

Abstract
Microtubules dramatically change their dynamics and organization at the entry into mitosis. Although this change is mediated by microtubule-associated proteins (MAPs), how MAPs themselves are regulated is not well understood. Here we used an integrated multi-level approach to establish the framework and biological significance of MAP regulation critical for the interphase/mitosis transition. Firstly, we applied quantitative proteomics to determine global cell cycle changes in the profiles of MAPs in human and Drosophila cells. This uncovered a wide range of cell cycle regulations of MAPs previously unidentified. Secondly, systematic studies of human kinesins highlighted an overlooked aspect of kinesins: most mitotic kinesins suppress their affinity to microtubules or reduce their protein levels in interphase in combination with nuclear localization. Thirdly, in-depth analysis of a novel Drosophila MAP (Mink) revealed that the suppression of the microtubule affinity of this mitotic MAP in combination with nuclear localization is essential for microtubule organization in interphase, and phosphorylation of Mink is needed for kinetochore-microtubule attachment in mitosis. Thus, this first comprehensive analysis of MAP regulation for the interphase/mitosis transition advances our understanding of kinesin biology and reveals the prevalence and importance of multi-layered MAP regulation.
Microtubules are universally found in eukaryotic cells and are involved in diverse processes including cell division, polarity, and intracellular transport. A striking feature of microtubules is that they change their dynamics and organization depending on cellular contexts. Proteins that interact with microtubules, collectively called microtubule-associated proteins (MAPs),1 are considered to play a major role in determining microtubule dynamics and organization.
Although MAPs in general lack recognizable sequence motifs, many MAPs from various sources have been successfully identified by means of biochemical purification followed by mass spectrometry (1–4). However, functional analysis is more problematic, as hundreds of MAPs can interact with microtubules. In addition, multiple MAPs have functional redundancy (5–7), making their biological function often difficult to determine, which results in their importance being grossly underappreciated. Furthermore, it is challenging to understand how MAPs collectively determine the diverse organization and dynamics of microtubules in different cells.
One of the most dramatic changes of microtubule organization is found at the transition from interphase to mitosis. During mitosis, microtubules are much more dynamic and are organized into a dense bipolar structure, the spindle, whereas microtubules in interphase are less dynamic and are arranged in a radial array. This transition is rapid and is thought to reflect mainly a change in the activities of both motor and nonmotor MAPs (8); however, we do not have sufficient knowledge of how MAPs themselves are regulated. It is crucial to identify and understand the regulation of MAPs whose activities change in the cell cycle, and how they collectively change microtubule dynamics and organization. Misregulation of such MAPs could interfere with chromosome segregation or cell polarity and potentially contribute to oncogenesis (9). Also, this misregulation can be used to elucidate important functions that are masked due to functional redundancy.
We hypothesize that some proteins bind to microtubules only during mitosis and are released from microtubules in interphase. The binding of such proteins to spindle microtubules in mitosis could collectively trigger the formation of the functional spindle, and, of equal importance, removing such proteins from microtubules at the mitotic exit could be essential for disassembling the spindle and proper organization and/or function of interphase microtubules. Conversely, some proteins may bind to microtubules specifically during interphase. No studies have been reported that systematically identify proteins whose microtubule-binding activities change between interphase and mitosis.
Here we report a combined approach integrating three levels of analyses to gain insights into how MAPs are regulated as a whole to drive microtubule reorganization at the transition between interphase and mitosis. Firstly, we applied proteomics to determine the quantitative change of the global MAP profile between mitosis and interphase in both human and Drosophila cells. Secondly, we systematically analyzed the human kinesin superfamily for cell cycle localization in relation to microtubule association to gain insight into the general principle of MAP regulation in the cell cycle. Thirdly, we focused on one novel Drosophila MAP to understand the molecular mechanism and biological significance of MAP regulation. This integrated approach has provided the framework of MAP regulation critical for the interphase/mitosis transition.
EXPERIMENTAL PROCEDURES
Molecular and Protein Techniques
Gateway molecular cloning technology was used to generate entry and expression clones of Mink protein. The destination vectors pAWG and pAGW were used for the expression of Mink GFP N- or C-terminus fusion proteins under the actin5C promoter. Mink truncations were created using four different methods. C-terminal regions were generated using the gateway cloning system. N-terminal regions of MINK were created via the introduction of premature stop codons using the Quick Change XLII site-directed mutagenesis kit (Agilent, Santa Clara, USA). Internal deletions were created in two ways. Firstly, an additional EcoRI cleavage site was introduced at the end of the region to be deleted. Digestion with EcoRI and self-ligation generated the desired deletions. Secondly, samples underwent PCR amplification of the gateway expression clone with primers flanking the deletion region and carrying an enzyme restriction site. Restriction digestion and self-ligation resulted in the desired plasmid. This second method with the inclusion of the human PKIα NES coding sequence within the forward primer was also used for the generation of NES fusion proteins. All Mink expression plasmids were sequenced to confirm that no undesired mutations were introduced.
Cell Culture
HeLa cells were cultured in custom serum-free Dulbecco's modified Eagle's medium without l-arginine or l-lysine (Athenaes, Baltimore, MD) for 15 to 21 passages. Medium was supplemented with either 12C- or 13C-arginine and lysine at 0.12 mm and 0.26 mm, respectively (Cambridge Isotopes Laboratories, Tewksbury, USA). To create mitotically enriched cell populations, cultures were incubated with 0.1 μg/ml nocodazole for 16 to 18 h.
Drosophila Schneider S2 cells used for Mink localization and transient transfection were cultured in Schneider media (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (Invitrogen) at 27 °C. Drosophila cells used for microtubule co-sedimentation were cultured in SILAC conditions for 10 to 25 passages (10). Media was made following Sigma Schneider media, omitting arginine, lysine, and yeast extract. Yeast extract at 200 mg/ml was dialyzed overnight using a pore size of 1200 MW against 0.9% NaCl. Media was supplemented with 10% dialyzed FCS (Invitrogen) and dialyzed yeast extract to 2 mg/ml and either 12C- or 13C-arginine and lysine at 3.4 mm and 2.8 mm, respectively. To create mitosis-enriched cell populations, cultures were incubated with 12 μg/ml colchicine for ∼24 h.
In order to observe the localization of GFP-Mink and its variants, expression plasmids were transfected using Effectene transfection reagent (Qiagen, Hilden, Germany) and were cultured for 48 h. Images of live transfected cells were obtained using a microscope (Axiovert, Carl Zeiss, Jena, Germany) attached to a spinning-disc confocal head (Yokogawa, Tokyo, Japan) controlled by Volocity (PerkinElmer Life Sciences) using a laser intensity of 3% and an exposure of 250 ms so expression levels could be assessed. The total GFP signal in a circle with a diameter three times that of the nucleus was measured for each cell, and the background signal was subtracted from it.
For RNA interference, S2 cells were treated with dsRNA corresponding to mink or the negative control β-lactamase gene as previously described (11). Primers used for Mink were forward CGACTCACTATAGGGAGAGGTCGCCAAGAAGCAGAA and reverse CGACTCACTATAGGGAGAGGTTCATCCAGGCCATCA.
Cytological Analysis
To determine the cell cycle localization of mammalian kinesins, cDNAs were obtained as IMAGE clones. We transiently transfected the fluorescently tagged kinesins using Effectene (Qiagen). HeLa Kyoto cell lines expressing GFP-Kif4a, GFP-CENPE, and GFP-Kif23 (mouse) were obtained from Mitocheck. Images were acquired on a DeltaVision Core microscope (Applied Precision, Spokane, Washington) equipped with a CoolSnap HQ2 camera. For live cell imaging, 6 to 12 z sections were acquired at steps of 0.5 to 1 μm using a 60× 1.3 numerical aperture Olympus U-Plan Apochromat objective lens with 1 × 1 binning.
