

Abstract
Much like the host cells they infect, viruses must also regulate their life cycles. Herpes simples virus type 1 (HSV-1), a prominent human pathogen, uses a promoter-rich genome in conjunction with multiple viral trans-activating factors. Following entry into host cells, the virion-associated outer tegument proteins pUL46 and pUL47 act to increase expression of viral immediate–early (α) genes, thereby helping initiate the infection life cycle. Because pUL46 has gone largely unstudied, we employed a hybrid mass spectrometry-based approach to determine how pUL46 exerts its functions during early stages of infection. For a spatio-temporal characterization of pUL46, time-lapse microscopy was performed in live cells to define its dynamic localization from 2 to 24 h postinfection. Next, pUL46-containing protein complexes were immunoaffinity purified during infection of human fibroblasts and analyzed by mass spectrometry to investigate virus-virus and virus-host interactions, as well as post-translational modifications. We demonstrated that pUL46 is heavily phosphorylated in at least 23 sites. One phosphorylation site matched the consensus 14-3-3 phospho-binding motif, consistent with our identification of 14-3-3 proteins and host and viral kinases as specific pUL46 interactions. Moreover, we determined that pUL46 specifically interacts with the viral E3 ubiquitin ligase ICP0. We demonstrated that pUL46 is partially degraded in a proteasome-mediated manner during infection, and that the catalytic activity of ICP0 is responsible for this degradation. This is the first evidence of a viral protein being targeted for degradation by another viral protein during HSV-1 infection. Together, these data indicate that pUL46 levels are tightly controlled and important for the temporal regulation of viral gene expression throughout the virus life cycle. The concept of a structural virion protein, pUL46, performing nonstructural roles is likely to reflect a theme common to many viruses, and a better understanding of these functions will be important for developing therapeutics.
Viruses, like the host cells they infect, face the challenge of regulating their own gene expression from shortly after cell entry to the end stages of viral egress. Indeed, viruses have evolved a slew of diverse mechanisms to regulate their life cycles, often requiring temporally regulated virus–virus and virus–host interactions. One prominent example is the herpesvirus family, a group of large, double-stranded DNA viruses distinguished by their high degree of temporal regulation of gene expression (1–3). Herpes simplex virus-1 (HSV-1)1 is a prominent human pathogen, infecting ∼60% of the adult population, triggering lesions of the buccal and gingival mucosa, disseminated neonatal infections, and fatal encephalitis (4). The HSV-1 virion itself is structured for regulation of gene expression; between the inner DNA-containing capsid and outer glycoprotein-studded membrane lies a proteinaceous tegument layer containing several proteins critical for the initiation of viral gene transcription from immediate–early (α) promoters (5–7).
The progression of HSV-1 infection requires the temporal expression of viral genes, which is tightly controlled by viral proteins and their interactions with viral and host factors. Following entry into host cells and virion uncoating, the tegument protein pUL48 and its modulating proteins pUL46 and pUL47 play key roles in expression of immediate–early α genes. The α-trans-inducing factor pUL48 (VP16/α-TIF), in conjunction with cellular factors Oct-1 and HCF-1 (6), initiates the first wave of gene expression by binding to α-cis promoter sequences, resulting in transcription of the five α genes (ICP0, ICP4, ICP22, ICP27, and ICP47). These α genes are multifunctional, serving to block host antiviral defenses, hijack critical host pathways, and trans-activate expression of early (β) viral genes, which in turn promotes late (γ) viral gene expression (1, 3), and reviewed in (8). However, the picture is not so simple; leaky late viral genes (γ1) are expressed very early during infection and possess a variety of functional roles, whereas true late genes (γ2) are expressed much later and tend to perform structural roles in assembly and packaging (1). Previous evidence suggests that the leaky late (9) proteins pUL46 (VP11/12) and pUL47 (VP13/14) both modulate pUL48 function. Although pUL46 and pUL47 lack trans-activation activity themselves, cotransfection of pUL46 or pUL47 and pUL48 results in two- to threefold higher α promoter expression when compared with transfection of pUL48 alone (7, 10–12). Recent evidence has demonstrated that pUL47 might facilitate mRNA export and therefore enhance pUL48-induction expression of α genes (13).
In contrast, nonstructural functions for pUL46 during early stages of infection have not yet been clearly defined. Previous studies on pUL46 functions have largely focused on virion assembly and packaging during late stages of infection. Interestingly, localization studies have demonstrated that pUL46 is predominantly cytoplasmic (12, 14, 15), whereas pUL48 and pUL47 both undergo nucleo-cytoplasmic shuttling (16), suggesting that pUL46 is likely not involved in mRNA export. In conjunction with the expression profile of pUL46 as a high abundance, leaky late protein expressed during early stages of infection, these observations suggest that pUL46 may have other nonstructural roles in addition to modulation of pUL48-dependent trans-activation. Therefore, it is likely that other host and viral proteins that directly or indirectly interact with pUL46 are involved in its regulation. Yeast 2-hybrid studies have identified pUL46 as involved in assembly and packaging via interactions with several viral capsid proteins: pUL17 (17), pUL18, pUL19, pUL38, and viral packaging protein pUL25 (18). Furthermore, studies have demonstrated that pUL46 is essential for PI3K-Akt signaling, and that this occurs via interaction with the lymphocyte-specific Src family kinase Lck in T cells (19–21). However, the yeast 2-hybrid approach does not replicate normal cellular conditions, raising the likelihood of false positives, and T cells cannot be infected by cell-free HSV-1 in vitro. Therefore, to date no previous work has focused on identifying host or viral interacting partners of pUL46 in directly permissive cells. As such, the molecular players involved in pUL46-dependent modulation of pUL48 α-trans-activating activity, and more generally, in other nonstructural roles of pUL46, remain currently unknown. Furthermore, although pUL46 has long been known to be phosphorylated (22), knowledge regarding the precise site-localization or functions of these modifications remains limited.
Recent developments in mass spectrometry-based proteomics provide the opportunity to significantly expand our current knowledge about the identity and functions of virus-host and virus–virus interactions. Work from our and other labs has shown that affinity purifications at different stages of infection allow identification of spatial-temporal protein interactions, while simultaneously providing the necessary enrichment of the virus bait for confident detection of post-translational modifications (PTMs) (23). We have successfully implemented proteomic-based approaches for characterizing dynamic interactions following infections with Sindbis virus (24), human cytomegalovirus (HCMV) (25–27), and pseudorabies virus (PRV) (28). Overall, these studies have identified mechanisms involved in virus-induced modulation of host cell pathways, demonstrating the promise that proteomic approaches have for defining critical pathways in the virus life cycle and pathogenesis. However, currently very few proteomic studies have focused on HSV-1 infection, and, therefore, there is a need to better define virus protein interactions at specific stages of infection.
Here, we used a hybrid mass spectrometry approach to characterize the role and regulation of pUL46 during the HSV-1 life cycle. To our knowledge, this represents the first mass spectrometry-based proteomic study focused on pUL46, leading to the first evidence of a viral protein being targeted by another viral protein for proteasome-mediated degradation during HSV-1 infection. Using a GFP-tagged-pUL46-expressing HSV-1 strain, we integrated proteomic analysis and localization studies. Live-cell imaging revealed that pUL46 is expressed within distinct perinuclear and cytoplasmic punctae. We identified previously unreported pUL46 interactions with both viral and host proteins, as well as numerous pUL46 phosphorylations. Importantly, we identified, validated, and temporally defined the pUL46 interaction with the viral E3 ubiquitin ligase ICP0. We characterize the function of this interaction, demonstrating that pUL46 is targeted for degradation in an ICP0-dependent, proteasome-mediated manner during HSV-1 infection. Together, our results led us to propose a model in which pUL46 levels are tightly controlled by ICP0 and important for the temporal regulation of viral gene expression. Therefore, pUL46 possesses both structural and nonstructural roles at different stages of infection, with proteasomal degradation and phosphorylation modulating pUL46 abundance, interactions, and localization. These results contribute to the current understanding of the roles of virion-associated proteins in temporal regulation of viral gene expression during early stages of infection.
MATERIALS AND METHODS
Viruses and Cells
Three HSV-1(17+) strains were grown from bacterial artificial chromosomes (BACs) (Fig. 1A). The GFP control HSV-1 contained the wild-type genome in addition to free GFP expressed under the control of an MCMV promoter inserted between the ORFs of UL55 and UL56 (29). The HSV-1 (17+)Lox-VP11/12GFP, expressing the pUL46-GFP (Döhner, Sandbaumhuter, Sodeik, to be published elsewhere), contained the wild-type genome with the UL46 reading frame replaced with an identical reading frame except for a monomeric GFP sequence inserted 10 codons upstream of the C terminus (following the same cloning strategy described in (14)). The HSV-1(17+)Lox-UL37mGFP contained the wild-type genome with the UL37 reading frame replaced with an identical reading frame except for a monomeric GFP sequence inserted 2 codons upstream of the C terminus (30). All three constructs, provided by the Sodeik lab (Hannover Medical School, Hannover, Germany), had been generated from a parental wild-type BAC, HSV1(17+)Lox, using a traceless Red recombination system ((30–34) and Pohlmann N, Müther, Döhner and Sodeik, to be published elsewhere). To probe the interaction between pUL46 and ICP0, two additional HSV-1 (17+) strains were used. The ICP0 C116G/C156A mutant (ring finger/RF-dead) HSV-1 was constructed from a wild-type HSV-1 genome by PCR amplification, mutagenesis, and re-insertion by homologous recombination (35). Both wild-type and RF-dead HSV-1 were generously provided by Bernard Roizman (University of Chicago, Chicago, IL). BACs were maintained in E. coli strain GS1783 (31), and BAC DNA was harvested with a modified mini-prep protocol. Briefly, cells were pelleted, resuspended in CMPS (50 mm Tris pH 8.0, 10 mm EDTA pH 8.0, 200 μg/ml RNase A), lysed in alkaline SDS (1% SDS, 0.2 m NaOH), and neutralized with 3 m KAc pH 5.5. Bacterial DNA was precipitated away with isopropanol, and BAC DNA was precipitated with absolute ethanol and resuspended in distilled water. Vero cells were electroporated with BAC DNA, and cell-associated virus was harvested as a P0 stock by scraping, sonication in MNT buffer (30 mm MES, 100 mm NaCl, 20 mm Tris), and centrifugation to remove debris. P0 stock was expanded to infect additional fresh Vero cells and harvested similarly as a P1 stock that was used for experimentation. Unless where indicated, human foreskin fibroblasts (HFFs) were used as primary cells for experiments. Both HFFs and Veros were maintained in Dulbecco's modified Eagle medium (DMEM, GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Gemini Bio-Products, Irvine, CA) and 1% penicillin/streptomycin (P/S, GIBCO). During inoculation and infection, infected cells were maintained in DMEM supplemented with 2% FBS and 1% P/S.
