

Abstract
Pre-eclampsia (PE) is a serious complication of pregnancy with potentially life threatening consequences for both mother and baby. Presently there is no test with the required performance to predict which healthy first-time mothers will go on to develop PE. The high specificity, sensitivity, and multiplexed nature of selected reaction monitoring holds great potential as a tool for the verification and validation of putative candidate biomarkersfor disease states. Realization of this potential involves establishing a high throughput, cost effective, reproducible sample preparation workflow. We have developed a semi-automated HPLC-based sample preparation workflow before a label-free selected reaction monitoring approach. This workflow has been applied to the search for novel predictive biomarkers for PE.
To discover novel candidate biomarkers for PE, we used isobaric tagging to identify several potential biomarker proteins in plasma obtained at 15 weeks gestation from nulliparous women who later developed PE compared with pregnant women who remained healthy. Such a study generates a number of “candidate” biomarkers that require further testing in larger patient cohorts. As proof-of-principle, two of these proteins were taken forward for verification in a 100 women (58 PE, 42 controls) using label-free SRM. We obtained reproducible protein quantitation across the 100 samples and demonstrated significant changes in protein levels, even with as little as 20% change in protein concentration. The SRM data correlated with a commercial ELISA, suggesting that this is a robust workflow suitable for rapid, affordable, label-free verification of which candidate biomarkers should be taken forward for thorough investigation. A subset of pregnancy-specific glycoproteins (PSGs) had value as novel predictive markers for PE.
The identification of clinically relevant plasma biomarkers with diagnostic and/or predictive value continues to challenge the proteomics field. Whereas once the biomarker pipeline was described as a two part discovery and validation process, there is increasing consensus that an intermediate step is required in which the proteins identified in the discovery phase are technically verified in 50 to 200 samples. This verification step identifies false positives from the discovery phase and allows prioritization of proteins to be taken into large-scale clinical validation studies (1). Although commercial ELISA kits may be used in this phase, these are unavailable for many proteins, are expensive, and may lack specificity. In addition, sample requirements may be too high to perform ELISA on all candidates, especially if many proteins are identified as potential markers by low powered, high penetration discovery workflows.
Selected reaction monitoring (SRM)1 mass spectrometry has great potential as an alternative verification method (2–6) as it can be multiplexed, customized, and is highly specific. This potential has not been exploited to date, largely because of technical issues developing a low-cost, reproducible workflow encompassing plasma and serum preparation and LC/MS analysis with the capability to measure protein levels reproducible in hundreds of samples. With traditional stable isotope dilution SRM (SID-SRM), the high cost of accurately quantified, purified stable isotope encoded peptides or proteins may be prohibitive for the verification of multiple peptides from many proteins. Label-free relatively quantitative methods are increasingly popular in discovery proteomics but to a much lesser extent in targeted SRM studies (7, 8).
For any SRM method, sample preparation workflows must balance the extent of enrichment and fractionation to enable quantification of lower abundance proteins, against increased technical variability (which is influenced by the number of sample handling steps) and reduced multiplexed potential as a consequence of fractionating peptides from the protein of interest into several distinct fractions. It is also essential that the true technical variation in the workflow is quantitatively evaluated from freezer to MS analysis, rather than just the variation within the LC-SRM part of the experiment. As a paradigm for a label-free SRM assay, we developed our workflow and applied it to the verification of candidate biomarkers that indicate the risk of pre-eclampsia (PE).
PE affects 2–8% of pregnancies, and is characterized by hypertension and proteinuria, which may progress to severe maternal complications or death (9). Because delivery of the infant is the only effective intervention, a third of babies are born premature and fetal or newborn mortality is increased three- to 10-fold (10). Its complex etiology involves abnormal placentation, an altered immune response and a sensitized maternal vascular endothelium (11). Prediction of the condition in early pregnancy would allow prevention strategies, such as low dose aspirin, to be targeted to high risk women. In first-time pregnant women, a group particularly at risk, biomarkers continue to fall short of a test that would be useful or cost effective in clinical practice (12–14). Better-performing novel biomarkers are required.
The aim of this study was to identify candidate predictive biomarkers for PE and then develop a verification assay using mass spectrometry to determine whether these should be taken forward into more extensive and expensive validation studies. Initial discovery experiments were employed using a pooled sample iTRAQ approach using two different MS platforms to increase plasma proteome coverage. Among the set of proteins discovered, we then developed a label-free SRM assay for relative quantification of CXCL7 (Platelet basic protein; PBP) and members of the Pregnancy specific glycoprotein (PSG) family in a 100-sample set from the international SCreeningfOr Pregnancy Endpoints (SCOPE) study (www.scopestudy.net). Our workflow allowed the specificity and linearity of response for each peptide to be determined, along with true technical variability. Although absolute concentration and LOD/LOQ cannot be calculated using this approach, we aimed to test the hypothesis that a label-free SRM approach could provide a rapid, robust, and efficient screen of candidate plasma biomarkers.
EXPERIMENTAL PROCEDURES
A flow diagram describing the analytical steps is provided in Fig. 1.
Fig. 1.

Flowchart detailing the experiments performed in this study.
SCOPE Sample Collection
Early pregnancy plasma samples were acquired via the SCOPE Study (Australian and New Zealand Clinical Trials Registry ACTRN12607000551493), a prospective cohort study of nulliparous women. Full ethical approval was obtained and all women gave written informed consent. Full inclusion and exclusion criteria are described in North et al. (15). A research midwife interviewed and examined the participants at 14–16 and 19–21 weeks gestation. At the time of interview, data were entered on a web-accessed central database with a complete audit trail (MedSciNetAB) and pregnancy outcomes were prospectively tracked with data available in >99% of participants. PE was defined as systolic blood pressure ≥140 mm Hg or diastolic blood pressure ≥90 mm Hg, or both, on at least two occasions four hours apart after 20 weeks' gestation but before the onset of labor, or postpartum, with either proteinuria (24 h urinary protein ≥300 mg or spot urine protein:creatinine ratio ≥30 mg/mmol creatinine or urine dipstick protein ≥++) or any multisystem complication of pre-eclampsia. Women recruited in the Australian or New Zealand centers (n = 3182) were included in this study, of whom 178 (5.6%) developed PE. Venipuncture was performed at 14–16 weeks gestation according to a standardized protocol. Plasma samples were collected into EDTA-Vacutainer̈ tubes (Becton, Dickinson) centrifuged at 1500 × g at 4 °C for 10 min, plasma layer aliquotted and stored at −80 °C. Samples were processed within 4 h of collection. Plasma samples were defrosted on ice and maintained below 4 °C during experiments wherever possible. Using a customized software package, samples from women with early onset PE (EO-PE, delivery <34 weeks) and late onset PE (LO-PE, delivery ≥34 weeks) were selected. Controls were randomly selected from women who had an uncomplicated pregnancy (delivered at ≥37 weeks of an appropriately grown fetus in the absence of any medical or obstetric complications) at the same center as the cases. The sample numbers used in the different phases of the study are outlined in Fig. 1 and the demographic data for this population are shown in Table I.
