (33). Our identification of this well-known complex offers confidence in the approach. Several more such “positive controls” were found high on the list, including multiple chains of the immunoglobulin G (IgG) complex (34) at #5, #8, #12, etc.; the known assembly of inter-α-trypsin inhibitor heavy chains H1 and H2 at #14 (35); and the non-covalent association of the complement factor C8 α- and β-subunits (36) at #9. Among distinct proteins (i.e. those that are not subunits of each other), we also recapitulated known interactions. For example, apoA-I and apoA-II (#3), the two highest abundance proteins in density-defined HDL, have been shown to reside together on HDL particles (37). In addition, we also confirmed the interaction of apoL-I and haptoglobin-related protein in the trypanosome lytic factor fraction of HDL (38). The list also includes associations between various members of the complement pathway (numbers 18, 23, and 39). However, the bulk of pairings on this list represent potentially novel protein–protein interactions. It will be important to begin to biochemically confirm these associations through immunological techniques and eventually assign functional relevance to these relationships.
Taking our analysis a step further, Fig. 8 shows a network representation of the top 3% (80) of the putative protein pairings identified in this study. Each node represents a protein that was tracked in all three separation techniques. The edges connecting them reflect a co-separation correlation score of greater than 1.80 between the indicated proteins. Several isolated networks are present that could be envisioned to represent compositions of lipoprotein particles beyond simple pairings of proteins. The three fibrinogen subunits formed a clear subnetwork with apoB-100, suggesting that fibrinogen can preferentially associate with LDLs, an assertion that may explain their known co-precipitation during hemorrheological interventions for the treatment of acute vascular occlusion (39). As expected, multiple subunits of IgG were also found to associate in a subnetwork. IgG has been identified in several proteomic studies of lipoproteins (15) and persisted during the CSH treatment performed here. Other subnetworks included (a) paraoxonase with prothrombin and vitronectin and (b) several components of the complement system, including inter-α-trypsin inhibitor heavy chains H1 and H2 (but not H3 and H4) and complement components C4-b, C1s, and C5. Most of the other proteins identified fell into a larger network. Of these, about 11 proteins seemed to form centers with at least seven connections to other proteins (in orange in Fig. 8). These included apoA-II, apoM, apoA-IV, extracellular matrix protein 1, alpha-1β-glycoprotein, α-2-antiplasmin, hemopexin, inter-α-trypsin inhibitor 4, heparin cofactor 2, complement factor B, and histidine-rich glycoprotein.
Fig. 8.

Network of the top-scoring pairs identified in this analysis. The protein pairs scoring in the top 3% (80 proteins with composite scores above 1.80) are shown as a network as represented by the Cytoscape software package using the organic layout (43). Network “hubs” that exhibit seven or more connections to other proteins are shown in orange. Other subnetworks are shown in distinct colors.
Although space limitations preclude a detailed discussion of each of these identified associations, some are worth noting in the context of our hypothesis that phospholipids act as a platform for the assembly of specific proteins in order to facilitate their function. The subnetwork shown in pink in Fig. 8 contains complement C1s, which is important in the formation of the C1 complex, a serine protease responsible for the cleavage and subsequent activation of various downstream components in the classical complement cascade (40). One immediate downstream target is C4, cleavage of which produces the C4b fragment, which itself has further downstream effects in the complement cascade with direct consequences on inflammation and tissue injury. Thus, the presence of both of these factors on the same lipoprotein particle is biologically plausible, as the molecules would not have to diffuse in three dimensions prior to interacting. Even more interesting is recent work suggesting that inter-α-trypsin inhibitor (IaI) is a complement-activation inhibitor that may directly interact with several complement components, at least in murine models (41). Given that the IaI H1 and H2 subunits were also present in this subnetwork, one can imagine a lipoprotein particle that contains at least two active complement components that are held in check by IaI. Indeed, the vWA domains on the IaI heavy chains may directly bind to C4 (41), perhaps preventing its cleavage by C1. Given the potentially disastrous consequences of unchecked complement activation, the co-localization of key complement factors and their relevant inhibitors on specific PL platforms in the plasma may allow for optimal and timely regulation of the pathway.