Immunostaining of S2 cells and embryos was carried out as previously described (11, 12). Antibodies against α-tubulin (mouse DM1A, 1:250, Sigma), GFP (rabbit 1:500, Molecular Probes, Eugene, OR), histone-H3-phsophate (rabbit 1:500, Upstate, Charlotteville, VA), CLIP190 (sheep 1:50; see Ref. 11), Mink peptide (rabbit 1:50), and Mink (rabbit 1:50) were used as primary antibodies. Cells were visualized using a Zeiss Axioplan 2 microscope, and images were captured with a charge-coupled device camera (Hamamatsu, Hamamatsu, Japan) controlled by Openlab 2.2.1 software (PerkinElmer Life Sciences) or using a Plan-Apochromat lens (63×, 1.4 numerical aperture, Zeiss, Jena, Germany) attached to an Axiovert 200 m (Zeiss) with a confocal scan head (LSM5 Exciter, Zeiss).
The mitotic index was determined by counting the frequencies of phospho-H3(Ser10) positive cells. In the case of S2 cells, we immunostained an aliquot from the exact same cell population used for each SILAC experiment, as the mitotic index can vary greatly in each experiment. For HeLa cells, in which robust mitotic arrest can be induced, we immunostained cells cultured in exactly the same conditions, although they were not the identical cell population used for the SILAC experiments.
Microtubule Co-sedimentation
Arrested “light” and cycling “heavy” SILAC cultured cells were mixed 1:1 based on cell number and lysed in BRB80x buffer (80 mm Pipes-KOH pH 6.8, 1 mm MgCl2, 1 mm Na3EGTA, 1 mm DTT, 1 mm PMSF, Roche Complete EDTA-free protease inhibitor mixture tablets) supplemented with 15 mm Na3VO4, 10 mm p-nitrophenyl phosphate, and 1 μm okadaic acid. After one 20-min incubation at 30 °C, the lysate was cleared of debris using ultracentrifugation. The extract was split into two portions, and taxol-stabilized microtubules (7.5 μg/108 cells) were added to one and colchicine to the other. Microtubules were pre-polymerized from tubulin (>99% pure; Cytoskeleton, Denver, CO). Microtubules were incubated for 30 min at room temperature to allow MAP binding and then pelleted twice through a 50% sucrose cushion.
For immunoblot verification of the SILAC results, microtubule co-sedimentation was carried out as described above using interphase and mitotic extracts separately. Protein extracts and microtubule pellets were separated via SDS-PAGE and analyzed via imunoblotting using antibodies against CAMSAP2 (rabbit, 1:1000; Proteintech 17880–1-AP, Chicago, IL), Eg5 (mouse, 1:100, Santa Cruz A-1, Santa Cruz Biotechnology, Santa Cruz, CA), Kid (rabbit, 1:100; Bioscience A310–366A), and Aurora B (rabbit, 1:1000; Abcam Ab13824, Cambridge, UK).
Proteomics/Mass Spectrometry
Microtubule pellets were fractionated using a NuPAGE 4–12% Bis-Tris gel with MOPS running buffer and visualized using a Colloidal Blue Staining Kit (Invitrogen). Gel regions, excluding tubulin, were excised and the proteins were reduced/alkylated and in-gel digested using trypsin following standard protocols (13). Peptides were desalted using C18 StageTips (14) and loaded directly onto a column needle self-packed with ReproSil-Pur C18-AQ material (3 μm; Dr Maisch, GmbH, Beim Bruckle, Germany) at a flow rate of 0.7 μl/min. S2 peptides were separated using an Agilent 1100 binary nanopump LC system with an HTC Pal autosampler (CTC) using a two-step linear gradient of 0%-20%-80%-80% B over either 35, 4, and 2 min or 75, 13, and 10 min, depending on sample complexity (mobile phases were (A) 5% acetonitrile, 0.5% acetic acid and (B) 99.5% acetonitrile, 0.5% acetic acid). Peptides were eluted into an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) using a flow rate of 300 nl/min and a spray voltage of 1.8 kV. HeLa peptides were separated using ultra-high-performance liquid chromatography (nanoACQUILTY, Waters, Milford, MA) using a multiple-step linear gradient of 1%-5%-32%-35%-85%-85% B in 1, 119, 5, 5, and 5 min. (Mobile phases were (A) 0.1% formic acid and (B) 99.9% acetonitrile, 0.1% formic acid.) Peptides were eluted into an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) using a flow rate of 600 nl/min and a spray voltage of 2.2 kV.
Database Search and Data Analysis
Peak lists were generated from Thermo Raw files using MaxQuant (versions 1.2.2.5 and 1.3.0.5 (15)) with default SILAC parameters and with the addition of quantitation using unique peptides. Default MaxQuant parameters are described below. The fixed modification considered was carbamidomethylation (C), with variable modifications of oxidation (M), acetyl (Protein-N term), and phospho (STY) sites also considered. A minimum peptide length of six and score of 0 were used. No protein cutoff was employed after MaxQuant analysis. Peptide and protein false discovery rates were set at 0.01. The mass tolerance for precursor ions was 20 ppm for the first search and 6 ppm for the second search, and the mass tolerance for fragment ions was 0.5 Da. The maximum number of modifications per peptide was five, the maximum precursor charge was 7, the maximum number of labeled amino acids was three, and two missed cleavages were allowed. Searches were conducted against the UniProt/sProt Drosophila melanogaster database (November 30, 2011, with 18,780 entries) or human database (July 13, 2012, with 36,849 entries, and April 3, 2012, with 35,961 entries). To avoid the distortion of normalization by light-labeled peptides derived from exogenously added microtubules, we calculated the median H/L protein ratio after excluding exogenous proteins (tubulin and trypsin) and contaminants and used this median H/L ratio for normalization. All raw data from MaxQuant are included in supplemental Tables S4–S6.
To map the protein interaction network, the 270 identified proteins were submitted to the STRING database, version 9.0 (16). The STRING confidence score was set at >0.9 (the highest confidence), and experiments, databases, and textmining were used for active prediction methods. Phosphopeptides were identified and quantified using MaxQuant (versions 1.2.2.5 and 1.3.0.5) (15). Absolute phosphate occupancy was calculated from the phosphopeptide I/M (Interphase/Mitosis) ratio, the non-phosphopeptide I/M ratio, and the protein I/M ratio (17). Relative phosphate occupancies were calculated as the I/M ratio of a phosphopeptide divided by the I/M ratio of the corresponding proteins, without requiring the I/M ratio of the corresponding non-phosphopeptide.
Analysis of Nuclear Protein Loss
In order to analyze the loss of nuclear proteins during extract preparation, the first pellet after cell lysis was resuspended in the same volume as the extract. Equal volumes of extract and pellet were fractionated via SDS-PAGE and analyzed by means of immunoblotting using Dbr1 (rabbit; Proteintech 16019–1-AP), Pol II (rabbit; Santa Cruz N20), and PCNA (rabbit, 1:100; Santa Cruz FL-261) primary antibodies and Licor anti-rabbit 680 (1:20,000) and anti-mouse 800 (1:20,000) secondary antibodies. Secondary antibodies were visualized using Licor Odyssey (v3.0.30). Images were processed using Volocity (PerkinElmer Life Sciences). Brightness and contrast were uniformly adjusted without changing image features, and band intensities were measured.
To analyze whether nuclear protein ratios were skewed by protein loss during extract preparation in our SILAC experiments, we looked at the I/M ratios of proteins (HeLa SILAC 1) assigned as “nuclear” in Uniprot. All proteins were binned into ratio groups, and the distribution of nuclear proteins was analyzed.