Table I. Demographic data for discovery and verification cohorts. Values are median (range). Differences between groups assessed by Mann Whitney; values in bold represent (p < 0.005) cases vs controls. Twelve women in the EO-PE discovery sample set are also included in the EO-PE verification sample set, 1 woman in the discovery control group is also included in the verification control group (occurred by chance during random selection).
| Study characteristics | Discovery |
Verification |
|||
|---|---|---|---|---|---|
| Control n = 24 | Early-onset PE n = 12 | Control n = 42 | Early-onset PE n = 16 | Late-onset PE n = 42 | |
| Maternal characteristics | |||||
| Age (years) | 28 (14–40) | 30 (24–40) | 28 (17–35) | 30 (20–40) | 26 (16–40) |
| BMI | 25 (17–45) | 27 (21–36) | 25 (19–42) | 27 (21–36) | 26 (20–50) |
| Systolic BP (15w) | 104 (90–130) | 116 (100–140) | 102 (90–128) | 121 (102–142) | 110 (96–130) |
| Diastolic BP (15w) | 62 (50–74) | 74 (70–92) | 60 (48–80) | 76 (54–98) | 69 (56–86) |
| Maximum Systolic BP (mmHg) | 120 (100–132) | 172 (140–200) | 120 (100–136) | 176 (140–200) | 155 (105–220) |
| Maximum Diastolic BP (mmHg) | 75 (60–88) | 108 (90–125) | 70 (50–88) | 109 (90–140) | 100 (70–125) |
| Gestation at diagnosis (weeks+days) | 32 + 5 (24 + 1–33 + 5) | 31 + 5 (24 + 1–33 + 5) | 37 + 1 (34 + 3–41 + 2) | ||
| Birth outcomes | |||||
| Gestation at delivery (weeks+days) | 40 + 3 (38 + 2–41 + 6) | 32 + 5 (28 + 4–33 + 6) | 40 + 0 (37 + 6–41 + 6) | 32 + 4 (27 + 3–33 + 6) | 37 + 7 (34 + 3–41 + 2) |
| Birthweight (g) | 3700 (3080–4580) | 1380 (980–2660) | 3663 (2850–4530) | 1553 (740–2660) | 3063 (1635–4415) |
| Birthweight Centile | 54 (27–96) | 1 (0–94) | 60 (16–100) | 3 (0–94) | 31 (0–99) |
iTRAQ Discovery Experiments
A subset of plasma samples was used for the iTRAQ experiments. A pool of early onset PE cases (EO-PE; n = 12) and two control pools (n = 12 each) were created. A reference pool containing equal amounts of all samples was also made. The pooled plasma samples were prepared for relative quantification by iTRAQ using two parallel workflows as outlined in the supplementary material. Briefly, pooled plasma samples were immunodepleted using either the SepproÏgY 14 -SuperMix Liquid Chromatography Column system (Genway, San Diego, USA) or the Multiple Affinity Removal LC Column - Human 14 (MARS 14; Agilent, UK). Depleted plasma was concentrated and exchanged into 0.5 m TEAB, digested using trypsin and labeled using iTRAQ8-plex reagent (Applied Biosystems, UK) according to the manufacturer's instructions. The reference pool and case pools were processed in duplicate from start to finish, thus providing an assessment of the technical variability of the workflow. The control pools were processed in parallel and the variation between these two control pools was used to assess biological variability and define the range outside of which proteins were considered changed in abundance. iTRAQ-labeled samples were pooled and fractionated using high pH reversed phase chromatography (3 μm Extend-C18 column; 4.6 × 100 mm; Agilent, Santa Clara, CA). Fractions were then were subjected to reversed-phase peptide fractionation using a Acclaim PepMap 100 C18 column (3 μm, 1 mm × 150 mm; Dionex, CA, USA) with 5 μm Acclaim PepMap μ-Precolumn (300 μm × 5 mm; Dionex) run on a UltiMate pump (LC Packings, Amsterdam, The Netherlands) either online to a QStar XL qTOF (AB Sciex), or fractions were spotted onto a MALDI target and analyzed using a 5800 MALDI TOF-TOF (AB Sciex).
Data were interrogated using ProteinPilot version 3.0 (AB Sciex) using default settings and a ‘thorough’ search effort. Peptide identifications were made using the Paragon algorithm (16) searching against the International Protein Index for Human Proteins (version 3.59, 160,248 entries). A 95% confidence interval cut-off was used for significant peptides, and single peptide identifications were included in grouped protein identifications. Three data sets were produced. False discovery rate (FDR) was calculated by searching all peptide data against a concatenated database containing both forward and reversed protein sequences. To ensure data analyzed was of the highest quality, the data sets were filtered to remove low confidence (<95%) protein identifications. The FDR for the filtered IgY 14-Supermix QSTAR and 5800 data sets was 0.61% and 0.63% respectively and 1.63% for the MARS 14–5800 data set. An average relative quantification ratio (index sample:reference pool) was calculated for each protein using default ProteinPilot settings.
Candidate biomarkers were identified following the application of stringent criteria defined by assessing the mean fold change between technical replicates (two identical pools) and control samples (two biologically distinct pools). Proteins were only considered as candidate biomarkers where the protein was identified in all three data sets, reproducibly quantified in the technical replicate samples (≤ ±2 S.D. mean fold change) and where the average fold change was greater for case/control than control/control (> ±2 S.D. mean fold change).
SRM Methods
Plasma Immunodepletion
Plasma samples were thawed on ice and centrifuged at 10,000 × g for 10mins at 4 °C. Supernatants were removed to fresh tubes then vortexed. Forty microliters of plasma was diluted with 120 μl MARS A Buffer (Agilent, UK). Each sample was injected onto a 4.6 × 100 mm MARS Hu-14 immunodepletion column (Agilent) using the manufacturer's recommended method and proprietary buffers with collection into glass vials using a 1200 series liquid chromatography system (Agilent) with autosampler and fraction collector held at 4 °C.
Following a batch of immunodepletion runs (n = 24), 5 μl of 500 mm dithiotreitol was added to each sample (approx 850 μl) before incubation at 40 °C for 30mins. Proteins were alkylated by addition of 10 μl of 500 mm iodoacetamide and incubation at room temperature in the dark for one hour.
Protein Desalting and Fractionation
To desalt and concentrate depleted plasma, a reversed-phase HPLC step was performed on a 4.6 × 50 mm mRP-C18 column (Agilent) using a 1200 liquid chromatography system with chilled autosampler, column oven, UV detector, and fraction collector. This afforded the opportunity to also perform fractionation of the proteins to increase sensitivity by further reducing background. Forty-five microliters of a 2% trifluoroacetic acid in acetonitrile solution was added to each depleted plasma sample, which was subsequently fractionated using the gradient shown in supplemental Table S4. Fraction collection was triggered at 4.8mins with eight 0.6min fractions collected vertically (A to H) in a 96-well plate (Nunc V96PP, Thermo). The column was cleaned in between each sample by injecting 500 μl of a 6 m urea + 1% (v/v) acetic acid solution and running a short gradient with a 4min 100% B wash step.