Fig. 1.
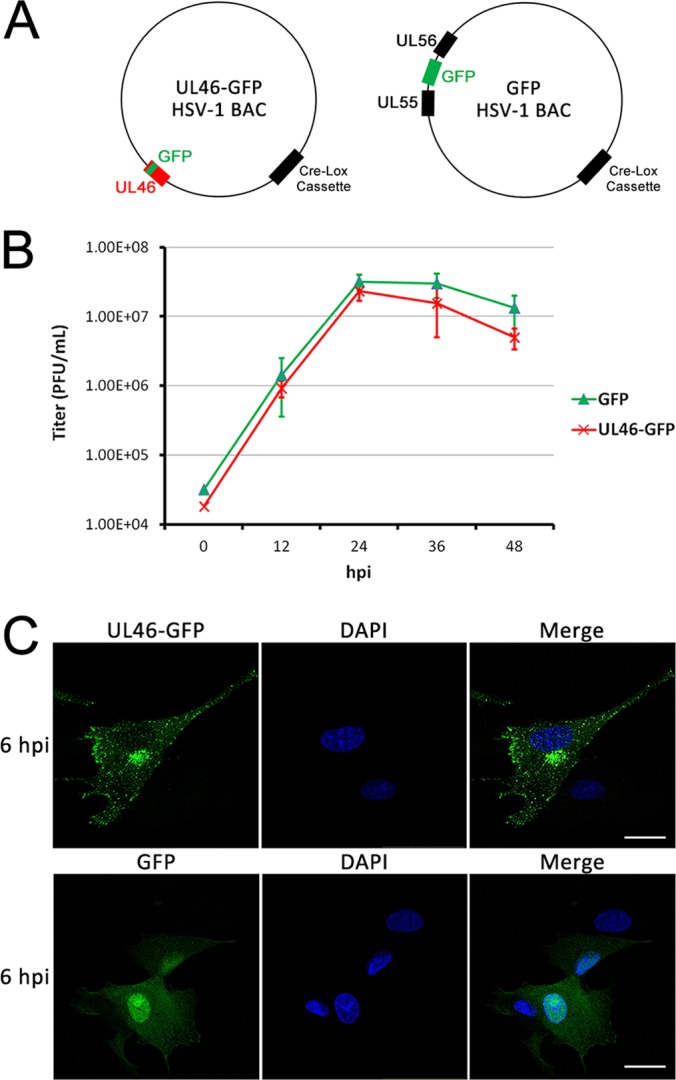
Characterization of a GFP-tagged pUL46 HSV-1 virus. A, Schematic of the BACs encoding for pUL46-GFP or free GFP-expressing HSV-1. B, pUL46-GFP HSV-1 grows with similar kinetics to free GFP-expressing HSV-1. Multistep growth curves were performed for both viruses in HFF cells. Cell-associated virus was harvested and titered at each time point by plaque assay. Error bars indicate S.E. from three biological replicates. C, pUL46-GFP accumulates to perinuclear and cytoplasmic punctae. Cells were infected with pUL46-GFP HSV-1 at an MOI of 0.5 and fixed at 6 hpi for direct fluorescence imaging. Scale bar demarcates 25 μm.
Antibodies
For all immunoaffinity isolations, an in-house rabbit polyclonal α-GFP (36) was used, with 5 μg of antibody conjugated per mg of magnetic bead (M-270 epoxy beads, Invitrogen, Carlsbad, CA), as previously described (37). For immunofluorescence studies, we used a mouse monoclonal α-ICP0 (1:10000, Virusys Corporation) and a goat α-mouse monoclonal antibody conjugated to Alexa Fluor 568 (1:2000, Invitrogen). Reciprocal co-immunoprecipitations were performed with mouse monoclonal α-ICP0 antibody (described above) at 1 μg per 20 μg of Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Antibodies used for Western blotting included rabbit polyclonal α-pan 14-3-3 (1:200, clone K-19, Santa Cruz Biotechnology), mouse monoclonal α-GFP (1:1000, clones 7.1 and 13.1, Roche), mouse monoclonal α-ICP0 (1:8000, described above), mouse monoclonal α-tubulin (1:200, clone B-7, Santa Cruz Biotechnology), mouse monoclonal α-tubulin (1:5000), mouse monoclonal α-ICP8 (1:1000, clone 10A3, Santa Cruz Biotechnology), mouse monoclonal α-DNA-PKCS (1:200, clone G-4, Santa Cruz Biotechnology), and mouse monoclonal α-ubiquitin (1:200, clone P4D1, Santa Cruz Biotechnology).
Cold-synchronized Viral Infection
For microscopy experiments and low MOI infections at early time points when synchronicity is essential, cold synchronization was performed to ensure that viruses entered at the same time. For mass spectrometry experiments, our initial infection used cold synchronization as well; however, following our observation that a good synchronicity of infection was achieved at the high MOI of 10 and at 6 hpi, further biological replicates were performed without cold synchronization. Cells were pre-chilled for 15 min at 4 °C, and then incubated at 4 °C with virus in CO2-independent DMEM (GIBCO), supplemented with 2% FBS and 0.1% (w/v) BSA for 1 h to allow for adsorption but not entry. Excess media was aspirated to remove nonadsorbed virus, and cells were incubated at 37 °C for 1 h with fresh DMEM supplemented with 2% FBS and 1% P/S to allow entry of adsorbed virus. After entry, viruses that had not entered were inactivated with a rapid citrate buffer wash (40 mm citrate, 135 mm NaCl, 10 mm KCl, pH 3.0) before incubation in DMEM supplemented with 2% FBS and 1% P/S.
Live-cell Imaging
HFFs were synchronously infected with HSV1-pUL46-GFP at a multiplicity of infection (MOI) of 10 pfu/cell. Images were captured on a Nikon Ti-Eclipse epifluorescent inverted microscope from 2 h post infection (hpi) to 24 hpi. Images were viewed and analyzed by ImageJ.
Immunoaffinity Purification of pUL46 With Viral and Host Protein Interactions
For mass spectrometry-based experiments, infected cells were harvested at 6 or 14 hpi by scraping and cryogenic preservation in PVP-HEPES buffer (20 mm Na-HEPES, 1.2% (w/v) PVP, pH 7.4) containing 1/200 (v/v) phosphatase inhibitor cocktails 2 and 3 (Sigma-Aldrich) each, and 1/100 (v/v) protease inhibitor mixture (Sigma-Aldrich). Because starting material was greater than 0.2 g, cells were frozen drop wise in liquid nitrogen, and cryogenically lysed into cell powder using a Retch MM301 Mixer Mill (Retch), as previously described (38). Differential lysis and wash conditions used for immunoaffinity purification can distinguish weak from strong interactions by sampling different subcellular compartments. Here, cells or cell powder were further lysed for 20 min using (1) mild buffer - 20 mm K-HEPES pH 7.4, 110 mm KOAc, 2 mm MgCl2, 0.1% (v/v) Tween, 1 μm ZnCl2, 1 μm CaCl2, 250 mm NaCl, 0.5% (v/v) Triton X-100, 4 μg/ml DNase, 1/200 (v/v) each phosphatase inhibitor cocktails 2 and 3, 1/100 (v/v) protease inhibitor mixture, or (2) stringent buffer - 20 mm K-HEPES pH 7.4, 0.1% (v/v) Triton X-100, 0.05% (w/v) sodium deoxycholate, 0.03% (w/v) sodium N-lauryl-sarcosine, 1 mm DTT, 4 μg/ml DNase, 1/200 (v/v) each phosphatase inhibitor cocktails 2 and 3, 1/100 (v/v) protease inhibitor mixture. The stringent buffer was modified from a buffer that had been previously been optimized for extraction of proteins from vesicles and membranes (39), because our microscopy studies of pUL46 suggested that it accumulated within perinuclear and cytoplasmic punctae. Samples were subject to homogenization by Polytron (Kinematica). Insoluble material was pelleted by centrifugation at 8000 × g for 10 min at 4 °C, and pUL46-GFP was immunoaffinity purified from the supernatant using a 1 h incubation at 4 °C with magnetic beads (M-270 epoxy beads, Invitrogen) conjugated to in-house rabbit polyclonal α-GFP antibody (36). Beads were washed 6 times with either 20 mm K-HEPES pH 7.4, 110 mm KOAc, 2 mm MgCl2, 0.1% (v/v) Tween, 1 μm ZnCl2, 1 μm CaCl2, 500 mm NaCl, 0.5% (v/v) Triton X-100 or with 20 mm K-HEPES pH 7.4, 0.1% (v/v) Triton X-100, 0.05% sodium deoxycholate, 0.03% sodium N-lauryl-sarcosine, 1 mm EDTA to remove nonspecific interactions. Co-isolated proteins were eluted from beads with TEL buffer (160 mm Tris, 1.3% (w/v) LDS, 0.34 mm EDTA), and reduced and denatured with 100 mm DTT at 70 °C for 10–20 min. For smaller scale experiments where proteins were detected by Western blot rather than mass spectrometry, less than 0.2 g of cells were used. The same protocol was followed, except that frozen cell droplets were not ground by Retch MM301 Mixer Mill nor subjected to homogenization by Polytron because the starting material was too small. To test the presence of pUL46 ubiquitination, a more stringent isolation of pUL46-GFP or GFP was performed to ensure enrichment of the bait. Lysis conditions were as follows: 50 mm Tris pH 6.8, 150 mm NaCl, 0.5% (w/v) sodium deoxycholate, 1% (w/v) Triton X-100, 1/100 (v/v) protease inhibitor mixture. Wash conditions were as follows: 50 mm Tris pH 6.8, 0.05% (w/v) sodium deoxycholate, 0.1% (w/v) Triton X-100.
Mass Spectrometry-based Proteomic Sample Preparation
Samples were prepared for mass spectrometry analysis using a filter assisted sample preparation (FASP) for in-solution digestion as described previously (40, 41). Proteins eluted from beads were buffer exchanged with 0.1 m TrisHCl, pH 8.0, 8 m urea (UB) in 30 kDa Millipore filters (Millipore). Proteins were alkylated with 100 mm iodoacetamide (IAA) for 45 min, buffer-exchanged into 50 mm ammonium bicarbonate, and digested with 100 μl of 5 ng/μl trypsin in ammonium bicarbonate overnight at 37 °C. Peptides were concentrated by vacuum centrifugation and diluted to 1% (v/v) TFA. Acidified peptides were desalted by StageTips (42) constructed from Empore C18 material (3 m). Desalted peptides were concentrated to 2 μl and then diluted to 10 μl with 1% formic acid. Peptides were either analyzed directly or stored at −20 °C before nLC-MS/MS analysis.