To check nuclear disruption during extract preparation, cells were lysed in two ways: using 0.5% Triton X-100 with homogenization to maintain nuclei, and by sonication as used in the preparation of extract for microtubule co-sedimentation. Cells were stained before and after lysis with DAPI (1:50), and images were taken with an Axioplan 2 microscope (Zeiss) attached to a charge-coupled device camera (Hamamatsu) controlled by OpenLab 2.2.1 software (Improvision, Warwick, UK) and were processed with Photoshop (Adobe, San Jose, CA).
RESULTS
SILAC-based Quantitative Proteomics to Identify Cell-cycle-regulated MAPs
Our first goal was to systematically identify MAPs whose microtubule-binding activity or protein level differed in mitosis and interphase in human and Drosophila cells. We applied a SILAC-based quantitative proteomics approach (18) (Fig. 1A) to the human HeLa cell line and the Drosophila S2 cell line. One cell population labeled with light isotope (12C) using normal arginine and lysine was enriched for mitotic cells via incubation with a microtubule-destabilizing drug. An asynchronous population labeled with heavy isotope (13C) was used as the interphase-dominated population, although it contained various cell cycle stages. The mitotic index of HeLa cells routinely reaches roughly 90%, compared with about 5% in an asynchronous population, and that of S2 cells reached 20% to 25%, compared with 2% in particular experiments. This relatively inefficient mitotic enrichment in S2 cells is due to the general nature of Drosophila cell lines and slow growth in the SILAC medium, but the enrichment achieved was predicted to be sufficient for identifying mitosis-specific MAPs.
Fig. 1.

A quantitative proteomics approach to identify cell-cycle-regulated MAPs. A, the schematic outline of the SILAC-based method used to identify cell-cycle-regulated MAPs. Mitosis-enriched cells labeled with light isotopes were mixed with interphase-dominated cells labeled with heavy isotopes. Taxol-stabilized microtubules were added to soluble extracts and sedimented through a sucrose cushion. The microtubule pellet was analyzed by a mass spectrometer. B, C, I/M ratios of individual proteins in microtubule fractions derived from interphase- and mitosis-dominated cell populations in two independent experiments in human HeLa cells (B) or in Drosophila S2 cells (C). Proteins that reproducibly gave I/M ratios of >2 or <0.5 are labeled with names.
Equal numbers of differentially labeled cells enriched in mitosis and interphase were mixed, and a soluble cell lysate was prepared. We confirmed that nuclei were disrupted before centrifugation and that nuclear proteins were not specifically lost during lysate preparation (supplemental Fig. S1). Taxol-stabilized microtubules were added and co-sedimented through a sucrose cushion. In the absence of microtubules, few proteins were sedimented. Throughout the purification, protein phosphorylation was maintained using our optimized method (supplemental Fig. S2). Proteins in the microtubule pellet were digested with trypsin after fractionation by an SDS gel. Resulting peptides were identified and quantified via mass spectrometry (LC-MS/MS).
Identification of Cell-cycle-regulated MAPs from Humans
We successfully quantified 472 and 456 proteins in microtubule fractions from two independent microtubule co-sedimentation experiments in HeLa cells. Of these, 280 proteins were common to both (Fig. 1B, supplemental Table S1). The median ratio of peptides labeled with heavy isotopes to those labeled with light isotopes in microtubule fractions was calculated for each protein. This ratio (which we call the I/M ratio here) represents the protein abundance in microtubule fractions from the interphase-dominated population relative to fractions from the mitosis-enriched one. In this definition, a low I/M ratio means a mitosis-specific (or enriched) MAP, and a high I/M ratio means an interphase-specific (or enriched) MAP. In this assay, these cell cycle differences can be due to changes in the proteins' affinity to microtubules and/or in protein abundance. Results from the two independent biological experiments were well correlated (R2 = 0.64; Fig. 1B), indicating good reproducibility.
Among the 280 proteins successfully quantified in both experiments, 32 proteins reproducibly showed clear characteristics of mitosis-enriched MAPs (I/M ratios less than 0.5 in both experiments), and 12 proteins reproducibly showed characteristics of interphase-enriched MAPs (I/M ratios greater than 2 in both experiments). Those proteins that show clear cell cycle changes in microtubule association include proteins whose microtubule-binding activities are known to change between mitosis and interphase, such as Eg5 (mitosis specific; I/M = 0.2) (19) and MAP4 (interphase specific; 3.3) (20). These results confirm that we have successfully developed a method that systematically identifies MAPs with cell-cycle-dependent microtubule binding. In addition, we identified proteins previously unknown to be regulated in the cell cycle. These include C2orf44 (mitosis specific; I/M = 0.1), which was previously uncharacterized, and two members of the Patronin family, CAMSAP2 and CAMSAP3 (interphase specific; 5.2/5.1), which are known to stabilize minus ends of non-centrosomal microtubules in epithelial cells (21). Our SILAC results were confirmed by immunoblotting of selected proteins with high or low I/M ratios (supplemental Fig. S3). The large majority of MAPs have I/M ratios close to 1. These I/M ratios appear to show some correlation between the two experiments. This raises the interesting possibility that many MAPs show a subtle change in their microtubule association between mitosis and interphase.
As phosphorylation is the major means of protein regulation (17, 22), we also estimated the absolute phosphate occupancy of 78 sites on 25 proteins in the microtubule fraction in mitosis and interphase (supplemental Fig. S4, supplemental Table S3). As no phosphopeptide enrichment was used, these likely represent a very small fraction of all phosphorylations. Most have a high occupancy (>50%) in mitosis and/or interphase (supplemental Fig. S4A). Consistent with previous reports (17), the phosphorylation at most sites increased in mitosis and matched Cdk target motifs ([pS/pT]P), some of which further matched the polo box binding motif (S[pS/pT]P). Others matched the Aurora or Polo target motif ([R/K]X[pS/pT], [D/E]X[pS/pT]). Multiple phosphopeptides found on the same protein often showed widely varied ratios between mitosis and interphase and are potentially phosphorylated by different kinases (supplemental Fig. S2B). These observations suggest multi-layered phospho-regulation on MAPs.
A change in protein–protein interaction is another way to regulate protein function and localization. To gain insight into cell cycle regulation, known protein–protein interactions were mapped among the 280 MAPs reproducibly quantified in our study with reference to their I/M ratios (supplemental Fig. S4C). A total of 308 interactions involving 146 MAPs were found. Proteins that form a complex tended to have similar I/M ratios, but some interactions were found between MAPs with very different I/M ratios. These interactions between a cell-cycle-regulated MAP and a more constitutive MAP would potentially be targets of cell cycle regulation and include the interaction between the kinesin Kif23 (I/M = 0.3) and the constitutive spindle protein Septin 9 (I/M = 1.4) (23).
Cell Cycle Localization of Human Kinesins
Our proteomics study reproducibly quantified changes in microtubule association between mitosis and interphase (I/M ratios) for 18 human kinesins (16 kinesin complexes). Kinesins play crucial roles in various steps of mitosis (24), but previous human studies have varied a lot in focus and depth, with comparative studies being very successful for Drosophila kinesins (6, 25). To systematically assess and compare the cell cycle localizations of these human kinesins expressed in cycling cells side by side, 13 kinesins including all those with low I/M ratios were tagged with GFP and expressed in HeLa cells. We observed distinct cell-cycle-dependent localizations (Figs. 2A and 2B) that were largely consistent with previous reports on individual kinesins (26–40). During the course of this study, the first systematic comparative study was reported for human kinesins, and it is consistent with our results (41).
Fig. 2.