Although our analysis has identified many potential protein pairings in PL-containing plasma particles, there are some potential limitations of our approach. First, levels of protein detection by the mass spectrometer varied among the different methods. If a given protein was not detected in one of the three methods, it was dropped from the analysis. Thus, our data may miss potential interactions, particularly among lower abundance proteins. Second, the heterogeneous nature of lipoproteins resulted in a reduction of the apparent resolution of each separation technique with respect to any given protein. Indeed, even high-resolution techniques such as two-dimensional electrophoresis show wide size and charge heterogeneity when probed for individual HDL proteins (18). This may have led to some false positives in Table I that resulted from the coincidental co-elution of proteins. However, we believe that our use of three orthogonal separation techniques minimized this issue.
As our analysis did not use density-based separations of plasma lipoproteins, we refrained from associating specific protein networks with density-centric definitions of lipoproteins (i.e. HDL, LDL, etc.). However, it is well known from proteomics studies on ultracentrifugally isolated lipoproteins that most of the proteomic diversity lies in the HDL density range (1.063–1.210 g/ml) (42). Given that most of the protein pairs identified in Table I are known to reside primarily in this density range, our data challenge the notion that HDL is essentially a single entity with numerous interchanging protein components. The specific co-separation of certain protein pairs, or even networks of proteins, suggests that some HDL subspecies exist that maintain stable compositions likely to carry out distinct functions. This concept is highly significant with respect to developing pharmacological approaches designed to enhance HDL function. Under the single-entity scenario, it may be possible to raise HDL levels generically and thereby achieve improvements in a majority of its functions. However, if individual species perform distinct functions, it may be most advantageous to pharmacologically raise only certain ones to achieve benefits, particularly if altering other subspecies might have deleterious effects on other important functions, such as host defense. The recent failures of niacin (5) and two different cholesteryl ester transfer protein inhibitors (6, 7), all capable of significantly raising HDL cholesterol levels, have cast doubt on the idea that raising HDL cholesterol in the generic sense can translate to protection against CVD predicted from observational epidemiology. Given the huge functional and compositional pleiotrophy in HDL, it may be that individuals have a “portfolio” of HDL subspecies that are individually tasked to different functions across lipid metabolism, inflammation, anti-oxidation, and host defense. In such a case, it may be wise to specifically identify and target those subspecies that are most relevant to CVD protection. Thus, continued exploration of the functional and compositional heterogeneity of HDL, and indeed of all lipoproteins, is needed.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the invaluable assistance of Lance Morton for developing custom software and macros for dealing with the large data sets.
Footnotes
* This work was supported by NIH grants HL104136 (W.S.D.) and HL111829 (J.L.L.). A.S.S. was supported by a Procter Scholar Grant from Cincinnati Children's Hospital Medical Center. S.M.G. was supported by a pre-doctoral fellowship from the Great Rivers Affiliate of the American Heart Association.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CH
- cholesterol
- CSH
- calcium silica hydrate
- CVD
- cardiovascular disease
- HDL
- high-density lipoprotein
- IaI
- inter-α-trypsin inhibitor
- IEF
- isoelectric focusing
- LDL
- low-density lipoprotein
- MS
- mass spectrometry
- PL
- phospholipid
- VLDL
- very low-density lipoprotein.
REFERENCES
- 1. Grundy S. M., Cleeman J. I., Merz C. N., Brewer H. B., Jr., Clark L. T., Hunninghake D. B., Pasternak R. C., Smith S. C., Jr., Stone N. J. (2004) Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Arterioscler. Thromb. Vasc. Biol. 24, e149–e161 [DOI] [PubMed] [Google Scholar]
- 2. Mackness B., Mackness M. (2012) The antioxidant properties of high-density lipoproteins in atherosclerosis. Panminerva Med. 54, 83–90 [PubMed] [Google Scholar]