Plasma Protein Digestion
Following a batch of fractionation runs (n = 24), fractions were dried in a speed-vac concentrator (Savant SPD131DDA, Thermo Scientific) until a vapor pressure of 300 mTorr was achieved corresponding to complete dryness. Each sample was resuspended in 50 μl of 50 mm ammonium bicarbonate, 5% (v/v) acetonitrile with 0.75 μg trypsin (sequencing grade, Sigma, UK). Samples were vortexed well, placed in a 37 °C oven, and vortexed every hour for 4 h. At 4 h an additional 50 μl of 50 mm ammonium bicarbonate, 5% (v/v) acetonitrile with 0.75 μg trypsin was added to each sample before incubation for 16 h at 37 °C. 5 μl of 4% (v/v) formic acid was added to each sample to terminate digestions, which were then centrifuged at 11,000 × g for 5mins before supernatants were transferred to fresh 96-well plates.
SRM Transition Selection and Optimization
A spectral library of human plasma MS/MS data was generated on an Agilent 6530 QTOF, searched using Mascot (Matrix Science, UK) and imported into Skyline software (17) where a library was built. Because of the high sequence identity of PSG family members, to select specific peptides their sequences were aligned and tryptic peptides selected manually based on their uniqueness to individual family members. BLAST searching was carried out (http://www.uniprot.org/blast/) to ensure that peptides shared no similarity to other proteins likely to be found in human plasma. No exclusion criteria based on peptide sequences were set. For peptides that were required to attain isoform specificity (or a sufficient number of unique peptides per protein), and for which no high quality spectrum was found in the library, crude synthetic peptides were purchased (JPT Technologies, Berlin, Germany) and MS/MS sequences for these peptides obtained and used for selection of optimal transitions.
SRM methods were established for PBP and PSG-family members. We initially monitored 6–8 y-ion transitions per peptide to ensure specificity with the criteria that >5 y-ions with the same elution profile and in the same ratios as the spectral library, and at predicted retention times were observed. Optimal collision energies and cell acceleration voltages were experimentally determined for each peptide by running replicates of each transition with ramping of the collision energy by 2 and 4 eV in either direction and the acceleration voltage at 1, 4, or 7. The settings that gave the highest intensity response for that transition were selected as optimal. From these data, a minimum of three abundant transitions were identified and subsequently used in further analyses, where a summing of all transitions was used to generate quantification data for each peptide.
A test pool of plasma from pregnant donors was immunodepleted, fractionated and digested as described. Preliminary SRM assays for the PSG family (23 peptides) and PBP (5 peptides) were used to analyze each fraction to determine where these proteins were detected. PSG peptides were distributed across fractions B, C, and D; these fractions were combined into a 1.5 ml microcentrifuge tube (before drying) to facilitate measurement of all PSG family members in a single assay for the remainder of the study. All PBP peptides were detected only in fraction A.
LC-SRM Method
LC-SRM analysis was performed on a 1290 uHPLC – 6460 QqQ LC/MS platform with a JetStream electrospray source (Agilent). Peptides were separated on a 250 × 2.1 mm, 2.7 μm particle size Poroshell 120 column (Agilent) with mobile phases A: Water + 0.1% (v/v) formic acid and B: acetonitrile + 0.1% (v/v) formic acid. A post-column valve enabled the loading and wash phases to be diverted to waste. A linear gradient of 8–35% B over 23 mins was applied at a flow rate of 150 μl/min with column oven at 50 °C. Source parameters were as follows: Gas temp: 250 °C, Gas flow: 10l/min, Nebuliser: 40psi, Sheath gas temp: 300 °C, Sheath gas flow: 10l/min.
Peptide retention times and optimized collision energy values were supplied to MassHunter (vB03.01, Agilent Technologies) to establish a Dynamic MRM scheduling method based on input parameters of 500ms cycle times and 1.2min retention time windows. Min/max dwell times were established by the software and data was analyzed using the Skyline software package (17). Data was inspected manually to identify any errors in the integration process and the total area under the peak for each peptide (summed from the areas of all transitions per peptide) was exported for analysis in Microsoft Excel and Prism (Graphpad Software, La Jolla, CA). %CV of the biological and technical replicates was used as a measure of variance and was calculated using the standard calculation of %CV = (s.d./mean)*100.
SRM Method Development
Twelve replicate plasma samples were immunodepleted and digested as previously described. Dried samples were then resuspended in 100 μl acetonitrile 0.2% (v/v) formic acid. These replicate sample preparations were used to determine the coefficient of variation of the SRM assay for each peptide; the technical variability of peptide FTFTLHLETPKPSISSSNLNPR was high and therefore this peptide was not included beyond this stage (supplemental Fig. S1). A further three peptides FQQSGQNLFIPQITTK, SDPVTLNLLPK, and IYPSFTYYR were omitted from the final assay because they were shared between many PSG isoforms and offered no additional information above those that remained. Full details of the final dynamic SRM transitions are provided in supplemental Tables S7 and S8. Minimum and maximum dwell times for the final assay were 38.17 ms and 163.17 ms respectively for the PSG peptides, 41.95 ms and 163.17 ms for the PBP peptides. In order to assess the linearity of the SRM assays, volumes of 1, 5, 10, 20, and 40 μl were injected in duplicate.
The pregnancy-specific nature of the PSG peptides was exploited and used to assess the specificity and quantitative sensitivity of the SRM assay. Relative quantification of several PSG peptides was performed in serial dilutions of 15 week plasma with PSG negative male plasma. Each of these samples were prepared in triplicate and processed in a randomized order.
Clinical Sample Analysis
One-hundred clinical samples were processed across four batches along with eight full replicates of the same plasma sample (two per batch), which allowed a true assessment of technical variability over the entire workflow/experiment to be made. Following immunodepletion, reduction/alkylation, and protein fractionation, the 108 samples were digested simultaneously. SRM analyses for PSG and PBP peptides were performed, alongside 11 injections of a pooled sample from the same vial to assess technical variation within the LC-MS and blank injections (loading buffer only), which confirmed that no carryover was evident. Kruskal-Wallis test (Dunns correction for multiple comparisons) was used to determine differences between sample groups (significance p < 0.05). All transitions described in supplemental Tables S7 and S8 were used for quantification; no minimum threshold for inclusion was set and no outliers were removed during data analysis.
NAP-2 ELISA
As no commercial ELISA against PBP is available, an ELISA that recognizes NAP-2 was used (RayBiotech, Norcross GA, USA). This ELISA measures a proteolytic fragment of PBP containing all peptides quantified in the iTRAQ discovery and SRM experiments. Two microliters of EDTA plasma was diluted 1 in 8100 in a two-stage dilution using the provided dilution buffer. The kit protocol was followed exactly with the primary sample incubation occurring overnight at 4 °C with gentle shaking. Technical replicates were randomly spread across the three plates used in the assay. All case/control samples were diluted and assayed in duplicate.
Correlation Analysis
A visual correlation analysis was generated using the Corrgram tool within an in-house R-based statistical toolbox. Implementation based on Sarkar (18).