Cell cycle localization of human kinesins quantified in the study. Localization of kinesins in metaphase (A) or in interphase (B). GFP-tagged kinesins expressed in HeLa cells were detected in live cells. C, the name, alternative name, kinesin family, and I/M ratios for each kinesin from the two experiments and a summary of the protein localization in mitosis and interphase. MT, microtubule; KLC, kinesin light chain; KHC, kinesin heavy chain. A square bracket indicates that a complex formed (KHC/KLC, Kif3a/Kif3b). ND, not determined in this study.
As our proteomics results predicted, two kinesins with very low I/M ratios, Kif15 (a kinesin-12 family member; I/M = ∼0.1) and Eg5 (a kinesin-5; I/M = 0.2), showed spindle microtubule association in metaphase and no association in interphase. Kif2c (a kinesin-13; I/M = 0.4) was enriched at spindle poles and kinetochores in metaphase and showed microtubule plus end association in interphase, which is unlikely to be detectable by means of microtubule co-sedimentation. Cenp-E (a kinesin 7; I/M = 0.5) showed spindle association in metaphase (kinetochore localization in prometaphase) but no microtubule association in interphase. Kinesin I (KHC/KLC, a kinesin-1; I/M = 0.7/0.7) and Kif14 (a kinesin-3; I/M = 0.4) showed no or very weak association with microtubules in both metaphase and interphase. Finally, Kif2a (a kinesin-13 with a relatively high I/M value of 0.9), expected to be a constitutive MAP based on our data, showed strong spindle pole association in metaphase and clear microtubule association in interphase.
Five kinesins, Kif18a (a kinesin-8; I/M = 0.2), KifC1 (a kinesin-14; I/M = 0.3), Kif23 (a kinesin-6; I/M = 0.3), Kif18b (a kinesin-8; I/M = 0.6), and Kid (a kinesin-10; I/M = 0.7), showed spindle microtubule association in metaphase and nuclear localization in interphase. Kif20a (a kinesin-6; I/M = 0.3) was also localized to the nucleus in interphase. It did not associate with the spindle in metaphase, but it associated with the spindle midzone in anaphase. Interestingly, most of these kinesins have low I/M ratio, revealing a general regulation (a combination of nuclear localization and a reduced affinity to microtubules) by which kinesin-microtubule association is prevented during interphase.
Overall, many kinesins are prevented from interacting with microtubules in interphase through the suppression of microtubule binding, often in combination with nuclear localization.
Klp61F and the Novel Protein Mink Were Identified as Mitosis-specific MAPs in Drosophila
For Drosophila S2 cells, we successfully quantified 190 and 187 proteins in microtubule fractions from two independent experiments, and 121 proteins were common to both (supplemental Table S2). Furthermore, there was significant overlap with the proteome of MAPs purified from Drosophila embryos (2), confirming that a substantial proportion of identified proteins were genuine MAPs.
From S2 cells, three proteins, Klp61F (kinesin-5 family), CG11120, and CG3523, reproducibly gave low I/M ratios (<0.5) as estimated from multiple unique peptides in both experiments (Fig. 1C). Despite the low mitotic index achievable in S2 cells, we were able to identify a small set of mitotic MAPs. As previously published (6), our proteomics results confirmed that the microtubule-binding property of Klp61F is cell cycle regulated. We then focused on CG11120, as it had not been previously characterized. We called the CG11120 protein Mink (named after a nocturnal animal only active at a certain time of day, and also as an abbreviation of mitotic spindle and nuclear protein; see below) and studied it in detail to uncover the role of its cell cycle regulation.
We first examined the subcellular localization of Mink in S2 cells. Transiently expressed GFP-tagged Mink associated uniformly with spindle microtubules during mitosis and was restricted to the nucleus in interphase (Fig. 3A). We also observed the localization via immunostaining using newly generated antibodies (Fig. 3B, supplemental Fig. S5). Mink localized to the nucleus in interphase, to spindle microtubules from prometaphase to anaphase, and again to the nucleus from telophase onward. The depletion of Mink by RNAi abolished these antibody signals (Fig. 3C). This established that endogenous Mink localizes to spindle microtubules and the interphase nucleus.
Fig. 3.
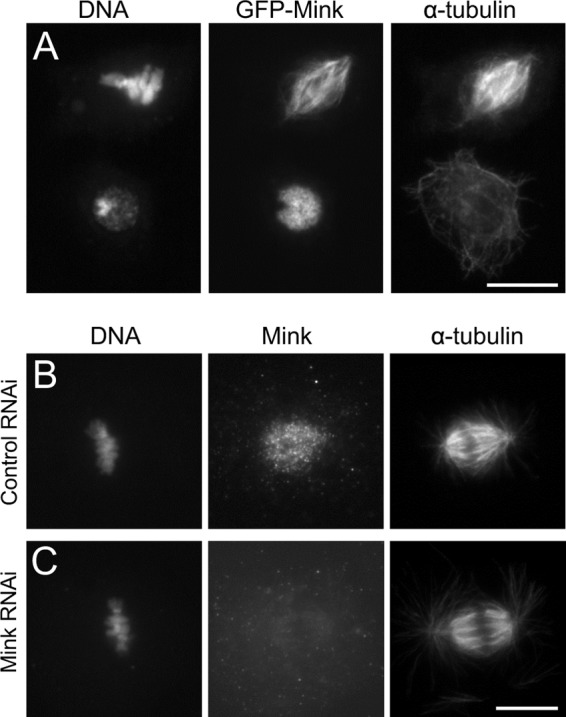
Mink localizes to spindle microtubules in mitosis and the nucleus in interphase. A, transiently expressed GFP-Mink localizes to spindle microtubules in mitosis and to the interphase nucleus in S2 cells. Bar = 10 μm. B, C, immunostaining of a mitotic S2 cell using antibodies against Mink, α-tubulin after control, or Mink RNAi. Bar = 10 μm.
Mink Has a Region Homologous to Human NuSAP that Is Separate from Nuclear and Spindle Localization Regions
To identify the domain structure of Mink, we first examined its sequence similarity to other proteins using BLAST (Fig. 4A). A region close to the C terminus (550–600) had weak but significant similarity to proteins in various animals, including human NuSAP (nucleolar spindle-associated protein). Like Drosophila Mink, human NuSAP was identified as a mitosis-specific MAP in our HeLa study and localizes to the spindle in mitosis and the nucleus in interphase (42). It is required for proper spindle formation (42, 43). An additional region at the C terminus (664–748) has some similarity to uncharacterized proteins in mosquitos. Further regions share similarities with proteins in other Drosophila species, including the N-terminal region (33–169).
Fig. 4.

The domain structure of Mink. A, conserved regions of Mink and a protein sequence alignment among Mink, human NuSAP, and a mosquito protein (Anopheles gambiae AGAP004345). Identical residues are marked. B, a summary of the subcellular localization of truncated Mink proteins. “I” and “M” indicate interphase and mitosis. “Nuc,” “chr,” and “MT” indicate localization to nucleus, chromosomes, and microtubules, respectively. “–” indicates diffuse localization. C, immunostaining of mitotic S2 cells transiently expressing GFP-fused truncated Mink proteins using antibodies against GFP and α-tubulin. The inset shows a merged image with Mink in green, tubulin in red, and DNA in blue. Bar = 10 μm. D, a summary of the subcellular localization of truncated Mink proteins. Localization locations in parentheses indicate weak localization. E, immunostaining of mitotic S2 cells transiently expressing GFP-fused truncated Mink proteins using antibodies against GFP and α-tubulin. Bar = 10 μm. F, the C-terminal region of Mink directly interacts with microtubules in vitro. MBP-fused Mink (548–754) or MBP alone was produced in bacteria and incubated with tubulin. After microtubules had been polymerized using taxol and GTP, microtubules were spun down. For the control, taxol and GTP were omitted. Supernatants (sup) and pellets (ppt) were analyzed via Western blot using an antibody against MBP.