Nanoliquid Chromatography-Tandem Mass Spectrometry Analysis
For each biological replicate, desalted samples were analyzed on an LTQ Orbitrap Velos or LTQ Orbitrap XL coupled online to a Dionex Ultimate 3000 UHPLC (both from Thermo Fisher Scientific). Peptides were injected directly onto the analytical column (Acclaim PepMap RSLC; 1.8 μm, 75 μm × 25 cm) and separated by nanoLC at 250 nL/min over a 3 h discontinuous acetonitrilereverse-phase gradient using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in 97% acetonitrile (mobile phase B) as follows: 4 to 16% B over 90 min, and 16% to 40% B over 90 min. nLC-separated peptides were introduced into the mass spectrometer by nano-electrospray ionization and peptide precursors were analyzed by either a data-dependent method, as briefly described. A single data acquisition cycle consisted of a full scan mass spectrum (m/z = 350 - 1700) in the Orbitrap (resolution = 30,000), and fragmentation of the 10 (XL) or 20 (Velos) most intense ions (min. signal = 1E3) by collision-induced dissociation (CID) in the linear ion trap. The instrument settings for both Velos and XL configurations were as follows, except as noted: predictive AGC disabled and lock-mass enabled (Velos only); FT MS target values of 1E6; IT MSn (tandem MS) target values of 5E3 (XL) or 1E4 (Velos); FT MS maximum injection time of 500 ms, IT MSn max injection time 100 ms (XL) or 125 ms (Velos). CID was performed at an isolation width of 2.0 Th, normalized collision energy of 30, and an activation time of 10 ms (Velos) or 30 ms (XL), with dynamic exclusion enabled (repeat count, 1; exclusion duration, 45–60 s; exclusion mass width, 7 ppm.
Database Search and Peptide-spectrum Assignment Scoring
Acquired spectra were separately searched by Proteome Discoverer (PD ver. 1.3)/SEQUEST (ver. 1.2) against tryptic forward and reverse UniProt FASTA containing human and herpesvirus sequences appended with common contaminants (21, 569 sequences). Search parameters used included, full trypsin specificity; maximum missed cleavages, 2; precursor tolerance, 10 ppm; fragment tolerance, 0.5 Da; carbamidomethylation of Cys as a static modification, oxidation of Met, and phosphorylation of Ser, Thr, and Tyr residues as variable modifications. Peptide spectral matches were assembled into protein groups by Scaffold (ver. 3.6.5, Proteome Software, Inc.) using the PeptideProphet and ProteinProphet algorithms (43, 44) followed by X!Tandem (GPM 2010.12.1.1) re-search using the subset database parameter and additional variable modification of deamidation of asparagine/glutamine (+1 Da), oxidation of tryptophan (+16 Da), acetylation of lysine (+42 Da), and pyro-Glu formation of peptide N-terminal glutamate (−17 Da). Proteins groups were reported that had at least two unique peptides/protein in at least one biological sample, and Peptide and protein confidence filters were selected to limit peptide and protein global false discovery rates to < 1% based on reverse database assignments. Protein isoforms were automatically distinguished by Scaffold using unique peptides. Viral proteins were manually inspected and accession numbers were matched to the HSV-1 17+ strain when necessary. Proteins passing these filters were exported as “unweighted spectrum counts” for analysis by the Significance Analysis of INTeractions (SAINT) algorithm (45), as detailed below. For phosphorylation analysis, the Percolator algorithm was used to calculate posterior error probabilities (PEP) and q-values for forward database peptides-spectrum matches. Top-ranked peptide-spectrum matches were filtered to q-values < 0.01, estimating < 1% global FDR at the peptide level. Phosphosite localization probabilities for phosphopeptide MS2 spectra were calculated by the phosphoRS (ver. 2.0) algorithm. All site assignments > 0.89 were then manually inspected to ensure accuracy in site localization.
Label-free Spectral Counting Analysis Using SAINT
SAINT statistically models spectral count distributions for each protein in the experimental isolations versus the corresponding control isolations to distinguish between specific and nonspecific interactions (45). For the stringent isolation conditions, SAINT probability scores of 2 biological replicates were averaged. As we predicted that milder conditions would capture more transient and weaker interactions, three biological replicates were performed, and the highest 2 of 3 SAINT probability scores for each protein were averaged. Proteins were considered as putative specific interacting partners only if the average SAINT probability exceeded 0.9 in either the mild or stringent isolation conditions.
Immunofluorescence Microscopy
HFFs were seeded sparsely on glass coverslips and infected with pUL46-GFP HSV-1 at a MOI of either 0.5 or 0.25 PFU/cell, both resulting in low MOIs sufficient for visualizing both infected and neighboring uninfected cells. Cells were fixed at 3, 6, and 14 hpi with 2% (v/v) paraformaldehyde (PFA) for 15 min at room temperature. Cells were permeabilized for 15 min with 0.1% (v/v) Triton X-100 in PBS and washed 3 times with wash buffer (0.2% (v/v) Tween-20 in PBS). Blocking was performed for 1 h with a blocking solution (2% (w/v) BSA, 2.5% (v/v) human serum, 0.2% (v/v) Tween in PBS), optimized (supplemental Fig. S1) to avoid high-affinity, nonspecific binding of antibodies to the HSV-1 Fc receptor (46). Although pUL46-GFP was detected by direct fluorescence to avoid excess use of antibodies, other proteins were detected by indirect immunofluorescence. Cells were protected from light with aluminum foil and incubated for 45 min with the appropriate primary antibody, and unbound antibody was removed with 3 washes with wash buffer. Cells were incubated for 1 h with the appropriate secondary antibody, and unbound antibody was removed with 3 washes of wash buffer. Nuclei were stained with 1.33 μg/ml DAPI (Sigma-Aldrich) for 15 min, and unbound dye was removed with 3 washes. Images were captured on a Leica SP5 confocal microscope with a 63× oil immersed objective. Images were viewed and analyzed by LAS AF Lite software (Leica Microsystems).
Reciprocal Immunoaffinity Purification
Reciprocal immunoaffinity purification was used to validate interactions identified from the AP-MS/MS studies. At 6 hpi, infected cells were harvested by scraping and cryogenic preservation in PVP-HEPES buffer (described above) containing 1/200 (v/v) phosphatase inhibitor cocktails 2 and 3 (Sigma-Aldrich) each and 1/100 (v/v) protease inhibitor mixture (Sigma-Aldrich). Cells were lysed under mild conditions (described above) for 20 min. Insoluble material was pelleted by centrifugation at 8000 × g for 10 min at 4 °C, and protein complexes were immunoaffinity purified for 1 h at 4 °C from the supernatant with 20 μl of primary antibody and Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology), which had been pre-incubated together overnight at 4 °C. Agarose beads were washed 2 times under mild conditions (described above) and 2 additional times with PBS to remove nonspecific protein–protein interactions, and protein complexes were eluted from beads with electrophoresis sample buffer, denatured at 95 °C for 10–20 min, and separated from beads by centrifugation.
RESULTS
pUL46GFP fusion HSV-1 Strain is Functional
GFP fusion proteins as fluorescent probes and as tags for immunoaffinity purification have emerged as powerful means for integrating knowledge regarding protein localization and interactions (36, 47), and as effective tools for studying interactions in the context of viral infection (24–28). To use a similar approach for identifying pUL46 interactions during infection, virus stocks of HSV-1 (17+ strain) were generated by electroporation of BACs (provided by the Beate Sodeik lab), into Vero cells (Fig. 1A). The HSV1(17+)Lox-VP11/12GFP virus strain has been engineered to contain a GFP sequence fused in-frame near the C terminus of the UL46 gene (HSV1(17+)Lox-VP11/12GFP in the same manner as described for a HSV1(F) strain before (14)). HSV-1 expressing a freely diffusible EGFP under the control of an MCMV promoter (GFP HSV-1, (29)) was used as a control. Multi-step viral growth curves were performed to ensure that the viral growth rates for the HSV1(17+)Lox-VP11/12GFP and the control GFP HSV-1 were similar, and therefore, that the functions of pUL46 were not affected by tagging. Primary human foreskin fibroblasts (HFFs), which are permissive targets for HSV-1 infection, were infected with either HSV1(17+)-VP11/12GFP (expressing pUL46-GFP) and HSV1(17+)-GFP (expressing GFP alone) at a low multiplicity of infection (MOI) of 0.05. Infected cells were harvested every 12 h for 48 h post-infection (hpi), and virus production was measured by plaque assay. The results demonstrate that HSV1(17+)-VP11/12GFP and HSV1(17+)-GFP grew at a comparable rates, suggesting that the UL46-GFP fusion does not impair viral function or growth (Fig. 1B). We next confirmed that the localization of pUL46-GFP at a middle stage of infection was similar to previous reports for both the tagged and untagged viral protein (12, 14, 15). At 6 hpi, pUL46-GFP was predominantly cytoplasmic (Fig. 1C). pUL46-GFP-containing cytoplasmic punctae were particularly intense within perinuclear regions (Fig. 1C) that are similar to sites of secondary envelopment and previously observed structures that act as scaffolds for assembly in virus-infected cells (30, 33). In contrast, free GFP expression was diffusely localized to both the cytoplasm and nucleus, suggesting that perinuclear accumulation of pUL46-GFP was not because of the GFP fusion. Altogether, these results for virus growth and pUL46 localization demonstrate that the tagged pUL46-GFP HSV-1 virus strain is functional and can be used for characterizing pUL46 localization and interactions.
Life Cell Imaging Demonstrates a Dynamic Localization of pUL46-GFP to Perinuclear and Cytoplasmic Puncta Throughout Viral Infection
To gain a temporal view of pUL46-GFP localization during infection and specifically to determine whether the perinuclear puncta remain throughout the virus life cycle, we performed live-cell imaging. HFFs were synchronously infected with pUL46-GFP HSV-1 at a high MOI of 10 and analyzed by time-lapse epifluorescence microscopy from 2 to 24 hpi (supplemental Movie S1). The high MOI was utilized to ensure a synchronous infection of all cells in the fields of view, and an accurate depiction of the different stages of infection. pUL46-GFP remained predominantly localized within cytoplasmic puncta throughout the virus life cycle (Fig. 2 and supplemental Movie S1). In agreement with the expression of pUL46 as a leaky late protein, we were able to detect it in most cells as early as 3 hpi (Fig. 2), and this localization likely derives from newly synthesized pUL46-GFP rather than pUL46-GFP from incoming virions. Between 0 and 2 hpi, we were able to observe fluorescence from pUL46-GFP at a much higher MOI of 100 (supplemental Fig. S2), in agreement with data from other groups (14, 48). In this case, virion-associated pUL46-GFP also began to accumulate in the cytoplasm at perinuclear regions similar to those observed at middle and late stages of infection. The retention of these structures throughout the viral life cycle suggests that they are likely important for viral-host dynamics and warrant further study.
Fig. 2.