Sample Size Calculations
iTRAQ discovery experiments were performed on pooled samples containing 12 individuals, this is the minimal number acceptable for a discovery experiment (19). Given the fold changes observed in the iTRAQ studies for the proteins of interest (≥1.3), a minimum sample size of 16 per group was necessary for the SRM assays to determine a significant difference between groups (α = 0.05; β = 0.80).
RESULTS
iTRAQ Based Discovery Phase
Pooled plasma samples taken at 15 ± 1 week from women who developed EO-PE (n = 12) or had uncomplicated pregnancies (n = 24; 2 pools of n = 12) were processed for analysis using iTRAQ labeling to enable protein level comparison from tryptic fragment MSMS analyses. A total of 502 proteins were identified across three iTRAQ discovery experiments (319 proteins in the IgY 14-SuperMix- QSTAR data set, 331 in the IgY 14-SuperMix-5800 data set and 189 in the MARS 14–5800 data set) (Fig. 2A). Within each data set, the standard deviation of the log2 iTRAQ ratios for the replicate samples was calculated as an indication of the variability between technical replicates in each data set. This gave values of 0.19, 0.27 and 0.13 for the IgY 14-SuperMix-QSTAR, IgY 14-SuperMix-5800 and MARS 14–5800 data sets respectively. To identify the candidate biomarkers with the highest confidence, we initially only considered proteins identified and quantified in all three replicate experiments. A total of 113 proteins were identified in all three data sets and following application of stringent candidate protein selection criteria, two proteins; platelet basic protein (PBP, CXCL7) and pregnancy-specific beta-1-glycoprotein 9 (PSG9) were prioritized for further assessment in the SRM-based verification phase (Fig. 2B). In total, 113 proteins were altered in abundance in any one of the iTRAQ data sets and this list is provided in supplemental Table S5. The compete iTRAQ data set is provided in supplemental Table S9, and all discovery data and spectra are available at www.scalpl.org/hank/ProteomeHexPage?me=Pre-eclampsia.
Fig. 2.

Summary of iTRAQ-based discovery phase. Across the three discovery experiments a total of 502 nonredundant plasma proteins were identified. A, Venn diagram: the number of identified proteins and number of changed proteins for each experimental data set used. The proteins found to be different between cases and controls for each data set are shown. B, Scatter plot of Log2iTRAQ ratios for the two replicate pooled EO-PE samples and the two pooled control samples in each of the three iTRAQ experiments. All values are expressed as the ratio of the index sample to the reference sample for each data set. Numbers in brackets represent the number of peptides identified for each protein.
SRM Method Development
To assess whether these changes could be observed in a significant proportion of patients and hence have genuine value as putative biomarkers, we developed a novel, label-free SRM workflow. As proof-of-principle, this was tested on two proteins from our discovery experiment, PBP and PSG9. PSG9 is a member of a large multigene family, which is expressed in a pregnancy-specific manner. Because we had also seen some evidence for dysregulation of PSG2 and 5 from our discovery analysis, 15 peptides from other PSG family members were also included in the assay. As part of this workflow, we employed a protein based fractionation step simultaneously with concentrating the MARS-14 flow through. This step theoretically reduces background (and hence should increase sensitivity) without requiring an additional sample preparation step. The reversed-phase protein concentration and fractionation protocol was highly reproducible based on overlaying UV chromatograms from replicate injections (Fig. 3) with a CV of 2.3% across 8 replicate plasma injections. A further advantage of a protein versus a peptide-level separation is that all peptides for a single protein are contained in the same fraction, such that only one fraction has to be analyzed to quantify several proteotypic peptides. MASCOT analysis of each fraction suggested that PBP eluted exclusively in the first fraction collected, whereas the PSG proteins were found exclusively in fractions 2, 3, and 4, and these data were confirmed by our optimized SRM method (data not shown). These fractions were selected for further analysis by SRM.
Fig. 3.

Visualization of SCOPE 100 sample preparation. Eight replicates of the same plasma sample were interspersed among the 100 case and control samples processed for LC-SRM analysis. UV chromatograms of the MARS-14 immunodepletion and mRP protein fractionation are recorded for each sample. The traces for all eight technical replicates (A) and 20 consecutive case/control samples (B) are shown overlaid to allow a visual assessment of technical versus biological variability. The fractions collected from the mRP runs are shown. By comparing the eight technical replicates with twenty randomly selected case/control samples it is evident that the technical variability in both chromatographic steps is significantly lower than the inherent biological variation.
To select optimal peptides for SRM analysis, it is critical that these peptides are reproducibly produced and detected following plasma tryptic digestion. To that end, we performed 12 replicate digests of the same plasma sample and assessed the reproducibility of the signal (expressed as %CV) for each peptide (supplemental Fig. S1). The peptide FTFTLHLETPKPSISSSNLNPR showed poor reproducibility (CV >20%) and was discarded at this point. To check for potential interferences from other peptides in the sample, the relative levels of the 6–8 initial transitions per peptide were monitored in a plasma sample and their ratios compared with the relative levels of product ions in an MS2 spectrum of the same peptide. Correlation using Skyline suggested that no other co-eluting peptide significantly affected any given transition. The relative ratio of the three final transitions (which, by definition, we also ensured have identical RT) was also assessed to ensure consistency in experimental runs.
Dilution curves were generated from 12 replicate digests for the peptide assays to determine the linear range of the assay. The dilution curves are shown in supplemental Fig. S2. The peptide assays were linear up to 20 μl. From this data it was determined that 20 μl of each plasma sample should be used in the SRM assays.
This method of confirming linearity does not necessarily confirm the linear change of a specific signal in a constant background (because by definition background and analyte are changing at the same rate). To test this, we exploited the fact that one of our targets was unique to the plasma of pregnant females. We performed a dilution series of early pregnancy plasma from pregnant donors with male plasma at 100%, 90%, 80%, 50%, 10%, and 0% of total volume. This analysis, performed in triplicate, demonstrated that all PSG peptides showed good linearity when plotted as dilution curves. This confirmed that the SRM was selectively measuring female specific proteins with linear responses (supplemental Fig. S3). The majority of peptides reached significance comparing 100% female plasma to 90% female plasma with male plasma, all peptides reach high significance (p < 0.01 or p < 0.001) when comparing median levels of the 100% to 80% groups. This demonstrates that our complete workflow is sufficiently robust and sensitive to detect relatively minor fold-changes in plasma protein concentrations.
Clinical Samples Analysis
SRM analyses for 19 PSG peptides and 5 PBP peptides were performed on 108 plasma samples (16 EO-PE, 42 LO-PE, 42 controls, eight full technical replicates of a pooled sample). The semi-automated HPLC-based approach for immunodepletion and protein fractionation required little manual handling and the UV chromatograms generated act as critical quality control points allowing assessment of the technical variability associated with replicate injections (n = 8, interspersed across the 108 injections) compared with the biological variability observed (Fig. 3). Over the course of the experiment, the mRP column (protein fractionation) backpressure increased by only four bars. In addition, blank injections of MARS buffer A gave no deflection on the chromatogram traces, confirming that little, if any, sample carryover was present (data not shown). Blanks were subjected to standard IDA analysis by LC-MS/MS, using the protocols described in the methods section, and no peptides could be identified. The use of urea injections as part of the mRP column cleaning between sample injections was critical for carry-over prevention and maintenance of column performance.