To determine which regions are responsible for Mink localization, truncated proteins were tagged with GFP and expressed in S2 cells (Fig. 4B). We found that two regions, the N-terminal 109 residues (1–109) and the C-terminal 132 residues, are redundantly responsible for nuclear localization in interphase (Fig. 4B). The C-terminal 132 residues (623–754) are also sufficient to associate with spindle microtubules in mitosis. Finer truncations of the C-terminal region showed that a small conserved region (707–748) was crucial for both localizations (Figs. 4D and 4E). Furthermore, a cryptic signal that directs association with the chromosomes was also found. The N-terminal 404 residues (1–404) were diffuse in mitotic cells, while the N-terminal 171 residues (1–171) were associated with the chromosomes (Fig. 4C). Western blots confirmed that the truncated proteins had roughly the predicted sizes in transfected cells, and the expression of the proteins was comparable between constructs. Furthermore, the expression of key truncations was examined via microscopy at the single-cell level, confirming that the defects were not due to differences in expression levels (supplemental Fig. S6). In summary, the truncation study identified an N-terminal nuclear localization region, a cryptic chromosome association signal, a NuSAP homology region, and a C-terminal nuclear and spindle localization region.
Microtubule-binding Activity of Mink Is Suppressed in Interphase Cells
To test the direct binding of Mink to microtubules, the region sufficient to localize to spindle microtubules in cells (residues 548–754) was produced as an MBP-fusion protein in bacteria. Taxol-stabilized microtubules (polymerized from pure tubulin) were incubated in the presence of MBP-Mink and co-sedimented. As controls, MBP alone was tested for microtubule co-sedimentation, and sedimentation was carried out without microtubules. Immunoblots showed that MBP-Mink (548–754), but not MBP alone, was able to bind microtubules (Fig. 4F). These results demonstrated that Mink can directly bind to microtubules without other proteins or eukaryote-specific post-translational modifications.
As our SILAC data indicated that Mink does not bind to microtubules in interphase, the microtubule-binding activity of Mink must be suppressed. Nuclear localization of Mink prevented us from cytologically confirming reduced microtubule-binding activity in interphase cells. To overcome this, we forced Mink to localize to the cytoplasm in interphase by fusing a nuclear export signal (NES) from human PKIα. Although it is still more concentrated in the nucleus, GFP-NES-Mink localizes considerably to the cytoplasm (Figs. 5A and 5B). Cytoplasmic GFP-NES-Mink failed to associate with microtubules in interphase, but it associated normally with spindle microtubules in mitosis. This confirmed that the microtubule affinity of Mink is suppressed in interphase.
Fig. 5.

Misregulation of Mink disrupts microtubule organization. A, a summary of the subcellular localization of NES-fused Mink and truncated proteins. “MT” in bold and followed by an asterisk indicates that Mink localized to microtubules and induced abnormal microtubule organization. B, confocal images of immunostained GFP-Mink, GFP-NES-Mink, GFP-NES-Mink (623–754), and GFP-NES-Mink (Δ109–171). GFP-NES-Mink localized in the cytoplasm without associating with microtubules. The spindle binding domain (623–754) fused with GFP-NES in the cytoplasm associated with microtubules and induced microtubule bundling. Bar = 10 μm. C, microtubule stability in cells with or without expression of GFP-NES-Mink (623–754). Cells were immunostained for α-tubulin after incubation with colchicine. D, the frequencies of cells with considerable amounts of microtubules after incubation with colchicine. Cells expressing GFP-NES-Mink (623–754) and cells not expressing GFP (–) in the same transfected cell population were counted for microtubule morphology. The differences are significant between them (p < 0.001, Chi-square test).
Association of Mink with Microtubules in Interphase Induces Microtubule Bundling
To determine how Mink microtubule association is suppressed in interphase, the spindle localization domain (623–754 residues) was forced to localize to the cytoplasm via the addition of the same NES (GFP-NES-Mink (623–754); Figs. 5A and 5B). This protein in the cytoplasm was able to bind to microtubules in interphase, demonstrating that the N-terminal region suppresses the microtubule-binding activity of the C-terminal region during interphase. In addition, extensive microtubule bundles were observed in the interphase cytoplasm at a high frequency (85%, versus 8% for untransfected cells; p < 0.001), even though the protein expression level was comparable to that of the full-length protein. As this truncated protein also associated with unbundled microtubules, the microtubule bundling itself was not a cause of this association. To assess microtubule stability, cells were treated with colchicine overnight, which induces the depolymerization of nearly all microtubules in untransfected cells. In contrast, most cells expressing the NES-fused spindle-binding domain retained a considerable amount of microtubules, indicating the hyperstabilization of microtubules (Fig. 5C).
In summary, in interphase Mink is confined in the nucleus away from microtubules, and its microtubule-binding activity is suppressed. These two mechanisms together ensure that Mink is prevented from interfering with microtubules during interphase.
Misregulation of Mink Disrupts Microtubule Organization
In order to define the region responsible for cell cycle regulation of the Mink microtubule-binding activity, further deletions were tested for the localization and microtubule bundling in interphase (Fig. 5A). NES-fused Mink lacking residues 57–232 bound to interphase microtubules and induced bundling, whereas NES-fused Mink lacking a smaller region (109–172) bound to interphase microtubules to some degree but did not induce extensive microtubule bundling. This indicates that this smaller region (109–172) is essential for suppressing microtubule binding in interphase, and flanking regions (57–109, 171–232) are required for the bundling activity. Spindle localization was not affected by these deletions.
Mink lacking these flanking regions (57–109 or 171–232) showed normal suppression of microtubule binding in interphase (Fig. 5A). Strikingly, in mitosis, the expression of either of them resulted in severe disruption of chromosome alignment and spindle organization (Figs. 6A and 6C). The spindle was long, thin, and often bent, and chromosomes were distributed all over the spindle. These two regions may act together to modulate the activity of Mink. This phenotype may be caused by antimorphic/dominant-negative proteins or hypermorphic/hyperactive proteins. To distinguish these possibilities, Mink protein was depleted from S2 cells by RNAi. Immunostaining showed a loss of Mink but no significant abnormalities relative to a control, including late mitotic or cytokinesis defects (supplemental Fig. S7). This suggests that deletion of the flanking regions misregulates or hyperactivates the Mink protein (in other words, it induces a gain-of-function effect), rather than making the protein antimorphic/dominant negative to inhibit the endogenous Mink protein.
Fig. 6.