- 3. Bandeali S., Farmer J. (2012) High-density lipoprotein and atherosclerosis: the role of antioxidant activity. Curr. Atheroscler. Rep. 14, 101–107 [DOI] [PubMed] [Google Scholar]
- 4. Kontush A., Chapman M. J. (2012) High-density Lipoproteins: Structure, Metabolism, Function and Therapeutics, Wiley & Sons, New York [Google Scholar]
- 5. Boden W. E., Probstfield J. L., Anderson T., Chaitman B. R., Desvignes-Nickens P., Koprowicz K., McBride R., Teo K., Weintraub W. (2011) Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 365, 2255–2267 [DOI] [PubMed] [Google Scholar]
- 6. Barter P. J., Caulfield M., Eriksson M., Grundy S. M., Kastelein J. J., Komajda M., Lopez-Sendon J., Mosca L., Tardif J. C., Waters D. D., Shear C. L., Revkin J. H., Buhr K. A., Fisher M. R., Tall A. R., Brewer B. (2007) Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 357, 2109–2122 [DOI] [PubMed] [Google Scholar]
- 7. Schwartz G. G., Olsson A. G., Abt M., Ballantyne C. M., Barter P. J., Brumm J., Chaitman B. R., Holme I. M., Kallend D., Leiter L. A., Leitersdorf E., McMurray J. J., Mundl H., Nicholls S. J., Shah P. K., Tardif J. C., Wright R. S. (2012) Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 367, 2089–2099 [DOI] [PubMed] [Google Scholar]
- 8. Austin M. A., King M. C., Vranizan K. M., Newman B., Krauss R. M. (1988) Inheritance of low-density lipoprotein subclass patterns: results of complex segregation analysis. Am. J. Hum. Genet. 43, 838–846 [PMC free article] [PubMed] [Google Scholar]
- 9. Austin M. A., Breslow J. L., Hennekens C. H., Buring J. E., Willett W. C., Krauss R. M. (1988) Low-density lipoprotein subclass patterns and risk of myocardial infarction. JAMA 260, 1917–1921 [PubMed] [Google Scholar]
- 10. Kontush A., Chantepie S., Chapman M. J. (2003) Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler. Thromb. Vasc. Biol. 23, 1881–1888 [DOI] [PubMed] [Google Scholar]
- 11. Gordon S. M., Deng J., Lu L. J., Davidson W. S. (2010) Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J. Proteome Res. 9, 5239–5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Asztalos B. F., Sloop C. H., Wong L., Roheim P. S. (1993) Two-dimensional electrophoresis of plasma lipoproteins: recognition of new apo A-I-containing subpopulations. Biochim. Biophys. Acta 1169, 291–300 [DOI] [PubMed] [Google Scholar]
- 13. Cheung M. C., Segrest J. P., Albers J. J., Cone J. T., Brouillette C. G., Chung B. H., Kashyap M., Glasscock M. A., Anantharamaiah G. M. (1987) Characterization of high density lipoprotein subspecies: structural studies by single vertical spin ultracentrifugation and immunoaffinity chromatography. J. Lipid Res. 28, 913–929 [PubMed] [Google Scholar]
- 14. Heinecke J. W. (2009) The HDL proteome: a marker—and perhaps mediator—of coronary artery disease. J. Lipid Res. 50 Suppl, S167–S171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shah A. S., Tan L., Lu L. J., Davidson W. S. (2013) The proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 54, 2575–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoofnagle A. N., Heinecke J. W. (2009) Lipoproteomics: using mass spectrometry-based proteomics to explore the assembly, structure, and function of lipoproteins. J. Lipid Res. 50, 1967–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shiflett A. M., Bishop J. R., Pahwa A., Hajduk S. L. (2005) Human high density lipoproteins are platforms for the assembly of multi-component innate immune complexes. J. Biol. Chem. 280, 32578–32585 [DOI] [PubMed] [Google Scholar]
- 18. Asztalos B. F., Schaefer E. J. (2003) High-density lipoprotein subpopulations in pathologic conditions. Am. J. Cardiol. 91, 12E–17E [DOI] [PubMed] [Google Scholar]
- 19. Davidson W. S., Silva R. A., Chantepie S., Lagor W. R., Chapman M. J., Kontush A. (2009) Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 29, 870–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gordon S., Durairaj A., Lu J., Davidson W. S. (2010) High-density lipoprotein proteomics: identifying new drug targets and biomarkers by understanding functionality. Curr. Cardiovasc. Risk Rep. 4, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y. (2011) The tandem affinity purification technology: an overview. Biotechnol. Lett. 33, 1487–1499 [DOI] [PubMed] [Google Scholar]