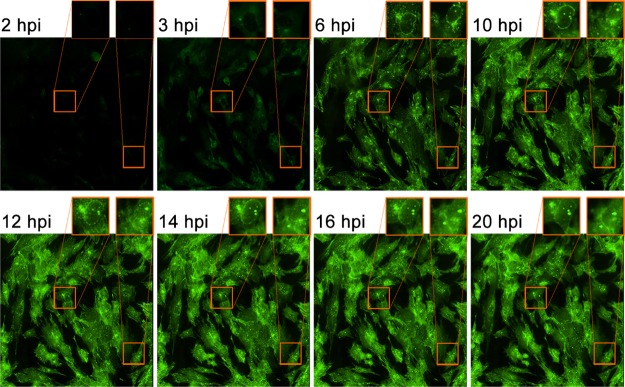
Live-cell imaging captures dynamic pUL46-GFP localization. HFFs were infected with pUL46-GFP HSV-1 at an MOI of 10 and imaged by epifluorescence time-lapse microscopy from 2 to 24 hpi. Magnified inserts indicate perinuclear accumulations.
Proteomic-based Approach Identifies pUL46 Interactions With Viral and Host Proteins During Infection
Perinuclear accumulations, likely sites of secondary envelopment, have been previously reported in the context of infection (30, 33), and are thought to contain host and viral proteins, including pUL46 (12, 14, 15). However, their composition remains poorly defined. Previous studies using a variety of methods have identified several viral protein interactions involving pUL46 (17, 18, 49, 50), but, to date, no proteomic approach has targeted pUL46 itself for interaction studies. Given our results for the temporal localization of pUL46-GFP, we selected the 6 hpi as a representative time point for characterizing pUL46 interactions. At this time point, nearly all infected cells demonstrated intense accumulation of pUL46 within cytoplasmic puncta and at perinuclear aggregates. Previous reports have implicated pUL46 in pUL48 modulation (7, 10–12), PI3K/Akt signaling (19–21), and assembly/packaging (17, 18, 51). Although the first two events occur early during infection and in the nucleoplasm, assembly of new virions occurs much later during infection in the cytoplasm. As a result, isolation of pUL46-containing complexes at 6 hpi would temporally separate these early nonstructural functions from later structural ones, and would provide information about pUL46 interactions that reflect these functions.
To characterize pUL46 interactions at 6 hpi, we employed an immunoaffinity purification approach that has been optimized for use with GFP tags (25, 28, 36). HFFs were infected at a high MOI of 10 with either pUL46GFP or GFP HSV-1, and harvested at 6 hpi. Two distinct lysis buffer conditions were optimized for the immunoaffinity purifications, one that we termed “mild condition,” which would provide a global view of pUL46 interactions, and a second one that we termed “stringent condition,” which we recently optimized and reported for studying interactions within vesicles (39), and which we predicted to give us a better access to puncta-containing interactions. pUL46-GFP- or GFP-containing protein complexes were isolated in triplicate or duplicate biological replicate experiments, respectively, subjected to in-solution digestion with trypsin, and analyzed by nLC-MS/MS on an LTQ Orbitrap Velos or an Orbitrap XL (Fig. 3A). Proteins and their respective spectral counts identified in pUL46-GFP and control GFP isolations were subjected to the Significance Analysis of INTeractome (SAINT) algorithm, which statistically models spectral count distributions to distinguish between specific and nonspecific interactions (45). For each identified protein, SAINT calculates an interaction specificity score, which allows selection of more unbiased thresholds for determining putative pUL46-GFP interactions. To focus on the most likely specific pUL46 interactions, we considered as putative interactions only proteins with SAINT probability scores above 0.9 (supplemental Tables S1 and S2). Indeed, this threshold retained the pUL46 bait and its previously reported interactions pUL21, pUS10, glycoprotein M (18), and pUS3 (50).
Fig. 3.

pUL46 associates with viral and host proteins following HSV-1 infection. A, Schematic of proteomic workflow for identification of specific interacting partners of pUL46. B, Spectral count comparison for pUL46 interactions observed using mild and stringent isolation buffer conditions. Viral and host proteins with SAINT probability scores greater than 0.9 were considered likely specific, and the ratios of spectral counts in the stringent versus mild conditions were plotted on a log2 scale. Select functional clusters of host and viral proteins are illustrated with circles and diamonds, respectively.
Based on the differential identification of putative pUL46 interactions under the mild or stringent conditions, we expect that (1) stable/strong pUL46 interactions would be found under both mild and stringent conditions, (2) weaker or transient interactions would be identified only under mild isolation conditions, and (3) the more stringent condition may provide access to an additional subset of pUL46 interactions. Indeed, the previously reported pUL46 interactions pUL21, pUS3, pUS10, and gM were identified in our isolations under both mild and stringent conditions. One previously unreported interaction that was also prominent in both mild and stringent conditions was the viral E3 ubiquitin ligase ICP0 (Fig. 3B). ICP0 was of particular interest because it is one of the five viral α proteins whose expression is partly controlled by pUL46; moreover, its function as an E3 ubiquitin ligase raises the possibility of regulation of pUL46 levels by ubiquitin-mediated degradation. Additionally, numerous proteins that play functional roles in phosphorylation were identified, including the viral kinases pUL13 and pUS3, host kinases CaMKIId and Src, signaling proteins Grb2 and GPSM1 in MAPK/ERK and G-protein signaling, respectively, and phosphatase subunits PP1CA and PP1CB. In agreement with a possible critical role for phosphorylation in regulating pUL46 functions, we identified phospho-binding 14-3-3 proteins enriched in the mild isolation condition, suggesting that these interactions may be weak or transient. To determine if these pUL46 interactions persist during HSV-1 infection, we performed immunoaffinity purifications of pUL46GFP or GFP alone as control at 14 hpi, using the workflow described above and a mild isolation condition (Fig. 4 and supplemental Tables S3 and S4). These analyses confirmed pUL46 interactions with viral structural proteins that were previously reported from yeast 2-hybrid or mass spectrometry studies, including pUL17, pUL48, pUL21, pUS3, pUS10, and gM. Additionally, several previously unreported interactions with viral structural proteins were observed as enriched at 14 hpi when compared with 6 hpi. These included the tegument proteins thymidine kinase (TK) and pUL41, the envelope proteins gI and gC, and the capsid protein pUL35, suggesting the presence of pUL46 within intermediate virion structures at later stages of infection. Importantly, our results demonstrated that the pUL46 interactions with ICP0 and the 14-3-3 proteins were consistent, being observed both at 6 and 14 hpi. Altogether, these pUL46 interactions point to an important role for post-translational modifications, such as ubiquitination and phosphorylation, in regulating pUL46 functions during HSV-1 infection.
Fig. 4.
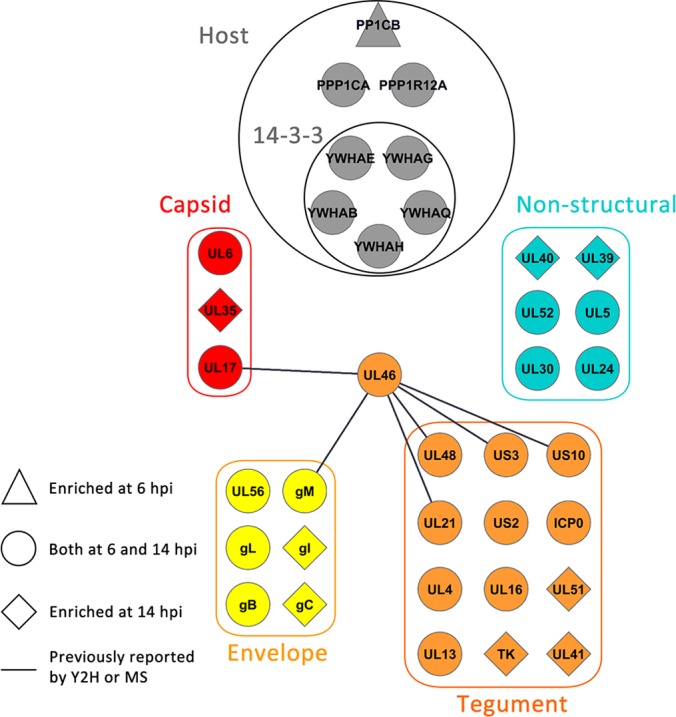
pUL46 associates with viral structural proteins at late stages of infection. Spectral count comparison for pUL46-GFP interactions observed at 6 hpi versus 14 hpi. Shapes indicate the enrichment of isolated viral or host proteins at the different time points. Select clusters of host and viral proteins are illustrated.
pUL46 is Heavily Phosphorylated Within Both Signature Host and Viral Kinases Motif
Until recently, no phosphorylation sites on pUL46 had been localized to specific residues. A recent study demonstrated that six phosphoserine/threonine residues cluster at the C terminus of pUL46 (52), whereas the tyrosine phosphorylation predicted in Vero and T cells (19–21) has not previously been localized to specific residues. Here, enrichment of pUL46 by immunoaffinity purification allowed us to confirm all previously reported sites and confidently identify 17 new phosphorylation sites, including 4 phosphotyrosine and 13 phospho-serine and -threonine sites (Table I and supplemental Fig. S4). Seven of these sites contain the minimal motif of the viral kinase pUL13 (S-P, (53)), which we identified as a previously unreported pUL46 interaction. Moreover, we confirmed previous results that the viral kinase pUS3 is a specific pUL46 interaction (Fig. 3B), and identified phosphosites matching the pUS3 consensus motif R-R-[R/A/V/P/S]-[pS/pT]-X-X (where X is any nonacidic residue) (54–56) at the previously unreported T431 phosphosite and the recently identified S554 site (52). The abundance of viral kinase motifs on pUL46, in conjunction with its proline-rich nature, may lend itself to hyper-phosphorylation. Indeed, it is known that other virion-associated proteins such as pUL36, pUL47, pUL48, and pUL49 dissociate from the capsid on phosphorylation, both by pUL13 and host kinases (57). Here, because we observed several phosphorylation sites on pUL46 at 6 hpi, well after virion uncoating, our results suggest that phosphorylation of pUL46 might mediate its interactions and functions at middle stages of infection.