Five percent of all 108 processed samples was taken and pooled post-digestion to provide a standard that was injected regularly throughout the analysis, allowing assessment of the variation due to the LC-SRM versus the variation observed across the entire sample preparation workflow. For each peptide analyzed, replicate LC-SRM injections (n = 11) were always associated with lower variation (CVs 4.1–9.9%) than the technical replicates (n = 8) spanning the whole workflow (CVs 12.8–24.5%). This confirms that the majority of the technical variation within this workflow was attributable to the sample preparation rather than the LC-SRM assay (Fig. 4).
Fig. 4.

Pregnancy specific glycoprotein (PSG) label-free SRM assay. Seven of the 19 PSG peptides measured in the PSG SRM assay are shown as scatter plots. The y axis represents total integrated area of three transitions per peptide. The “Rep SRM” group represents replicate injections of the same pooled digest, the “Tech Reps” group represents eight replicates of the same plasma sample processed in parallel through the whole workflow. The data points in the case and control samples each represent a unique plasma sample.
For three of the four PSG-9 specific peptides, there was a significant difference in the median levels between EO-PE (n = 16) and controls (n = 42) (p < 0.05), whereas the difference was not statistically significant for LO-PE (n = 42) (Table II, Fig. 4). The fourth PSG-9 peptide, SNPVILNVLYGPDLPR was also different between EO-PE and controls, although this difference was marginal (p = 0.051). Area under receiver-operator characteristics curves (AUROC) were calculated for each peptide to provide an additional metric of their ability to differentiate EO-PE versus control groups (Table II).
Table II. Summary of all peptide SRM data. Data for each peptide is shown as the median fold change. Comparison using Kruskal-Wallis (with Dunns correction for multiple comparisons). Rep SRM: Coefficient of variation (CV) for replicate LC-SRM injections (n = 11) Tech Rep: CV for technical replicates spanning the whole workflow (n = 8). EO: Early onset PE (n = 16); Con: Control (n = 42); LO: Late onset PE (n = 42). AUC: Area under receiver operator curve.
| PSG isoform | Rep SRM | Tech reps | EO:Con | LO:Con | P | AUC | 95% CI | |
|---|---|---|---|---|---|---|---|---|
| PSG peptides | ||||||||
| FQLPGQK | 1 | 6.65% | 14.29% | 1.13 | 0.94 | NS | ||
| YTAGPYECEIR | 1,2,3,4,8 | 5.18% | 13.82% | 1.40 | 1.18 | 0.09 | ||
| EDAGSYTLHIIK | 1,2,5,11 | 6.33% | 17.84% | 1.37 | 1.15 | NS | ||
| ILILPSVTR | 1,3,5,6,7,8,9 | 6.49% | 13.76% | 1.39 | 0.99 | NS | ||
| LPKPYITINNLNPR | 1,3,7 | 4.31% | 23.44% | 1.35 | 1.03 | NS | ||
| TLFLLGVTK | 1,8 | 5.48% | 13.09% | 1.08 | 0.95 | |||
| SDPVTLNLLHGPDLPR | 2 | 9.88% | 16.27% | 1.39 | 1.22 | 0.02 | 0.72 | (0.56–0.88) |
| TLFLFGVTK | 2,3 | 5.25% | 12.79% | 1.55 | 1.31 | 0.01 | 0.74 | (0.60–0.89) |
| FQLSGQK | 3,4,6,7,8,11 | 5.02% | 20.01% | 0.77 | 0.70 | NS | ||
| LFIPQITTK | 3,8 | 8.69% | 15.64% | 1.26 | 1.04 | 0.07 | ||
| LSIPQITTK | 4,5,7 | 6.42% | 19.48% | 1.08 | 0.93 | NS | ||
| SMTVEVSAPSGIGR | 5 | 5.54% | 15.74% | 1.82 | 1.34 | 0.01 | 0.75 | (0.59–0.90) |
| EVMEAVR | 6 | 8.82% | 18.91% | 1.05 | 1.03 | NS | ||
| SNPVTLNVLYGPDLPR | 6 | 8.17% | 24.56% | 1.28 | 1.05 | NS | ||
| YGPAYSGR | 7 | 6.78% | 19.65% | 0.43 | 0.41 | NS | ||
| LFIPQITR | 9 | 8.69% | 15.64% | 1.58 | 1.24 | 0.02 | 0.74 | (0.61–0.87) |
| SNPVILNVLYGPDLPR | 9 | 7.89% | 18.85% | 1.43 | 1.19 | 0.05 | 0.67 | (0.50–0.83) |
| IIIYGPAYSGR | 9 | 7.25% | 24.08% | 1.43 | 1.37 | 0.04 | 0.71 | (0.57–0.86) |
| LPIPYITINNLNPR | 9 | 4.14% | 19.45% | 1.43 | 1.31 | 0.03 | 0.72 | (0.57–0.86) |
| PBP peptides | ||||||||
| EESLDSDLYAELR | 6.83% | 105.97% | 1.16 | 1.06 | NS | |||
| NIQSLEVIGK | 7.74% | 32.25% | 1.44 | 1.13 | NS | |||
| GTHCNQVEVIATLK | 8.72% | 58.69% | 1.54 | 0.93 | NS | |||
| GKEESLDSDLYAELR | 6.28% | 43.55% | 1.62 | 0.88 | NS | |||
| KICLDPDAPR | 6.26% | 23.46% | 1.48 | 1.08 | NS |
The levels of peptides unique to PSG-1 and PSG-6, and peptides common to PSG-2, 3, 11 (e.g. TLFLFGVTK) were not different between cases and controls (Table II, Fig. 4). The median levels of the specific PSG-5 peptide (SMTVEVSAPSGIGR) were almost doubled in the EO-PE samples compared with the controls (p = 0.01), with a %CV for this peptide of 16% (Table II, Fig. 4). The AUROC value for this PSG-5 specific peptide SMTVEVSAPSGIGR peptide was 0.75.
Five peptides unique to PBP were selected based on our QToF MS/MS spectral library data and subsequent analysis to test reproducibility of generation from a plasma digest. Interestingly, two of these peptides contained missed cleavages (internal lysines) and one contained a cysteine. These would have been omitted using many of the conventional proteotypic peptide selection rules. However, these peptides appeared to be reproducibly generated and measured in replicate samples; KICLDPDAPR was the most reproducible PBP peptide, with %CV for full technical replicates of the same sample of 23.5%. Fully cleaved versions of these peptides were not observed in the discovery phase of the experiment. The median difference between EO-PE and controls reached significance (p = 0.048) for only one of the five PBP peptides measured (Table II).