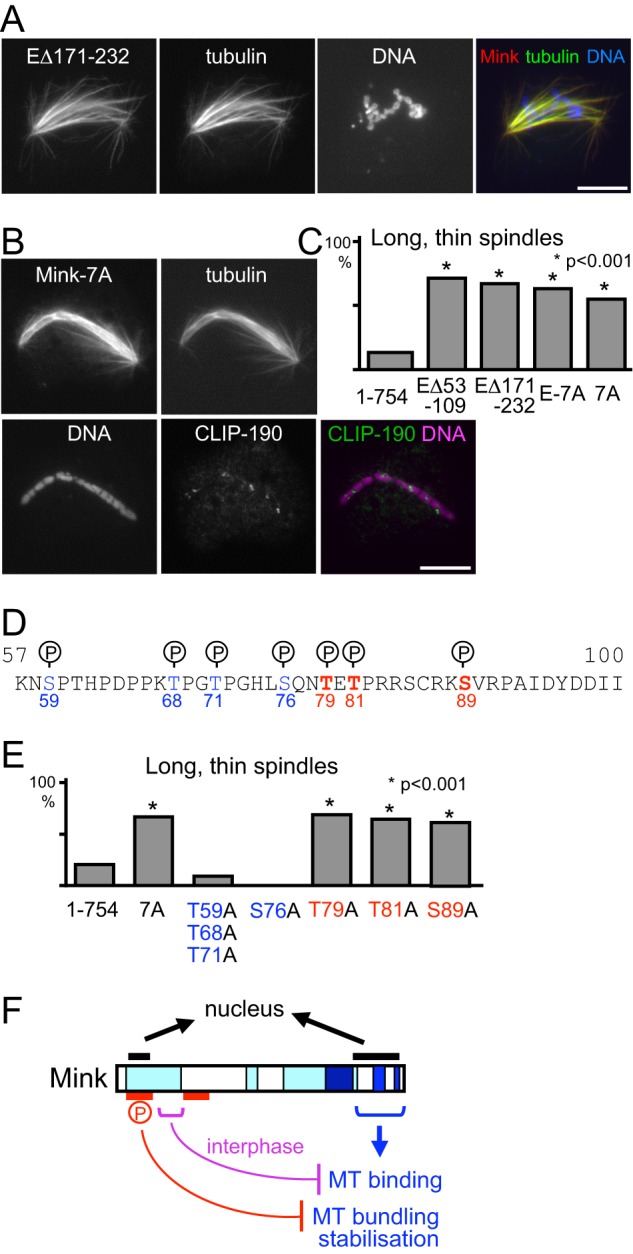
Phosphorylation of Mink is essential for kinetochore attachment to microtubules. A, B, Mink with a small deletion (A) or non-phosphorylatable Mink (B) disrupts kinetochore attachment to microtubules. Seven phosphorylated residues of Mink were substituted to alanine on non-phosphorylatable Mink (Mink-7A). S2 cells were immunostained for α-tubulin and GFP. The CLIP-190 protein that accumulates to unattached kinetochores was also stained for Mink-7A expressed cells. C, the frequencies of long, thin spindles among mitotic cells expressing two small deletions, non-phosphorylatable and the full-length wild-type Mink. The differences are significant between the full-length wild-type Mink and these Mink mutants (p < 0.001, Chi-square test). D, seven phosphorylation sites of Mink identified in global phosphoproteomics studies. Alanine mutations of red-colored residues induce the spindle defect, whereas those of blue-colored residues do not. E, the frequencies of long, thin spindles among mitotic cells expressing Mink with various phosphorylation sites substituted by alanine. The differences are significant between full-length wild-type Mink and the mutants containing T79A, T81A, or T89A, but not ones without these mutations (p < 0.001, Chi-square test). F, multi-layered regulation of Mink. Mink contains two nuclear localization signals. The C-terminal region directly binds to microtubules in mitosis but is suppressed by an N-terminal region (purple) during interphase. Flanking regions (red) and phosphorylation are required for the suppression of microtubule bundling and stabilization.
Phosphorylation Suppresses Mink Activity
Phosphorylation is considered to be a major mechanism of protein regulation. Previous global phosphoproteomics studies in Drosophila (44–46) identified a total of 25 sites of phosphorylation on Mink in S2 cells or embryos. There is a cluster of seven phosphorylation sites at residues 59–89 (Fig. 6D), and deletion of this region causes mitotic defects. To determine the role of phosphorylation at these sites, we mutated these residues to generate an alanine non-phosphorylatable mutant (Mink-7A or E-7A). The localization of Mink was not affected by the mutations (Fig. 6B), but chromosome alignment and spindle morphology were severely disrupted (Figs. 6B and 6C), as seen with the deletion of this region. The presence or absence of NES did not affect this phenotype. To test kinetochore attachment to microtubules, these cells were immunostained with an anti-CLIP-190 antibody. In normal cells, unattached kinetochores accumulate CLIP-190, as it is loaded onto kinetochores independently of microtubules and is removed along kinetochore microtubules (11). Misaligned chromosomes showed a discrete accumulation of CLIP-190, showing that the non-phosphorylatable Mink disrupts kinetochores' attachment to microtubules (Fig. 6B).
In order to precisely identify the residues responsible for the mitotic defects, these phosphorylation sites were mutated either singly or in combination. We found that a single non-phosphorylatable mutation at T79, T81, or S89 resulted in the mitotic defects (Figs. 6D and 6E). T81 and S89 match to strict consensus target motifs of Cdc2 kinase (TPxR) and Aurora B kinase (RxSφ), respectively, while T79 matches a broadened consensus target motif of Polo kinase (QxT) (47). This suggests that all of these sites must be phosphorylated by different kinases in order to suppress the activity of Mink during mitosis.
DISCUSSION
In this study we used a combination of three complementary approaches, starting from a global, proteome-wide analysis and progressing to a deeper analysis of the kinesin superfamily, and finally undertaking an in-depth study of a single MAP, Mink. First we applied quantitative proteomics to human and Drosophila cells to identify global quantitative changes of MAP profiles from interphase to mitosis. We then chose the kinesin superfamily for deeper analysis and found that the microtubule association of most human mitotic kinesins is suppressed in interphase by a combination of reduced affinity to microtubules and nuclear localization. Finally we focused our study on one novel Drosophila MAP, Mink, and revealed that reduced microtubule affinity and nuclear localization in interphase act as a “double locking” system to prevent this mitotic MAP from interfering with interphase microtubules. We also identified multiple phosphorylations on Mink that are essential for mitosis.
For this study, we established a SILAC-based proteomic method to measure the quantitative differences in profiles of MAPs between interphase and mitosis. A similar approach was previously used to identify cytokinesis-specific MAPs (3). Rather than separately preparing microtubule fractions, we first mixed cells from mitosis and interphase before isolating microtubule fractions. This pre-purification mixing provided high reproducibility and reliability as demonstrated by the high correlation of two independent experiments. One of the key optimizations here is the maintenance of protein phosphorylation, confirmed by phospho-specific antibodies and a high occupancy of phosphorylation sites. Other modifications were not actively maintained in this study, but the method can be easily adapted in the future. One limitation of our study was a failure to capture proteins that exclusively bind to dynamic ends, as taxol-stabilized microtubules were used.
Our study uncovered new, previously unknown cell cycle regulation of MAPs, in addition to MAPs already known to be cell cycle regulated. These include both mitosis-specific and interphase-specific MAPs. It should be noted that most of these MAPs are likely to change their affinity to microtubules between interphase and mitosis, but some may change in protein abundance too. Although post-translational modifications of tubulins are known to affect the binding of MAPs (48, 49), our assay could not have detected these proteins, as MAPs were co-sedimented with the same in vitro assembled microtubules in the same mixed cell lysates.
Furthermore, our results suggested an interesting feature of cell cycle regulation of the microtubule interactome. The regulation of MAP association to microtubules does not serve simply to turn “on or off” a small number of proteins. Instead, the microtubule associations of many MAPs change, to varying degrees, between mitosis and interphase. The contribution of each may be small, but MAPs may act collectively on microtubules to change their dynamics and organization. Single MAPs are often phosphorylated at multiple sites by different kinases with different dynamics, indicating that multi-layers of phospho-regulations generally act on MAPs to fine-tune their activity. Mapping of known interactions among MAPs showed that a MAP tends to interact with other MAPs with similar I/M ratios. This raises the possibility that many MAPs interact with microtubules as a complex, through other MAPs or cooperatively with other MAPs. There are also some interactions between MAPs with divergent I/M ratios, which could be modulated in a cell-cycle-dependent manner.