Table I. Identification of pUL46 phosphorylation during HSV-1 infection. Twenty-three total modifications are listed, including 17 previously unreported sites. All spectra were manually inspected to ensure correct site localization. Sites of ambiguity with two adjacent potential phosphosites are indicated by parentheses.
| Phosphosite(s) | Sequence(s) | z | Xc | PEP | phosphoRS (%) | Δppm | Reference |
|---|---|---|---|---|---|---|---|
| T18 | CLtPANLIR | 2 | 3.56 | 2.60E-05 | T(3): 100.0 | −4.53 | This study |
| S263 | FLPLGGsPEAPAETFAR | 2 | 5.21 | 2.30E-08 | S(7): 100.0 | −3.29 | This study |
| T431 (or T430) | RTtWSAGPPPDDMASGPGGHR | 3 | 5.61 | 1.20E-05 | T(3): 92.6; T(2): 6.9 | 0.28 | This study |
| S433 | TTWsAGPPPDDMASGPGGHR | 3 | 3.34 | 2.40E-06 | S(4): 99.7 | −4.09 | This study |
| T431/S433 | RTtWsAGPPPDDMASGPGGHR | 3 | 5.05 | 8.80E-10 | T(3): 94.6; S(5): 100.0 | −3.99 | This study |
| S443 | TTWSAGPPPDDMAsGPGGHR | 3 | 3.32 | 6.60E-04 | S(14): 100.0 | −4.35 | This study |
| S478 | LHsTPASTR | 2 | 2.66 | 1.50E-02 | S(3): 99.2 | 1.17 | 21 |
| S478/T479 | LHstPASTR | 2 | 3.51 | 1.70E-03 | S(3): 100.0; T(4): 100.0 | 0.09 | This study |
| S478/S481 | LHsTPAsTR | 2 | 3.2 | 4.90E-02 | S(3): 92.1; S(7): 92.0 | −4.01 | This study |
| T545 | RPPAAtCPLLVR | 3 | 2.38 | 1.80E-04 | T(6): 100.0 | −3.35 | This study |
| S554 | RAsLGSLDRPR | 2 | 2.33 | 8.90E-02 | S(3): 99.9 | −0.88 | 21 |
| S554/S557 | RAsLGsLDRPR | 2 | 2.98 | 2.30E-02 | S(3): 100.0; S(6): 100.0 | −2.94 | This study |
| Y579 (or T578) | VWGPAPEGEPDQMEATyLTADDDDDDAR | 3 | 5.26 | 1.50E-06 | Y(17): 99.3 | −0.96 | This study |
| T581 | VWGPAPEGEPDQMEATYLtADDDDDDAR | 3 | 6.21 | 7.00E-15 | T(19): 100.0 | −0.47 | This study |
| S611 | HAPYEDDEsIYETVSEDGGR | 3 | 4.28 | 1.80E-05 | S(9): 98.6 | −0.59 | This study |
| Y613 | HAPYEDDESIyETVSEDGGR | 3 | 4.76 | 4.20E-09 | Y(11): 100.0 | 0.42 | This study |
| T615 | HAPYEDDESIYEtVSEDGGR | 3 | 4.27 | 1.20E-06 | T(13): 99.9 | −0.52 | This study |
| S617 | HAPYEDDESIYETVsEDGGR | 3 | 5.58 | 2.00E-07 | S(15): 100.0 | 0.34 | 21 |
| Y606/S617 | HAPyEDDESIYETVsEDGGR | 3 | 4.33 | 1.70E-03 | Y(4): 99.3; S(15): 100.0 | −3.65 | This study |
| S611/S617 | HAPYEDDEsIYETVsEDGGR | 3 | 5.61 | 2.80E-07 | S(9): 100.0; S(15): 100.0 | −0.33 | This study |
| Y613/S617 | HAPYEDDESIyETVsEDGGR | 3 | 4.76 | 3.30E-08 | Y(11): 100.0; S(15): 100.0 | 1.03 | This study |
| T615/S617 | HAPYEDDESIYEtVsEDGGR | 3 | 3.64 | 3.00E-04 | T(13): 100.0; S(15): 100.0 | 0.5 | This study |
| Y606/T615/S617 | HAPyEDDESIYEtVsEDGGR | 3 | 4.32 | 7.50E-04 | Y(4): 92.2; S(9): 7.7; T(13): 100.0; S(15): 100.0 | −1.33 | This study |
| S611/Y613/S617 | HAPYEDDEsIyETVsEDGGR | 3 | 5.37 | 2.00E-05 | S(9): 100.0; Y(11): 100.0; S(15): 100.0 | −2.2 | This study |
| Y624 | VyEEIPWMR | 2 | 3.08 | 6.00E-05 | Y(2): 100.0 | −3.94 | This study |
| S647 | VYENVCVNTANAAPAsPYIEAENPLYDWGGSALFSPPGR | 3 | 6.71 | 3.60E-08 | S(16): 89.2; Y(18): 10.8 | −1.6 | This study |
| S666 | VYENVCVNTANAAPASPYIEAENPLYDWGGSALFsPPGR | 4 | 4.08 | 5.00E-05 | S(35): 100.0 | −0.7 | This study |
| S679 | TGPPPPPLsPSPVLAR | 3 | 3.3 | 7.60E-05 | S(9): 100.0 | 0.06 | 21 |
| S681 | TGPPPPPLSPsPVLAR | 2 | 4.11 | 6.00E-06 | S(11): 100.0 | −0.04 | 21 |
| S679/S681 | TGPPPPPLsPsPVLAR | 3 | 4.15 | 2.90E-07 | S(9): 100.0; S(11): 100.0 | −4.02 | This study |
Phospho-binding 14-3-3 Proteins are pUL46 Host Interacting Partners
The identification of pUL46 hyper-phosphorylation led us to hypothesize that these modifications are likely to be relevant for protein function. Interestingly, the S478 phosphoserine sites (Fig. 5B) matched the well-established 14-3-3 binding motif R-X-X-pS-X-P (58). 14-3-3 subunits comprise a family of proteins known to bind to other phosphorylated proteins to critically regulate their localizations and functions. Through these phospho-binding properties, 14-3-3 proteins play key roles in signaling, cell cycle, apoptosis, and transcription (reviewed in (59, 60)). To determine if the pUL46 interaction with 14-3-3 may be a conserved feature, we analyzed the corresponding sequences for diverse alpha herpes viruses. Indeed, the 14-3-3 binding motif in pUL46 is highly conserved among alpha herpes viruses, even across diverse species (Fig. 5C), suggesting an important functional relationship between pUL46 and 14-3-3. Interestingly, the average spectral counts detected for several of the 14-3-3 proteins, including isoforms gamma, epsilon, theta, eta, and beta/alpha, were 1.5–3.5-fold higher in the mild compared with the stringent isolation conditions (Fig. 5D). This suggests that the interaction between these proteins and pUL46 may be weak or transient, in agreement with the putative binding of 14-3-3 to the phosphorylated S478 site in pUL46.
Fig. 5.
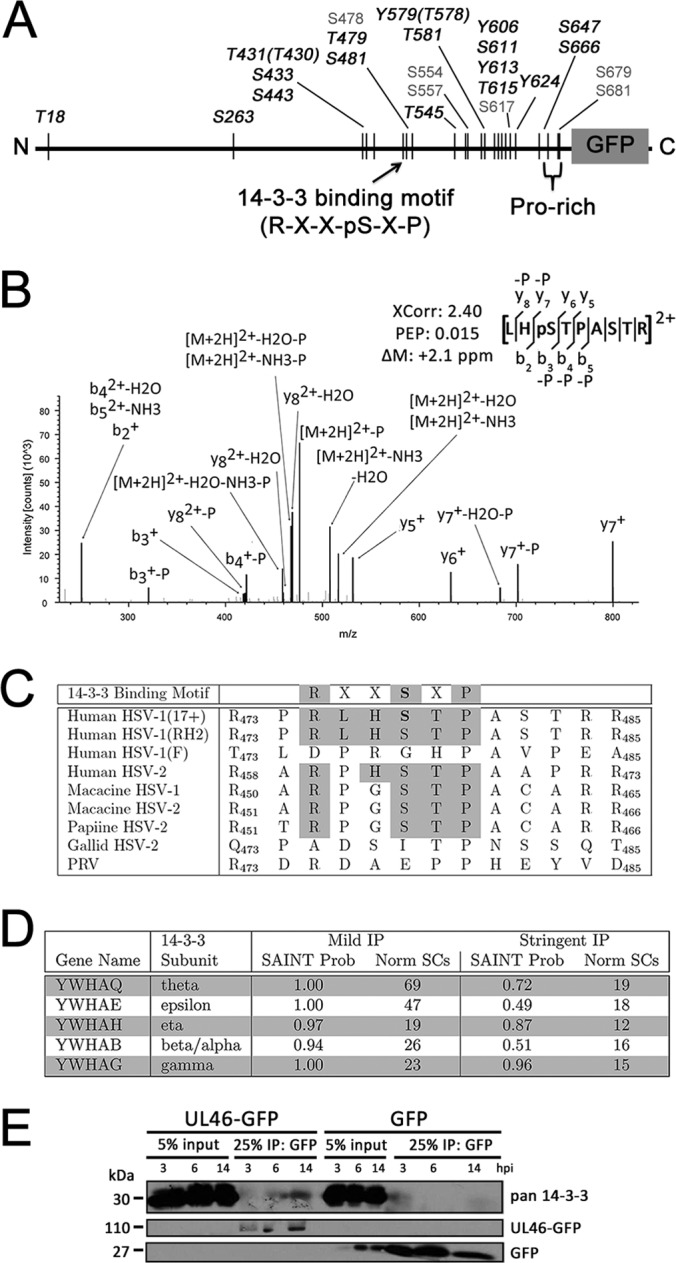
pUL46 is highly phosphorylated and associates with host 14-3-3 protein subunits. A, Schematic of phosphorylation sites identified in this study; black denotes novel and gray denotes previously reported modifications. B, Annotated MS2 spectra of a pUL46 phosphopeptide matching the putative 14-3-3 binding site. C, Sequence conservation of the pUL46 putative 14-3-3 binding motif site. D, Summary of proteomic data illustrating the co-isolation of 14-3-3 subunits with pUL46. SAINT probability scores and average spectral counts for both mild and stringent conditions are shown. E, The pUL46 interaction with 14-3-3 is observed at different stages of infection. HFFs were infected with either pUL46-GFP or GFP HSV-1 at an MOI of 10 and harvested at 3, 6, and 14 hpi. pUL46-GFP- or GFP-containing protein complexes were immunoaffinity purified with α-GFP antibody and separated by SDS-PAGE.
Given this potential for a dynamic, functional interaction between 14-3-3 and pUL46, we investigated their interaction at different time points of infection. In support of our proteomic data, 14-3-3 proteins were co-isolated with pUL46GFP at 3, 6, and 14 hpi, suggesting that this interaction is required throughout the course of infection. Given that both proteins are highly abundant in the cytoplasm, it is possible that 14-3-3 proteins modulate pUL46 localization to perinuclear aggregates or cytoplasmic punctae.
pUL46 Specifically Interacts With the Viral E3 Ubiquitin Ligase ICP0 Within Perinuclear Aggregates
Among the most stable pUL46 interacting partners identified as specific in both mild and stringent conditions was the viral E3 ubiquitin ligase, ICP0. ICP0 is a critical HSV-1 protein known to be necessary for the ubiquitin-mediated degradation of host defense factors during infection. As a viral α protein essential for wild-type HSV-1 growth, ICP0 is one of the first five proteins trans-activated by pUL48 in conjunction with pUL46 and pUL47, propagating downstream gene expression as part of the temporal cascade of HSV-1 gene expression. This interaction with ICP0 is particularly interesting because it suggests that pUL46 works to both enhance expression of this α protein (via modulation of pUL48), as well as interact with it following translation for some yet unknown function.