- 22. Dong M., Yang L. L., Williams K., Fisher S. J., Hall S. C., Biggin M. D., Jin J., Witkowska H. E. (2008) A “tagless” strategy for identification of stable protein complexes genome-wide by multidimensional orthogonal chromatographic separation and iTRAQ reagent tracking. J. Proteome Res. 7, 1836–1849 [DOI] [PubMed] [Google Scholar]
- 23. Hirowatari Y., Yoshida H., Kurosawa H., Doumitu K. I., Tada N. (2003) Measurement of cholesterol of major serum lipoprotein classes by anion-exchange HPLC with perchlorate ion-containing eluent. J. Lipid Res. 44, 1404–1412 [DOI] [PubMed] [Google Scholar]
- 24. Markwell M. A., Haas S. M., Bieber L. L., Tolbert N. E. (1978) A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 87, 206–210 [DOI] [PubMed] [Google Scholar]
- 25. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 26. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 27. Lacroix R., Dignat-George F. (2012) Microparticles as a circulating source of procoagulant and fibrinolytic activities in the circulation. Thromb. Res. 129 Suppl 2, S27–S29 [DOI] [PubMed] [Google Scholar]
- 28. Kunitake S. T., La Sala K. J., Kane J. P. (1985) Apolipoprotein A-I-containing lipoproteins with pre-beta electrophoretic mobility. J. Lipid Res. 26, 549–555 [PubMed] [Google Scholar]
- 29. Noble R. P. (1968) Electrophoretic separation of plasma lipoproteins in agarose gel. J. Lipid Res. 9, 693–700 [PubMed] [Google Scholar]
- 30. Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 [DOI] [PubMed] [Google Scholar]
- 31. Stahlman M., Davidsson P., Kanmert I., Rosengren B., Boren J., Fagerberg B., Camejo G. (2008) Proteomics and lipids of lipoproteins isolated at low salt concentrations in D2O/sucrose or in KBr. J. Lipid Res. 49, 481–490 [DOI] [PubMed] [Google Scholar]
- 32. Kunitake S. T., Kane J. P. (1982) Factors affecting the integrity of high density lipoproteins in the ultracentrifuge. J. Lipid Res. 23, 936–940 [PubMed] [Google Scholar]
- 33. McKee P. A., Mattock P., Hill R. L. (1970) Subunit structure of human fibrinogen, soluble fibrin, and cross-linked insoluble fibrin. Proc. Natl. Acad. Sci. U.S.A. 66, 738–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Capra J. D., Hopper J. E. (1976) Comparative studies on monotypic IgM lambda and IgG kappa from an individual patient—III. The complete amino acid sequence of the VH region of the IgM paraprotein. Immunochemistry 13, 995–999 [DOI] [PubMed] [Google Scholar]
- 35. Malki N., Balduyck M., Maes P., Capon C., Mizon C., Han K. K., Tartar A., Fournet B., Mizon J. (1992) The heavy chains of human plasma inter-alpha-trypsin inhibitor: their isolation, their identification by electrophoresis and partial sequencing. Differential reactivity with concanavalin A. Biol. Chem. Hoppe Seyler 373, 1009–1018 [DOI] [PubMed] [Google Scholar]
- 36. Steckel E. W., York R. G., Monahan J. B., Sodetz J. M. (1980) The eighth component of human complement. Purification and physicochemical characterization of its unusual subunit structure. J. Biol. Chem. 255, 11997–12005 [PubMed] [Google Scholar]
- 37. Cheung M. C., Albers J. J. (1984) Characterization of lipoprotein particles isolated by immunoaffinity chromatography. Particles containing A-I and A-II and particles containing A-I but no A-II. J. Biol. Chem. 259, 12201–12209 [PubMed] [Google Scholar]
- 38. Raper J., Fung R., Ghiso J., Nussenzweig V., Tomlinson S. (1999) Characterization of a novel trypanosome lytic factor from human serum. Infect. Immun. 67, 1910–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaeger B. R., Marx P., Pfefferkorn T., Hamann G., Seidel D. (1999) Heparin-mediated extracorporeal LDL/fibrinogen precipitation—H.E.L.P.—in coronary and cerebral ischemia. Acta Neurochir. Suppl. 73, 81–84 [DOI] [PubMed] [Google Scholar]
- 40. Schroeder H., Skelly P. J., Zipfel P. F., Losson B., Vanderplasschen A. (2009) Subversion of complement by hematophagous parasites. Dev. Comp. Immunol. 33, 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwartz G. G., Olsson A. G., Ballantyne C. M., Barter P. J., Holme I. M., Kallend D., Leiter L. A., Leitersdorf E., McMurray J. J., Shah P. K., Tardif J. C., Chaitman B. R., Duttlinger-Maddux R., Mathieson J. (2009) Rationale and design of the dal-OUTCOMES trial: efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am. Heart J. 158, 896–901 [DOI] [PubMed] [Google Scholar]
- 42. Vaisar T. (2012) Proteomics investigations of HDL: challenges and promise. Curr. Vasc. Pharmacol. 10, 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.