ELISA-based Quantification of PBP/NAP-2
To verify the relative quantitation of PBP/NAP-2 across our sample cohort, we also measured levels of the naturally occurring processing product of PBP, NAP2 using a commercially available ELISA kit. All peptides monitored in the SRM are present in the NAP-2 fragment. SRM quantification data were compared with measurements obtained using a commercial ELISA for NAP-2 in the same samples. The medians for the two data sets are comparable and the differences between PE and controls were not significant (Fig. 5A). There was a good correlation between the ELISA and SRM data, for example the integrated area of the NIQSLEVIGK peptide versus absolute NAP-2 levels in the ELISA showed a linear correlation with an R2 value of 0.863 (Fig. 5B). This demonstrates that our SRM-based assay has provided a reliable measurement of relative protein levels, across 100 plasma samples.
Fig. 5.
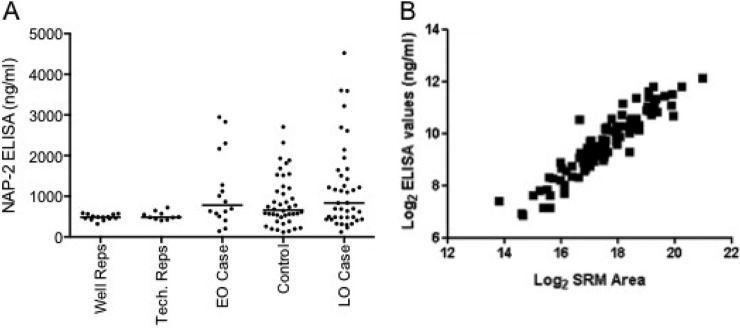
Measurement of Platelet Basic Protein by SRM and ELISA. A, ELISA measurement of NAP-2 (a proteolytic fragment of PBP encompassing all measured peptides) in the same 100 sample cohort. “Well Replicates” incorporates only ELISA variability (n = 14) whereas “Tech Replicates” incorporate both the dilution and ELISA variability (n = 10). Case and control groups are as SRM analysis, here each data point is the mean of each sample run in duplicate. B, Correlation analysis of NIQSLEVIGK peptide SRM versus NAP-2 ELISA for the 100 samples.
Correlation Analysis
A Corrgram was generated to visualize the correlation between all biochemical features measured in the study (supplemental Fig. S4). The MARS-14 peak area and height measure the amount of protein in each 40 μl plasma sample but do not correlate with any peptide features.
The lack of individual PSG-specific peptides representing all of the PSG family members makes it difficult to fully dissect the inter-relations between all of the members of this protein family, but it is clear that some PSG proteins behaved independently of each other. The four PSG-9 specific peptides correlated with each other almost perfectly (r = 0.95–0.99). Good correlation was observed between the ELISA and all PBP peptides, although this was not as strong as any of the correlations among the peptide SRM measurements. Of the five PBP peptides, the correlation between peptide (EESLDSDLYAELR) and the other four PBP peptides was the least good, despite the fact that this would have been the peptide most likely to have been selected according to common proteotypic peptide selection rules, whereas KICLDPDAPR, for example correlated very well with the ELISA data.
DISCUSSION
The aim of this study was to develop and validate a robust and reproducible plasma depletion/fractionation workflow with subsequent label-free SRM analysis to identify which candidate biomarkers from a discovery proteomics experiment should be further pursued in large scale verification and clinical validation studies. To meet this aim we developed an optimized sample preparation and analysis workflow that was successfully applied to the relative quantification of candidate biomarkers for the prediction of PE in a cohort of 100 women.
Identification of candidate biomarkers was achieved through the application of iTRAQ to pooled plasma collected from women in early pregnancy and who subsequently developed PE or had an event-free pregnancy. As this technique has modest proteome penetrance, pooled samples were used in this study. This strategy was selected as, although the pool can be skewed by very high (or low) expression in a small proportion of samples, it does allow increased fractionation (and therefore penetration) and analysis in a reasonable timeframe. Minor changes, which occur in a small proportion of patients, are averaged out, whereas strong biomarker candidates (relatively large fold changes in a high proportion of cases) are maintained. This allowed us to use three distinct workflows for iTRAQ analysis, increasing our coverage of the plasma proteome, and allowing us to combine the three iTRAQ experiments to improve confidence in the iTRAQ quantification. Quantification data for more than 500 proteins was obtained, with 113 proteins quantified in all three data sets. For this proof of principle verification experiment, we chose to focus on the two proteins that were consistently changed in abundance in PE in all three iTRAQ experiments according to stringent selection criteria. Additional candidate proteins were identified in two of the three discovery experiments and merit further investigation (supplemental Table S5), but as our eventual goal in this study was to develop robust workflow methodology, these were not investigated at this stage.
A common approach for the verification of plasma protein biomarkers is to select proteotypic peptides as analytical surrogates of the proteins of interest. This peptide selection is frequently performed using rules based on peptide features and in silico tools. Once selected, stable isotope labeled synthetic peptides are made, purified and accurately quantified prior to SID-SRM experiments (20). Although this approach may lead to the identification of peptides with the “best ionization potential” for a given protein, it does not necessarily select the peptides that are likely to translate to a robust assay for protein abundance. Such an assay requires peptides that are reproducibly generated during sample preparation, give a linear response in protein (not peptide) titration experiments, and provide strong correlation with other peptide measurements of the same protein. These features can only be determined experimentally and may differ according to each experimental workflow. Hence, for an experiment studying, say ten candidate proteins, where you may want to initially assess six peptide per protein in order to find those which give the most robust and reproducible responses, you would require 60 stable isotope labeled and purified and quantified peptides—a significant outlay, especially when most will be used for only one or two experiments. A recent approach, where crude synthetic peptides have been used to optimize the MS settings for specific peptides, provides a cheaper option (21). Our approach was to use stringent peptide selection criteria (e.g. we allowed Cys/Met containing and missed cleavage peptides) but to experimentally screen a larger number of peptides per protein. Peptides were selected based on their observation in standard LC-MS analyses of plasma. Additional peptides were identified from in silico digestion and crude synthetic sequences were obtained to aid in MS optimization. Replicate plasma samples were processed in order to filter peptides based on their reproducibility; interestingly peptides with Cys and Met residues and missed cleavage sites did not have noticeably higher CV values than those without. Indeed for the five peptides assayed from PBP, the one which correlates the least with the other four and a commercial ELISA is a “model” peptide, whereas peptides with missed cleavages and cysteine residues appear to be more precise surrogates for this protein in this case.