To support our proteomic results, we performed immunofluorescence studies to determine whether these proteins colocalize. First, we assessed their localization at the time of infection used in our immunoaffinity purification studies. Indeed, at 6 hpi in HFF cells, a strong colocalization of ICP0 and pUL46GFP was observed predominantly within the perinuclear aggregate, suggesting that these structures may be important for temporal regulation of gene expression (Fig. 6A). Next, to further validate their interaction, we performed reciprocal co-immunopurification of pUL46-GFP using an anti-ICP0 antibody coupled to Protein A/G-conjugated agarose beads. pUL46-GFP was co-isolated with ICP0 at 6 hpi following infection of HFF cells with pUL46-GFP, but not in control IgG isolations, suggesting that the ICP0-pUL46 interaction is specific and not an artifact of the GFP tag (Fig. 6B). Together, the colocalization and reciprocal isolation experiments confirmed the ICP0-pUL46 interaction at 6 hpi.
Fig. 6.

pUL46 specifically interacts with the viral E3 ubiquitin ligase ICP0. A, Colocalization of pUL46 with ICP0. HFFs were infected at an MOI of 0.5 and fixed at 6 h.p.i. for direct (pUL46-GFP) and immuno- (ICP0) fluorescence. Scale bar demarcates 25 μm. B, Reciprocal co-immunopurification of UL46-GFP by α-ICP0 antibody. HFFs were infected with pUL46-GFP HSV-1 at an MOI of 10 and harvested at 6 hpi. Cell lysates were incubated with beads conjugated to either α-ICP0 antibodies or control IgG, and co-immunoprecipitates were separated by SDS-PAGE. C, Dynamic interaction of pUL46 with ICP0. HFFs were infected with either pUL46-GFP or GFP HSV-1 at an MOI of 10 and harvested at 3, 6, and 14 hpi. GFP-containing protein complexes were immunoaffinity isolated with α-GFP antibody and separated by SDS-PAGE. D, Dynamic co-localization of pUL46 with ICP0. HFFs were infected at an MOI of 0.25 and fixed at 3 and 14 hpi. for direct (pUL46-GFP) and immuno- (ICP0) fluorescence. Scale bars demarcate 25 μm.
However, ICP0 is an immediate early viral protein expressed starting from very early time points of infection. To define the temporal nature of this interaction, we further investigated these proteins at very early, 3 hpi, and late, 14 hpi, time points of infection. Although ICP0 was well expressed at 3 hpi (Fig. 6C, input lanes), it poorly co-isolated with pUL46-GFP at this very early time of infection. In contrast, ICP0 was efficiently immunoaffinity purified with pUL46-GFP at both 6 and 14 hpi in agreement with our proteomic experiments. This interaction was not observed at any of these time points in HFF cells infected with the control free GFP HSV-1 strain (Fig. 6C, right GFP lanes), demonstrating the specificity of interaction for pUL46. Parallel microscopy studies supported our identification of this interaction because of strong colocalization at 14 hpi, but not at 3 hpi where diffuse ICP0 localization was observed (Fig 6D). Altogether, these results validate a previously unreported interaction between pUL46 and ICP0 at both middle and late stages of infection, suggesting that this interaction may be important for temporal control of viral gene expression.
pUL46 is Partially Degraded in an ICP0-Dependent, Proteasome-mediated Manner During HSV-1 Infection
As ICP0 is an E3 ubiquitin ligase and as pUL46 is known to be unstable in the absence of the viral kinase pUS3 (50), we hypothesized that pUL46 might be targeted for degradation in a proteasome-dependent manner by ICP0. However, ICP0 has so far been associated with targeting host proteins for degradation, not viral proteins. Early during infection, ICP0 serves as an essential viral factor in targeting host antiviral defenses for degradation via ubiquitination, including PML (61) and Sp100 (62) contained in ND10 bodies, the ubiquitin-specific protease USP7 (63), the DNA sensor IFI16 (64), and the catalytic subunit of the DNA-dependent protein kinase DNA-PKCS (65). Indeed, treatment of cells with the proteasome inhibitor MG132 before entry prevents ICP0-mediated degradation of host antiviral factors, and severely inhibits viral entry and replication (66, 67). A recent study showed that several viral proteins can be ubiqutinated in whole lysates of infected cells (52); however, no viral protein has been shown to be specifically targeted for degradation by ICP0.
As our results have validated the ICP0-pUL46 interaction, we sought to determine whether pUL46 is degraded in a proteasome-dependent manner during infection. HFFs were infected with pUL46-GFP HSV-1 at an MOI of 10, treated with 10 μm MG132 or PBS at 1 hpi, and harvested at 4.5, 6.5, 8, and 10 hpi (Figs. 7A and supplemental Fig. S3). Increased levels of pUL46GFP were readily apparent as early as 4.5 hpi in cells treated with MG132 (Fig. 7A), and these elevated levels were observed at times even late after infection (supplemental Fig. S3). In contrast, HFF cells infected with HSV-1 HSV1(17+)Lox-UL37GFP expressing pUL37GFP (30) show that this different tegument protein, is not degraded during viral infection. Under the same experimental setup, expression of pUL37-GFP remained constant regardless of mock or MG132 treatment, whereas the expression of DNA-PKCS, a positive control for ICP0-dependent degradation during viral infection (65), was rescued with MG132 (Fig. 7B). Additionally, we demonstrated that pUL46 is ubiquitinated, in agreement with our hypothesis that pUL46 is degraded in a proteasome-dependent manner. HFFs were infected with either HSV1(17+)Lox-UL46GFP or the control GFP HSV-1 at an MOI of 10, treated with 10 μm MG132 at 1 hpi, and harvested at 8 hpi. Subsequent immunoaffinity purification was performed with a stringent buffer to predominantly isolate pUL46-GFP or GFP. Western blotting against either GFP or ubiquitin revealed a slower-migrating diffuse band corresponding to ubiquitinated pUL46-GFP (Fig. 7C). These results demonstrate that proteasome-mediated degradation is specific to pUL46 and is neither a common feature of tegument proteins nor an artifact of the GFP tag.
Fig. 7.
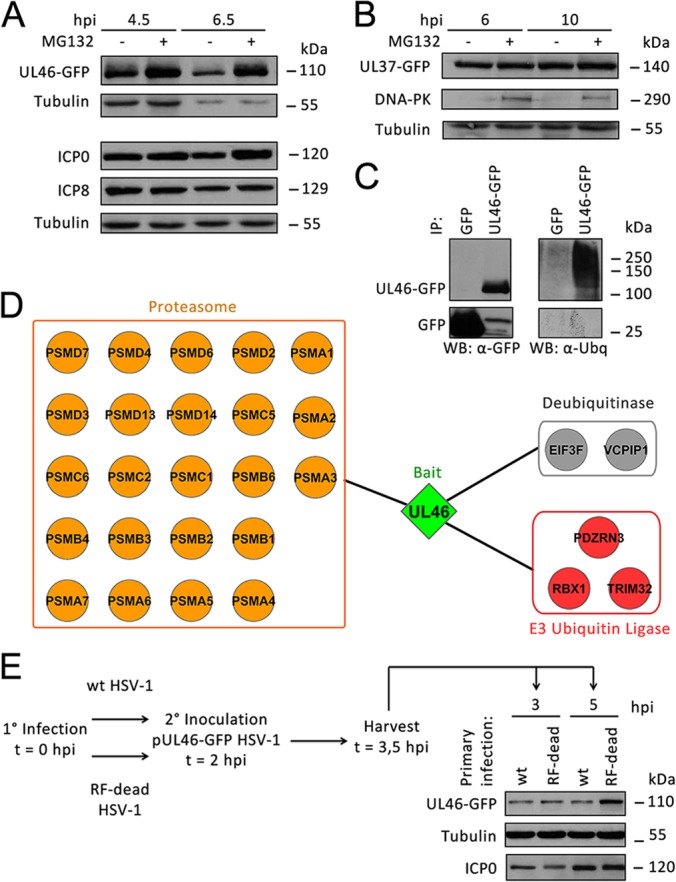
pUL46 is partially degraded by ICP0 in a proteasome-dependent manner during HSV-1 infection. A, Proteasomal degradation of pUL46. HFFs were infected with pUL46-GFP HSV-1 at an MOI of 10, treated with mock or 10 μm MG132 at 1 hpi, and harvested at 4.5 and 6.5 hpi. Whole cell lysates were separated by SDS-PAGE. B, pUL37-GFP is not degraded in a proteasome-dependent manner. HFFs were infected with pUL37-GFP HSV-1 at an MOI of 10, treated with mock or 10 μm MG132 at 1 hpi, and harvested at 6 and 10 hpi. C, pUL46 is ubiquitinated. HFFs were infected with HSV1-UL46-GFP or GFP HSV-1 at an MOI of 10, treated with 10 μm MG132 at 1 hpi, and harvested at 8 hpi. pUL46-GFP or GFP was immunoaffinity isolated using a higher stringency buffer, and eluates were separated by SDS-PAGE. D, Several proteasome subunits associate with pUL46 in infected cells treated with MG132. HFFs were infected with pUL46-GFP HSV-1 at an MOI of 10, treated with 10 μm MG132 at 1 hpi, and harvested at 6 hpi. Proteins co-isolated with pUL46-GFP were analyzed by nLC-MS/MS and visualized by Cytoscape. E, Catalytically active ICP0 is responsible for degradation of pUL46. HFFs were primarily infected with either wild-type or ICP0 RF-dead HSV-1 at an MOI of 10. At 2 hpi, HFFs were superinfected with pUL46-GFP HSV-1 at an MOI of 40, and cells were harvested at 3 and 5 hpi. Whole cell lysates were separated by SDS-PAGE.
Moreover, additional AP-MS/MS analyses further illustrated the association of pUL46 with the proteasome. HFFs were infected with pUL46-GFP HSV1 at an MOI of 10, treated with 10 μm MG132 at 1 hpi, and harvested at 6 hpi. pUL46-GFP-containing protein complexes were isolated by immunoaffinity purifications, and the co-isolated proteins were compared with those observed in the absence of MG132 (Fig. 3B). Several subunits of the proteasome were observed to associate with pUL46 following MG132 treatment (Fig. 7D and supplemental Tables S5 and S6). Altogether, these results indicate that pUL46-GFP is targeted for degradation in a proteasome-dependent manner during HSV-1 infection.