Among human MAPs quantified in our study, we further focused on the kinesin superfamily. Our determination of subcellular localization in mitosis and interphase combined with quantification of microtubule association provides valuable insight into the general regulatory mechanisms of human kinesins. Each kinesin shows a distinct pattern of localization and microtubule association, even among the same subfamily (for example, Kif18a and Kif18b). Interestingly, we found that a large proportion of human kinesins are prevented from binding to microtubules during interphase by the suppression of microtubule-binding activity, often in combination with nuclear localization. This regulation is likely to be crucial for preventing mitotic MAPs from interfering with microtubule organization and intracellular transport in interphase.
We also identified a novel mitosis-specific MAP, Mink, in Drosophila. Mink has a region homologous to human NuSAP and is the only protein with significant homology to NuSAP in Drosophila. Despite their limited similarity, Mink and NuSAP share many common features. NuSAP is one of the mitosis-enriched human MAPs identified in our HeLa cell study. Both localize to spindle microtubules and the interphase nucleus, have a domain that associates with mitotic chromosomes, and have microtubule-bundling and -stabilizing activity (42, 50). NuSAP is required for proper formation of the mitotic spindle (42, 43), whereas our RNAi and mutant analyses did not reveal an essential role of Mink in spindle formation or cell division. This essential function of Mink may be masked by the presence of other proteins with a similar function or alternative pathways in Drosophila.
Truncation analysis revealed three layers of regulation on Mink protein (Fig. 6F): (i) nuclear localization during interphase mediated by two regions; (ii) suppression of microtubule binding during interphase, which is mediated by a small region in the N terminus (containing potential phosphorylation sites by Cdk and Aurora); and (iii) suppression of Mink activity by phosphorylation, which is essential for proper kinetochore attachment to microtubules in mitosis. We demonstrated that these separate modes of regulation are essential for microtubule organization and function. The nuclear localization of Mink and the suppression of its microtubule binding during interphase are essential for preventing Mink from interfering with the microtubule network during interphase. We propose that these regulations act as a double-locking mechanism that ensures that Mink is kept away from interphase microtubules. This double-locking mechanism may be widespread, as we found that human kinesins with reduced microtubule-binding activity in interphase often show nuclear localization. Our study of Mink revealed an underappreciated role of cell cycle regulation by which mitotic MAPs are removed from microtubules at mitotic exit, and we propose that a double-locking mechanism is one way to ensure this.
Furthermore, th suppression of Mink activity by phosphorylation is essential for the proper attachment of kinetochores to microtubules. We have identified three sites, all of which must be phosphorylated in order for Mink to remain repressed. Cdc2/Cdk1 and Aurora B are kinases that potentially phosphorylate these sites. It is possible that these kinases together phosphorylate Mink at kinetochores during early mitosis to suppress the microtubule-bundling and -stabilization activity that interferes with kinetochore–microtubule attachment.
It was previously shown that the microtubule-binding activity of NuSAP is higher in the cytokinesis phase than in the M-phase (3). Consistently, Cdk1 phosphorylation of NuSAP at two sites reduces microtubule binding (51). In contrast, our proteomics results showed that microtubule binding of NuSAP is higher in M-phase (when Cdk1 is active) than in interphase. These results can be explained by a two-step activation: the initial activation in early mitosis, and further activation in cytokinesis. The Cdk1 phosphorylation site found by Chou et al. (51) may be equivalent to the Cdk1 site of Mink we identified, as our non-phosphorylatable mutation of Mink shows a prometaphase defect similar to the one caused by the non-phosphorylatable mutation of NuSAP (51).
In conclusion, our integrated approach, combining quantitative proteomics, a comparative study on kinesins, and an in-depth study of Mink, provides valuable insights into the general regulatory principles of MAPs. Firstly, our determination of cell cycle changes in the microtubule interactome provides a global view of MAP regulation and reveals that the microtubule association of many MAPs is regulated in the cell cycle to varying degrees. Secondly, MAPs often receive multi-layered cell cycle regulation that combines nuclear localization, a change in microtubule affinity, and phosphorylation by different kinases. These regulations work together to ensure that MAPs are activated and suppressed at the right time and the right place. Our study provides an important foundation for understanding how microtubule organization and dynamics are changed in the cell cycle through the collective regulation of the microtubule interactome.
Supplementary Material
Acknowledgments
We thank members of Rappsilber, Welburn, and Ohkura laboratories for support and discussion. We especially thank Sally Beard and Karen Wills for assisting in experiments, Jimi-Carlo Bukowski-Wills for critical reading of this manuscript, and Iain Cheeseman for support for the kinesin work. We also thank the Drosophila Genomic Research Center and Bloomington Drosophila Stock Center for providing reagents. HMS received a BBSRC PhD studentship.
Footnotes
* This work was funded by The Wellcome Trust (081849, 092076, 098030, 084229, 092076, 091020) and Cancer Research UK (C40377/A12840).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- GFP
- green fluorescent protein
- MAP
- microtubule-associated protein
- MBP
- maltose binding protein
- NES
- nuclear export signal
- SILAC
- stable isotope labeling with amino acids in cell culture.
REFERENCES
- 1. Sauer G., Körner R., Hanisch A., Ries A., Nigg E. A., Silljé H. H. (2005) Proteome analysis of the human mitotic spindle. Mol. Cell. Proteomics 4, 35–43 [DOI] [PubMed] [Google Scholar]
- 2. Hughes J. R., Meireles A. M., Fisher K. H., Garcia A., Antrobus P. R., Wainman A., Zitzmann N., Deane C., Ohkura H., Wakefield J. G. (2008) A microtubule interactome: complexes with roles in cell cycle and mitosis. PLoS Biol. 6, e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ozlü N., Monigatti F., Renard B. Y., Field C. M., Steen H., Mitchison T. J., Steen J. J. (2010) Binding partner switching on microtubules and aurora-B in the mitosis to cytokinesis transition. Mol. Cell. Proteomics 9, 336–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gache V., Waridel P., Winter C., Juhem A., Schroeder M., Shevchenko A., Popov A. V. (2010) Xenopus meiotic microtubule-associated interactome. PLoS One 5, e9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teng J., Takei Y., Harada A., Nakata T., Chen J., Hirokawa N. (2001) Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell Biol. 155, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goshima G., Vale R. D. (2005) Cell cycle-dependent dynamics and regulation of mitotic kinesins in Drosophila S2 cells. Mol. Biol. Cell. 16, 3896–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Groen A. C., Maresca T. J., Gatlin J. C., Salmon E. D., Mitchison T. J. (2009) Functional overlap of microtubule assembly factors in chromatin-promoted spindle assembly. Mol. Biol. Cell. 20, 2766–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manning A. L., Compton D. A. (2008) Structural and regulatory roles of nonmotor spindle proteins. Curr. Opin. Cell Biol. 20, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gordon D. J., Resio B., Pellman D. (2012) Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 13, 189–203 [DOI] [PubMed] [Google Scholar]
- 10. Bonaldi T., Straub T., Cox J., Kumar C., Becker P. B., Mann M. (2008) Combined use of RNAi and quantitative proteomics to study gene function in Drosophila. Mol. Cell. 317, 762–772 [DOI] [PubMed] [Google Scholar]