In comparison to conventional SID-SRM, the absence of isotopically labeled standards in our SRM workflow means that no correction or compensation for technical variation can be performed during data analysis. Although spiking labeled peptides into samples post-digestion allows the technical variation of the LC-SRM assay to be determined (and indeed corrected), other major sources of technical variation in sample preparation (e.g. proteolysis) may not be taken into account when interpreting results. Absolute concentrations of tryptic peptides are commonly determined using SID-SRM, but there is an inherent assumption that peptide levels perfectly correlate to protein levels, i.e. 100% digestion efficiency and 100% recovery throughout all processing steps. The use of isotope-labeled protein standards, e.g. PSAQ (22), is an elegant solution to address these issues but currently the effort required to generate multiple isotopically enriched, natively folded and purified proteins makes it impractical for an initial biomarker verification study. An alternative approach is QconCAT (23), although again this requires design, expression and purification of a synthetic protein, and quantitation is dependent on obtaining similar digestion efficiency in both the QconCAT and endogenous protein, despite differences in protein structure. An additional advantage of our label free approach therefore is that protein recovery throughout the workflow has to be reproducible but does not necessarily have to be 100%, as is the case for methods which attempt to determine absolute protein concentration in the sample. However, without the “safety net” of a spiked isotope-labeled internal standard, we must be aware of several factors crucial to the success of a label-free strategy: (1) minimize technical variability through optimization of every step, and (2) apply appropriate metrics to quantify true technical variation such that it can be observed in the context of the biological variation. We addressed these issues by using carefully optimized plasma immunodepletion and fractionation steps, employing a wide bore, higher flow rate liquid chromatography with more stable electrospray performance than nanoflow ESI and by incorporating appropriate technical replicates into the experimental design. Analysis of eight technical replicates spaced throughout our 100 sample analysis showed convincingly that this protocol can generate robust and reproducible measures of relative protein level and that differences in protein recovery, peptide digestion etc. were acceptable based on the replicate sample CV values. All steps were validated, including an assessment of the linear response and maximum loading capacity of the assays using stepped injections. In addition, the pregnancy-specific properties of the PSG proteins allowed us to confirm the linearity of response of the SRM assay by performing a series of dilutions of 15 week plasma from pregnant donors with a PSG negative male plasma matrix. This experiment is ideal because the male plasma affords us an analyte-free background for these spiking experiments, which cannot be guaranteed when spiking into nonpregnant female plasma; a 10–20% change in protein abundance was detected for each surrogate peptide. The absence of signal in the 100% male plasma sample also shows that these transitions were not subject to interference from endogenous plasma proteins.
The throughput of this assay is ∼100 samples/week (in the absence of liquid handling robotics); although this is not a high-throughput assay it is reasonable throughput for an initial biomarker verification study.
A main aim of this work was to demonstrate that this approach can help prioritize candidate biomarkers for more time consuming and expensive future studies, in other words providing a relatively tractable way of confirming which “candidate biomarkers” from a discovery experiment are worthy of the name. For example, measurement of PBP indicated there was no change in this protein in a verification sample set, despite its apparent differential expression in the discovery experiment. A third of women (4 from 12) from the EO-PE sample set used for the iTRAQ analysis showed very high levels of PBP in both the SRM and the ELISA experiments. This observation provides technical verification of the iTRAQ findings (which quantified the mean change in PBP between sample groups), but also demonstrated the need to verify discovery findings in modestly sized sample sets before proceeding to large-scale clinical validation studies. Elevated PBP levels in a subgroup of women also highlights the heterogeneity of clinical syndromes such as PE and suggests that candidate markers are likely to convey different predictive performance in different subgroups, or that panels of biomarkers may more powerful than single measurements of single compounds. The availability of an ELISA for PBP provided a useful opportunity to validate the SRM quantification performed in this study. In this sample set there was good correlation between the two platforms (r = 0.863), which is comparable to other studies (7) and provides further confirmation that the SRM technique described is fit-for-purpose.
The use of SRM assays to quantify members of the PSG family of proteins is an excellent example of the potential of mass spectrometry based assays to profile proteins with high sequence homology, i.e. those which pose the biggest challenge to antibody-based methods. Although PSG9 was the only PSG family member to be identified in all three iTRAQ experiments, we had some evidence of a change in PSG 2 and 5 (supplemental Table S5 and S9). This is the first time that these PSG family members have been implicated in pre-eclampsia disease development. Therefore, we took the opportunity to screen several PSG family members in our clinical sample set. PSG- 2, 5 and 9 levels differed between EO-PE and healthy controls whereas other PSG family members appeared unchanged. To our knowledge this study is the first to demonstrate that there may be some specificity in the regulation and/or activity of pregnancy specific glycoproteins in the context of pregnancy complications (24). Although further studies are required to assess the value of PSG peptides in the prediction of PE, these data define PSG-5 and PSG9 as potential biomarkers for future screening experiments, and allow us to perform appropriate power analyses such that we can determine the size of cohort which should be used for their future assessment.
A limitation of this label-free SRM approach is that it does not readily allow comparison of data sets from samples run at different times or locations, as can be achieved with absolute quantification against synthetic standards (25). However, we see this workflow as a rapid, affordable, and scalable intermediate stage in biomarker development to prioritize candidates from discovery experiments rather than a formal clinical validation (of the assay and the biomarker) that may be performed in several centers. This study has demonstrated that adequate reproducibility in LC-SRM analysis can be achieved using a 2.1 mm column with a standard uHPLC, but performance using a nanospray ESI has not been formally assessed. Pilot studies in our laboratory suggest that the using a combination of higher flow rate system with a 250 × 2.1 mm column packed with 2.7 μm Poroshell 120 causes a two- to fivefold reduction in sensitivity (the increased dilution of the sample at the source is largely offset by higher resolution chromatography and increased loading capacity) over our standard nanoflow system, but the gain is a much more reproducible separation and more robust ionization over a long period of time versus nanoflow chromatography/ESI. Given that the sample quantity is not limiting in the case of human plasma, this trade-off is one worth making. These observations are supported by a recent comparison where the use of an Agilent 1290 uHPLC operated at over a 150 × 2.1 mm column containing Zorbax Eclipse Plus C18 Rapid Resolution HD 1.8 μm particles was shown to be a better choice for plasma SRM than the nanochipcube interface with a 150 ×75 μm column packed with Zorbax 300SB-C18, 5 μm particles on the same instrument (26). In our study, only a small number of high ng/ml proteins were measured which has limited the assessment of the multiplexing potential of this SRM assay. Having established these principles for method development, however, the inclusion of additional SRM transitions is unlikely to be problematic.
In summary, we have demonstrated that with careful attention to experimental design, a label-free SRM approach to biomarker verification can determine the potential clinical value of candidate proteins across 100+ plasma samples. Of particular importance was the full assessment of technical variation and development of an assay that measured variation in peptide levels throughout the entire workflow, rather than the variation in levels of a peptide spiked into the sample at some point during the workflow. This study has demonstrated the capability of a label-free SRM assay in filling the gap between biomarker discovery and clinical validation studies and highlights specific PSG proteins as potential candidate biomarkers for the prediction of PE.
Supplementary Material
Acknowledgments
We would like to thank the pregnant women who participated in the SCOPE Study, the international study coordinator, Mrs. R. Taylor, the collaborating SCOPE PI, Professor G. Dekker (University of Adelaide) and the SCOPE Australia Country Project Manager Mrs. D. Healy.
Footnotes
* This work was supported by New Zealand's New Enterprise Research Fund, Foundation for Research Science and Technology; Health Research Council 04/198; Evelyn Bond Fund, Auckland District Health Board Charitable Trust; Australia's Premier's Science and Research Fund, South Australian Government. JM is also supported by Action Medical Research Endowment Fund and facilitated by the NIHR Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network. ADW is funded by Leukaemia Lymphoma Research and Cancer Research UK Experimental Cancer Medicine Centre and MJW was funded by the Experimental Cancer Medicine Centre Network. The study sponsors had no role in study design, data analysis or writing this report.