Finally, we tested whether ICP0 was responsible for the proteasome-mediated degradation of pUL46. As commercially available antibodies are not yet available for pUL46, we designed a modified pulse-chase approach to analyze the degradation of pUL46-GFP in the context of infection with either a wild-type HSV-1 or an ICP0 Ring Finger(RF)-dead HSV-1 strain. The ICP0 RF-dead HSV-1 contains C116G and C156A substitutions, which remove the catalytic cysteine residues in the ring finger domain, resulting in catalytically inactive ICP0 (35). The experimental workflow is illustrated in Fig. 7E. HFFs were infected with either wild-type (WT) or ICP0 RF-dead HSV-1 at a high MOI of 10 to saturate nuclear replication centers that can only transcribe from at most six to eight genomes (68). At 2 hpi, cells were superinfected with HSV1(17+)Lox-UL46GFP at an MOI of 40. The 2 hpi time point was selected as a time at which nuclear replication centers have been established (68) for the initial, primary infection with WT or ICP0 RF-dead, but before exclusion of superinfection (69, 70). Following the superinfection, cells were harvested at 3 hpi, at which time the entry of the secondary HSV1(17+)Lox-UL46GFP virus should be completed, and at 5 hpi, at which time ICP0 should already be partially cytoplasmic. As expression of only pUL46, and not pUL46-GFP, occurs, pUL46-GFP from incoming virions acts as a pulse that is “chased” by degradation. Indeed, pUL46 expression levels were substantially reduced in cells primarily infected with wild-type compared with ICP0 RF-dead HSV-1. These results indicate that a functional ICP0 is required for the targeting of pUL46 for proteasome-mediated degradation (Fig. 7E).
In summary, our results present the first evidence for the regulation of pUL46 levels by proteasome-mediated degradation, as well as the first demonstration that viral proteins can be targeted for degradation by ICP0. These results suggest that the temporal control of pUL46 is an important mechanism for modulating viral protein expression levels throughout the virus life cycle.
DISCUSSION
Here, we present a proteomic investigation of the interactions of the HSV-1 outer tegument protein pUL46, which led to the first evidence that an HSV-1 viral protein can be targeted for degradation by another viral protein. pUL46-containing assemblies were isolated under physiological abundance and localization in human primary fibroblasts, which are permissive to HSV-1 infection. We identified pUL46 interactions with both viral and host proteins, and showed that pUL46 is hyper-phosphorylated at a middle stage of infection. Additionally, time-lapse microscopy and functional assays provided temporal and spatial insights into pUL46 regulation. Overall, our results on pUL46 localization, post-translational modifications, interactions, and their functional characterizations, led us to propose a model in which pUL46 plays both nonstructural and structural roles at different time points during HSV-1 infection (Fig. 8). pUL46 protein levels are tightly controlled through interactions and PTMs and important for the temporal regulation of viral gene expression.
Fig. 8.
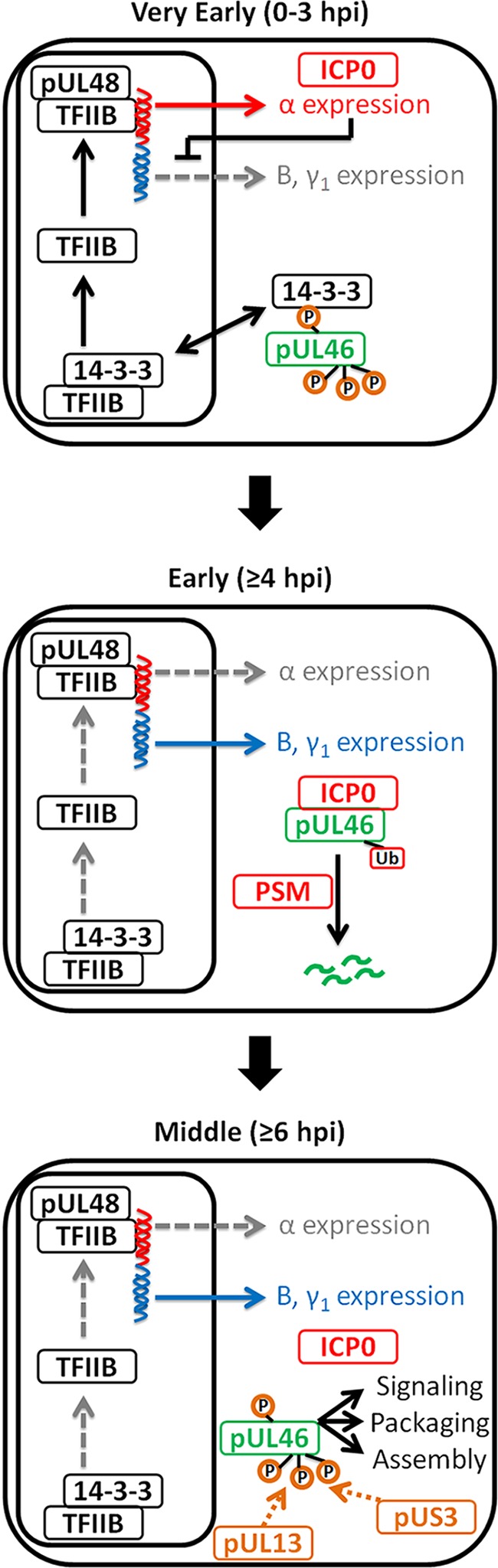
Model of pUL46 interactions and its regulation during HSV-1 infection.
One overarching theme highlighted by our proteomics studies was the regulation of pUL46 by phosphorylation (Fig. 8, upper and lower panels). Among the pUL46 interactions, we identified viral and host kinases, such as pUL13, pUS3, CamKIID, and Src. Furthermore, pUL46 was phosphorylated at 23 sites, 17 of which were novel. Several sites matched consensus motifs for the viral kinases pUL13 and pUS3, further supporting our identification of these proteins as specific pUL46 interactions. Our results add to the current knowledge regarding the regulation of herpesvirus tegument proteins by phosphorylation. Indeed, other tegument proteins, such as pUL36 (71), pUL47 (72), and pUL49 (73), are known to be phosphorylated, both inside the cell (22, 52, 74, 75) and in cell-free extracellular virions (57, 75). Further study investigating differences in modifications of cell-associated versus cell-free pUL46 will likely reveal differential functions during infection. We discovered that several 14-3-3 subunit proteins associate with pUL46 under mild isolation conditions, and that the pUL46 S478 phosphorylation site matches a conserved consensus 14-3-3 binding motif. This evidence suggests that the phosphorylation status may influence the functions or regulation of pUL46. The pUL46 phosphorylation may modulate its interactions in a temporal manner during infection. Additionally, phosphorylations can contribute to disordered protein structures, and it is known that such disordered regions are overrepresented among 14-3-3 binding partners (76). The possible use of this phosphorylation and 14-3-3 proteins for the regulation of pUL46 functions is intriguing. In previous work, 14-3-3 proteins were shown to be able to compete in vitro with pUL48 for binding to the TFIIB transcription complex (77) (Fig. 8, upper panel). If this is indeed the case in vivo, then 14-3-3 could inhibit the ability of pUL48 to trans-activate viral α genes. One hypothesis that may explain the mechanism by which pUL46 modulates pUL48 trans-activating activity could be that pUL46 can co-opt 14-3-3 proteins, thereby partly relieving 14-3-3 inhibition of pUL48. This would allow pUL48 full access to the TFIIB machinery required for α-trans-activation. Although this is one hypothesis for pUL46 functions during the very early stages of infection, pUL46 is likely to have other interactions during early or middle stages of infection.
Our localization and interaction studies identified a prominent pUL46 interaction with the α viral protein ICP0 during middle stages of infection. Though ICP0 is an E3 ubiquitin ligase, it has not yet been shown to target any viral proteins for proteasomal degradation. Here, we established that pUL46 is ubiquitinated during infection and that a catalytically active ICP0 protein is required for the targeting of pUL46 for proteasome-dependent degradation during HSV-1 infection (Fig. 8, middle and lower panels). Moreover, we demonstrated that this degradation was specific to pUL46 as another GFP-tagged tegument protein, pUL37-GFP, was not degraded. A recent study identified several ubiquitination sites on HSV-1 proteins (52), suggesting that ubiquitination of viral proteins may be more common than previously thought, though the authors hypothesized that those ubiquitinations serve as trafficking rather than degradation signals. Therefore, our study significantly adds to the emerging evidence of the functional roles played by viral protein ubiquitination. We present the first evidence that ICP0 targets a viral protein, pUL46, for proteasomal-mediated degradation, which suggests that partial degradation of viral proteins may be one mechanism to control expression levels throughout infection. In agreement with a tightly regulated balance of viral protein levels, several host de-ubiquitinases were found in our study to associate with pUL46. The host ubiquitin-specific protease USP9X, identified under mild isolation conditions, is of particular interest because it was shown to protect various host proteins from ubiquitin-mediated degradation (78). Another USP family member, USP7, was shown to protect ICP0 from auto-ubiquitination and degradation (79), suggesting that the ubiquitination of pUL46 may likely serve a role in regulating protein expression rather than trafficking.
ICP0 is known to be important for trans-activation of downstream β and γ viral gene expression, though the mechanisms have yet to be fully revealed (reviewed in (8)). Although α transcription levels are high during early infection, they decay rapidly during middle stages of infection, as β and γ transcription increases (80). HSV-1 mutants lacking both the virion host shutoff protein pUL41 and the other pUL48-modulating protein pUL47 overexpress α genes early during infection, thereby causing a delay in the expression of β and γ genes (81). We propose that, during very early stages of infection, pUL46 serves to increase the trans-activational activity of pUL48 to boost α gene expression (Fig. 8, upper panel). At middle stages of infection, ICP0 targets pUL46 for partial degradation to reduce the expression of α genes, so that β and γ1 expression can occur and propagate the viral life cycle (Fig. 8, middle panel). USP9X and other host de-ubiquitinases may act to protect pUL46 from total degradation during early and late stages of infection, when a certain level of pUL46 might be required for additional roles, such as in signaling, packaging or assembly (Fig. 8, lower panel). The complete elucidation of these mechanisms would require significant additional studies and are not the focus of the current work.
In summary, our study describes the first proteomic analysis of pUL46 interactions and phosphorylations, providing insight into nonstructural roles of pUL46 and its regulation during early and middle stages of infection. We demonstrate that the viral E3 ubiquitin ligase ICP0 protein targets pUL46 for degradation by the proteasome. Therefore, the levels of virion components, normally required for structural functions, are also finely regulated during the progression of an infection. It is rapidly becoming evident that virion-associated proteins play a diverse set of roles rather than simply serving as structural proteins during infections with various and quite diverse viruses. For example, the ebola virus matrix protein VP40 was recently found to adopt different conformations (82) and subsequently different oligomers (83, 84) in the cell, allowing it to perform a transcriptional role during early stages of infection (85) and a separate structural role during viral morphogenesis and budding (86). Indeed, many virions are highly complex and herpes viruses dedicate large amounts of genomic space to virion-associated proteins. This dual functionality of herpesvirus virion-associated proteins to (1) temporally regulate their genomes during early stages of infection and (2) structurally assemble virions during late stages of infection might be conserved across different viruses. A better definition of these mechanisms will be critical for understanding viral pathogenesis and for developing antivirals that disrupt specific stages of the viral life cycle.