- 11. Dzhindzhev N. S., Rogers S. L., Vale R. D., Ohkura H. (2005) Distinct mechanisms govern the localisation of Drosophila CLIP-190 to unattached kinetochores and microtubule plus ends. J. Cell Sci. 118, 3781–3790 [DOI] [PubMed] [Google Scholar]
- 12. Cullen C. F., Deak P., Glover D. M., Ohkura H. (1999) Mini spindles: a gene encoding a conserved microtubule associated protein required for the integrity of the mitotic spindle in Drosophila. J. Cell Biol. 146, 1005–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 14. Rappsilber J., Ishihama Y., Mann M. (2003) Stop and go extraction tips for matrix assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 15. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 16. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561-D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 18. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 19. Sawin K. E., Mitchison T. J. (1995) Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc. Natl. Acad. Sci. U.S.A. 92, 4289–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shiina N., Tsukita S. (1999) Mutations at phosphorylation sites of Xenopus microtubule-associated protein 4 affect its microtubule-binding ability and chromosome movement during mitosis. Mol. Biol. Cell. 10, 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka N., Meng W., Nagae S., Takeichi M. (2012) Nezha/CAMSAP3 and CAMSAP2 cooperate in epithelial-specific organization of noncentrosomal microtubules. Proc. Natl. Acad. Sci. U.S.A. 109, 20029–20034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spiliotis E. T., Kinoshita M., Nelson W. J. (2005) A mitotic septin scaffold required for mammalian chromosome congression and segregation. Science 307, 1781–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wordeman L. (2010) How kinesin motor proteins drive mitotic spindle function: lessons from molecular assays. Semin. Cell Dev. Biol. 21, 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goshima G., Vale R. D. (2003) The roles of microtubule-based motor proteins in mitosis: comprehensive RNAi analysis in the Drosophila S2 cell line. J. Cell Biol. 162, 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mayr M. I., Hümmer S., Bormann J., Grüner T., Adio S., Woehlke G., Mayer T. U. (2007) The human kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression. Curr. Biol. 17, 488–498 [DOI] [PubMed] [Google Scholar]
- 27. Stumpff J., von Dassow G., Wagenbach M., Asbury C., Wordeman L. (2008) The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev. Cell. 14, 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wordeman L., Mitchison T. J. (1995) Identification and partial characterization of mitotic centromere-associated kinesin, a kinesin-related protein that associates with centromeres during mitosis. J. Cell Biol. 128, 95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andrews P. D., Ovechkina Y., Morrice N., Wagenbach M., Duncan K., Wordeman L., Swedlow J. R. (2004) Aurora B regulates MCAK at the mitotic centromere. Dev. Cell. 6, 253–268 [DOI] [PubMed] [Google Scholar]
- 30. Moore A. T., Rankin K. E., von Dassow G., Peris L., Wagenbach M., Ovechkina Y., Andrieux A., Job D., Wordeman L. (2005) MCAK associates with the tips of polymerizing microtubules. J. Cell Biol. 169, 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Houliston E., Le Guellec R., Kress M., Philippe M., Le Guellec K. (1994) The kinesin related protein Eg5 associates with both interphase and spindle microtubules during Xenopus early development. Dev. Biol. 164, 147–159 [DOI] [PubMed] [Google Scholar]
- 32. Homma N., Takei Y., Tanaka Y., Nakata T., Terada S., Kikkawa M., Noda Y., Hirokawa N. (2003) Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 114, 229–239 [DOI] [PubMed] [Google Scholar]
- 33. Ganem N. J., Compton D. A. (2004) The KinI kinesin Kif2a is required for bipolar spindle assembly through a functional relationship with MCAK. J. Cell Biol. 166, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tokai N., Fujimoto-Nishiyama A., Toyoshima Y., Yonemura S., Tsukita S., Inoue J., Yamamoto T. (1996) Kid, a novel kinesin-like DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J. 15, 457–467 [PMC free article] [PubMed] [Google Scholar]
- 35. Kuriyama R., Kofron M., Essner R., Kato T., Dragas-Granoic S., Omoto C. K., Khodjakov A. (1995) Characterization of a minus end-directed kinesin-like motor protein from cultured mammalian cells. J. Cell Biol. 129, 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vanneste D., Takagi M., Imamoto N., Vernos I. (2009) The role of Hklp2 in the stabilization and maintenance of spindle bipolarity. Curr. Biol. 19, 1712–1717 [DOI] [PubMed] [Google Scholar]
- 37. Tanenbaum M. E., Macůrek L., Janssen A., Geers E. F., Alvarez-Fernández M., Medema R. H. (2009) Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr. Biol. 19, 1703–1711 [DOI] [PubMed] [Google Scholar]
- 38. Yen T. J., Li G., Schaar B. T., Szilak I., Cleveland D. W. (1992) CENP-E is a putative kinetochore motor that accumulates just before mitosis. Nature 359, 536–539 [DOI] [PubMed] [Google Scholar]
- 39. Gruneberg U., Neef R., Li X., Chan E. H., Chalamalasetty R. B., Nigg E. A., Barr F. A. (2006) KIF14 and citron kinase act together to promote efficient cytokinesis. J. Cell Biol. 172, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee Y. M., Kim E., Park M., Moon E., Ahn S. M., Kim W., Hwang K. B., Kim Y. K., Choi W., Kim W. (2010) Cell cycle-regulated expression and subcellular localization of a kinesin-8 member human KIF18B. Gene 466, 16–25 [DOI] [PubMed] [Google Scholar]
- 41. Maliga Z., Junqueira M., Toyoda Y., Ettinger A., Mora-Bermúdez F., Klemm R. W., Vasilj A., Guhr E., Ibarlucea-Benitez I., Poser I., Bonifacio E., Huttner W. B., Shevchenko A., Hyman A. A. (2013) A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat. Cell Biol. 15, 325–334 [DOI] [PubMed] [Google Scholar]
- 42. Raemaekers T., Ribbeck K., Beaudouin J., Annaert W., Van Camp M., Stockmans I., Smets N., Bouillon R., Ellenberg J., Carmeliet G. (2003) NuSAP, a novel microtubule associated protein involved in mitotic spindle organization. J. Cell Biol. 162, 1017–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vanden Bosch A., Raemaekers T., Denayer S., Torrekens S., Smets N., Moermans K., Dewerchin M., Carmeliet P., Carmeliet G. (2010) NuSAP is essential for chromatin induced spindle formation during early embryogenesis. J. Cell Sci. 123, 3244–3255 [DOI] [PubMed] [Google Scholar]
- 44. Bodenmiller B., Malmstrom J., Gerrits B., Campbell D., Lam H., Schmidt A., Rinner O., Mueller L. N., Shannon P. T., Pedrioli P. G., Panse C., Lee H. K., Schlapbach R., Aebersold R. (2007) PhosphoPep—a phosphoproteome resource for systems biology research in Drosophila Kc167 cells. Mol. Syst. Biol. 3, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gnad F., Ren S., Cox J., Olsen J. V., Macek B., Oroshi M., Mann M. (2007) PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhai B., Villén J., Beausoleil S. A., Mintseris J., Gygi S. P. (2008) Phosphoproteome analysis of Drosophila melanogaster embryos. J. Proteome Res. 7, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Santamaria A., Wang B., Elowe S., Malik R., Zhang F., Bauer M., Schmidt A., Silljé H. H., Körner R., Nigg E. A. (2011) The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol. Cell. Proteomics 10, M110.004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J., Verhey K. J. (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172 [DOI] [PubMed] [Google Scholar]
- 49. Dompierre J. P., Godin J. D., Charrin B. C., Cordelieres F. P., King S. J., Humbert S., Saudou F. (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ribbeck K., Groen A. C., Santarella R., Bohnsack M. T., Raemaekers T., Köcher T., Gentzel M., Görlich D., Wilm M., Carmeliet G., Mitchison T. J., Ellenberg J., Hoenger A., Mattaj I. W. (2006) NuSAP, a mitotic RanGTP target that stabilizes and cross-links microtubules. Mol. Biol. Cell 17, 2646–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chou H. Y., Wang T. H., Lee S. C., Hsu P. H., Tsai M. D., Chang C. L., Jeng Y. M. (2011) Phosphorylation of NuSAP by Cdk1 regulates its interaction with microtubules in mitosis. Cell Cycle 10, 4083–4089 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.