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S9.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S9.
Ethical approval: Ethical approval was obtained from local ethics committees [New Zealand AKX/02/00/364, Australia REC 1712/5/2008].
1 The abbreviations used are:
- AUROC
- Area under receiver-operator-characteristic
- CV
- coefficient of variance
- EO
- early onset (<34 weeks gestation)
- FA
- formic acid
- LO
- late onset (>34 weeks gestation)
- LOD
- limit of detection
- LOQ
- limit of quantification
- PBP
- platelet basic protein (also known as CXCL7)
- PE
- pre-eclampsia
- PSG
- pregnancy-specific glycoprotein
- SID
- stable isotope dilution
- SRM
- selected reaction monitoring.
REFERENCES
- 1. Carr S. A., Anderson L. (2008) Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory? Clin. Chem. 54, 1749–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makawita S., Diamandis E. P. (2010) The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin. Chem. 56, 212–222 [DOI] [PubMed] [Google Scholar]
- 3. Stahl-Zeng J., Lange V., Ossola R., Eckhardt K., Krek W., Aebersold R., Domon B. (2007) High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics 6, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 4. Kuzyk M. A., Smith D., Yang J., Cross T.J., Jackson A.M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 7. Zhi W., Wang M., She J. X. (2011) Selected reaction monitoring (SRM) mass spectrometry without isotope labeling can be used for rapid protein quantification. Rapid Commun. Mass Spectrom. 25, 1583–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang H. Y., Beer L. A., Barnhart K. T., Speicher D. W. (2011) Rapid verification of candidate serological biomarkers using gel-based, label-free multiple reaction monitoring. J. Proteome Res. 10, 4005–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilkinson H. (2011) Saving mothers' lives. Reviewing maternal deaths to make motherhood safer: 2006–2008. Br. J. Obs. GYN. 118:1402–1403; discussion 1403–1404 [DOI] [PubMed] [Google Scholar]
- 10. Centre for Maternal and Child Enquiries (CMACE) (2011). Perinatal Mortality 2009: United Kingdom. (CMACE, London: ). [Google Scholar]
- 11. Roberts J.M., Taylor R.N., Musci T.J., Rodgers G.M., Hubel C.A., McLaughlin M.K. (1989) Preeclampsia: an endothelial cell disorder. Am. J. Obstet. Gynecol. 161, 1200–1204 [DOI] [PubMed] [Google Scholar]
- 12. Myatt L., Clifton R. G., Roberts J. M., Spong C. Y., Hauth J. C., Varner M. W., Thorp J. M., Jr., Mercer B. M., Peaceman A. M., Ramin S. M., Carpenter M. W., Iams J. D., Sciscione A., Harper M., Tolosa J. E., Saade G., Sorokin Y., Anderson G. D. (2012) First-trimester prediction of preeclampsia in nulliparous women at low risk. Obstet. Gynecol. 119, 1234–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Myers J. E., Kenny L., McCowan L., Chan E., Dekker G., Poston L., Simpson N., North R. (2013). Angiogenic factors combined with clinical risk factors to predict preterm pre-eclampsia in nulliparous women: a predictive test accuracy study. Br. J. Obstet. Gyn., DOI: 10.1111/1471–0528.12195 [DOI] [PubMed] [Google Scholar]
- 14. Myers J. E., Tuytten R., Thomas G., Laroy W., Kas K., Vanpoucke G., Roberts C. T., Kenny L. C., Simpson N. A., Baker P. N., North R. A. (2013) Integrated proteomics pipeline yields novel biomarkers for predicting preeclampsia. Hypertension 61, 1281–1288 [DOI] [PubMed] [Google Scholar]
- 15. North R. A., McCowan L. M., Dekker G. A., Poston L., Chan E. H., Stewart A. W., Black M. A., Taylor R. S., Walker J. J., Baker P. N., Kenny L. C. (2011) Clinical risk prediction for pre-eclampsia in nulliparous women: development of model in international prospective cohort. BMJ 342, d1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 17. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sarkar D. (2008). Lattice: Multivariate Data Visualization with R (New York, U.S.A., Springer; ). [Google Scholar]
- 19. Mischak H., Allmaier G., Apweiler R., Attwood T., Baumann M., Benigni A., Bennett SE., Bischoff R., Bongcam-Rudloff E., Capasso G., Coon J. J., D'Haese P., Dominiczak A. F., Dakna M., Dihazi H., Ehrich J. H., Fernandez-Llama P., Fliser D., Frokiaer J., Garin J., Girolami M., Hancock W. S., Haubitz M., Hochstrasser D., Holman R. R., Ioannidis J. P., Jankowski J., Julian B. A., Klein J. B., Kolch W., Luider T., Massy Z., Mattes W. B., Molina F., Monsarrat B., Novak J., Peter K., Rossing P., Sánchez-Carbayo M., Schanstra J. P., Semmes O. J., Spasovski G., Theodorescu D., Thongboonkerd V., Vanholder R., Veenstra T. D., Weissinger E., Yamamoto T., Vlahou A. (2010). Recommendations for biomarker identification and qualification in clinical proteomics. Sci. Transl. Med. 2, 46ps42. [DOI] [PubMed] [Google Scholar]
- 20. Kroksveen A. C., Aasebo E., Vethe H., Van Pesch V., Franciotta D., Teunissen C. E., Ulvik R. J., Vedeler C., Myhr K. M., Barsnes H., Berven F. S. (2013) Discovery and initial verification of differentially abundant proteins between multiple sclerosis patients and controls using iTRAQ and SID-SRM. J. Proteomics 78, 312–325 [DOI] [PubMed] [Google Scholar]
- 21. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 22. Brun V., Dupuis A., Adrait A., Marcellin M., Thomas D., Court M., Vandenesch F., Garin J. (2007) Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 6, 2139–2149 [DOI] [PubMed] [Google Scholar]
- 23. Zhao Y., Jia W., Sun W., Jin W., Guo L., Wei J., Ying W., Zhang Y., Xie Y., Jiang Y., He F., Qian X. (2010) Combination of improved (18)O incorporation and multiple reaction monitoring: a universal strategy for absolute quantitative verification of serum candidate biomarkers of liver cancer. J. Proteome Res. 9, 3319–3327 [DOI] [PubMed] [Google Scholar]
- 24. Camolotto S., Racca A., Rena V., Nores R., Patrito L. C., Genti-Raimondi S., Panzetta-Dutari G. M. (2010) Expression and transcriptional regulation of individual pregnancy-specific glycoprotein genes in differentiating trophoblast cells. Placenta 31, 312–319 [DOI] [PubMed] [Google Scholar]
- 25. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Percy A. J., Chambers A. G., Yang J., Domanski D., Borchers C. H. (2012) Comparison of standard- and nano-flow liquid chromatography platforms for MRM-based quantitation of putative plasma biomarker proteins. Anal. Bioanal. Chem. 404, 1089–1101 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.