Supplementary Material
Acknowledgments
We thank the Sodeik lab (in particular Michaela Cappucci and Anja Pohlmann) for their contribution of the viral BACs used in this study, Bernard Roizman lab for generous contribution of the ICP0 mutant HSV-1 strain, Tal Kramer in the Lynn Enquist lab (Princeton University) for training on the use of time lapse microscopy, Yana Miteva in the Ileana Cristea lab (Princeton University) for training on the use of confocal microscopy, and J. Goodhouse for technical support (Microscopy Facility, Princeton University).
Footnotes
* This work was supported by NIH grants DP1DA026192 and R21AI102187, and HFSPO award RGY0079/2009-C to IMC, and to the Deutsche Forschungsgemeinschaft (German Research Council; Program Project Grants SPP1175 to R. Bauerfeind and B. Sodeik and SFB900, TP C2 to BS).
 This article contains supplemental Figs. S1 to S4, Movie S1, and Tables S1 to S6.
This article contains supplemental Figs. S1 to S4, Movie S1, and Tables S1 to S6.
1 The abbreviations used are:
- HSV-1
- herpes simplex virus type 1
- α
- immediate-early viral genes
- β
- early viral genes
- γ
- late viral genes
- CID
- collision induced dissociation
- GFP
- green fluorescence protein
- hpi
- hours post infection
- IP
- immunoaffinity purification
- MOI
- multiplicity of infection
- nLC
- nano-liquid chromatography
- PTMs
- post-translational modifications
- SAINT
- Significance Analysis of INTeractome.
REFERENCES
- 1. Honess R. W., Roizman B. (1974) Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14, 8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Post L. E., Mackem S., Roizman B. (1981) Regulation of alpha genes of herpes simplex virus: expression of chimeric genes produced by fusion of thymidine kinase with alpha gene promoters. Cell 24, 555–565 [DOI] [PubMed] [Google Scholar]
- 3. Roizman B., Kozak M., Honess R. W., Hayward G. (1975) Regulation of herpesvirus macromolecular synthesis: evidence for multilevel regulation of herpes simplex 1 RNA and protein synthesis. Cold Spring Harb. Symp. Quant. Biol. 39, 687–701 [DOI] [PubMed] [Google Scholar]
- 4. Whitley R. J., Roizman B. (2001) Herpes simplex virus infections. Lancet 357, 1513–1518 [DOI] [PubMed] [Google Scholar]
- 5. Batterson W., Roizman B. (1983) Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 46, 371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKnight J. L., Kristie T. M., Roizman B. (1987) Binding of the virion protein mediating alpha gene induction in herpes simplex virus 1-infected cells to its cis site requires cellular proteins. Proc. Natl. Acad. Sci. U.S.A. 84, 7061–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McKnight J. L., Pellett P. E., Jenkins F. J., Roizman B. (1987) Characterization and nucleotide sequence of two herpes simplex virus 1 genes whose products modulate alpha-trans-inducing factor-dependent activation of alpha genes. J. Virol. 61, 992–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajcani J., Durmanova V. (2000) Early expression of herpes simplex virus (HSV) proteins and reactivation of latent infection. Folia Microbiol. 45, 7–28 [DOI] [PubMed] [Google Scholar]
- 9. Hall L. M., Draper K. G., Frink R. J., Costa R. H., Wagner E. K. (1982) Herpes simplex virus mRNA species mapping in EcoRI fragment I. J. Virol. 43, 594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y., McKnight J. L. (1993) Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67, 1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y., Sirko D. A., McKnight J. L. (1991) Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kato K., Daikoku T., Goshima F., Kume H., Yamaki K., Nishiyama Y. (2000) Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145, 2149–2162 [DOI] [PubMed] [Google Scholar]
- 13. Dobrikova E., Shveygert M., Walters R., Gromeier M. (2010) Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol. 84, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Willard M. (2002) Rapid directional translocations in virus replication. J. Virol. 76, 5220–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murata T., Goshima F., Daikoku T., Inagaki-Ohara K., Takakuwa H., Kato K., Nishiyama Y. (2000) Mitochondrial distribution and function in herpes simplex virus-infected cells. J. Gen. Virol. 81, 401–406 [DOI] [PubMed] [Google Scholar]
- 16. Elliott G., Mouzakitis G., O'Hare P. (1995) VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69, 7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholtes L. D., Yang K., Li L. X., Baines J. D. (2010) The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein VP13/14. J. Virol. 84, 7642–7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee J. H., Vittone V., Diefenbach E., Cunningham A. L., Diefenbach R. J. (2008) Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378, 347–354 [DOI] [PubMed] [Google Scholar]
- 19. Zahariadis G., Wagner M. J., Doepker R. C., Maciejko J. M., Crider C. M., Jerome K. R., Smiley J. R. (2008) Cell-type-specific tyrosine phosphorylation of the herpes simplex virus tegument protein VP11/12 encoded by gene UL46. J. Virol. 82, 6098–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wagner M. J., Smiley J. R. (2009) Herpes simplex virus requires VP11/12 to induce phosphorylation of the activation loop tyrosine (Y394) of the Src family kinase Lck in T lymphocytes. J. Virol. 83, 12452–12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wagner M. J., Smiley J. R. (2011) Herpes simplex virus requires VP11/12 to activate Src family kinase-phosphoinositide 3-kinase-Akt signaling. J. Virol. 85, 2803–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lemaster S., Roizman B. (1980) Herpes simplex virus phosphoproteins. II. Characterization of the virion protein kinase and of the polypeptides phosphorylated in the virion. J. Virol. 35, 798–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miteva Y. V., Budayeva H. G., Cristea I. M. (2013) Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem. 85, 749–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cristea I. M., Carroll J. W., Rout M. P., Rice C. M., Chait B. T., MacDonald M. R. (2006) Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem. 281, 30269–30278 [DOI] [PubMed] [Google Scholar]
- 25. Moorman N. J., Sharon-Friling R., Shenk T., Cristea I. M. (2010) A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9, 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cristea I. M., Moorman N. J., Terhune S. S., Cuevas C. D., O'Keefe E. S., Rout M. P., Chait B. T., Shenk T. (2010) Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 84, 7803–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Terhune S. S., Moorman N. J., Cristea I. M., Savaryn J. P., Cuevas-Bennett C., Rout M. P., Chait B. T., Shenk T. (2010) Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 6, e1000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kramer T., Greco T. M., Taylor M. P., Ambrosini A. E., Cristea I. M., Enquist L. W. (2012) Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12, 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Snijder B., Sacher R., Ramo P., Liberali P., Mench K., Wolfrum N., Burleigh L., Scott C. C., Verheije M. H., Mercer J., Moese S., Heger T., Theusner K., Jurgeit A., Lamparter D., Balistreri G., Schelhaas M., De Haan C. A., Marjomaki V., Hyypia T., Rottier P. J., Sodeik B., Marsh M., Gruenberg J., Amara A., Greber U., Helenius A., Pelkmans L. (2012) Single-cell analysis of population context advances RNAi screening at multiple levels. Mol. Sys. Biol. 8, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sandbaumhüter M., Döhner K., Schipke J., Binz A., Pohlmann A., Sodeik B., Bauerfeind R. (2013) Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell Microbiol. 15, 248–269 [DOI] [PubMed] [Google Scholar]
- 31. Tischer B. K., Smith G. A., Osterrieder N. (2010) En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634, 421–430 [DOI] [PubMed] [Google Scholar]
- 32. Nagel C. H., Döhner K., Fathollahy M., Strive T., Borst E. M., Messerle M., Sodeik B. (2008) Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J. Virol. 82, 3109–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schipke J., Pohlmann A., Diestel R., Binz A., Rudolph K., Nagel C. H., Bauerfeind R., Sodeik B. (2012) The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86, 3682–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nygårdas M., Paavilainen H., Müther N., Nagel C. H., Röyttä M., Sodeik B., Hukkanen V. (2013) A herpes simplex virus-derived replicative vector expressing LIF limits experimental demyelinating disease and modulates autoimmunity. PloS One 8, e64200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lium E. K., Silverstein S. (1997) Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J. Virol. 71, 8602–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cristea I. M., Williams R., Chait B. T., Rout M. P. (2005) Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4, 1933–1941 [DOI] [PubMed] [Google Scholar]
- 37. Cristea I. M., Chait B. T. (2011) Conjugation of magnetic beads for immunopurification of protein complexes. Cold Spring Harb. Protoc. 2011, pdb.prot5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Conlon F. L., Miteva Y., Kaltenbrun E., Waldron L., Greco T. M., Cristea I. M. (2012) Immunoisolation of protein complexes from Xenopus. Methods Mol. Biol. 917, 369–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dokudovskaya S., Waharte F., Schlessinger A., Pieper U., Devos D. P., Cristea I. M., Williams R., Salamero J., Chait B. T., Sali A., Field M. C., Rout M. P., Dargemont C. (2011) A conserved coatomer-related complex containing Sec13 and Seh1 dynamically associates with the vacuole in Saccharomyces cerevisiae. Mol. Cell. Proteomics 10, M110.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 41. Tsai Y. C., Greco T. M., Boonmee A., Miteva Y., Cristea I. M. (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell. Proteomics 11, 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 43. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 44. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 45. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johansson P. J., Myhre E. B., Blomberg J. (1985) Specificity of Fc receptors induced by herpes simplex virus type 1: comparison of immunoglobulin G from different animal species. J. Virol. 56, 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheeseman I. M., Niessen S., Anderson S., Hyndman F., Yates J. R., 3rd, Oegema K., Desai A. (2004) A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev. 18, 2255–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morrison E. E., Stevenson A. J., Wang Y. F., Meredith D. M. (1998) Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J. Gen. Virol. 79, 2517–2528 [DOI] [PubMed] [Google Scholar]
- 49. Vittone V., Diefenbach E., Triffett D., Douglas M. W., Cunningham A. L., Diefenbach R. J. (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79, 9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsuzaki A., Yamauchi Y., Kato A., Goshima F., Kawaguchi Y., Yoshikawa T., Nishiyama Y. (2005) US3 protein kinase of herpes simplex virus type 2 is required for the stability of the UL46-encoded tegument protein and its association with virus particles. J. Gen. Virol. 86, 1979–1985 [DOI] [PubMed] [Google Scholar]
- 51. Murphy M. A., Bucks M. A., O'Regan K. J., Courtney R. J. (2008) The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376, 279–289 [DOI] [PubMed] [Google Scholar]
- 52. Bell C., Desjardins M., Thibault P., Radtke K. (2013) Proteomics analysis of herpes simplex virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J .Proteome Res. 12(4), 1820–1829 [DOI] [PubMed] [Google Scholar]
- 53. Cano-Monreal G. L., Tavis J. E., Morrison L. A. (2008) Substrate specificity of the herpes simplex virus type 2 UL13 protein kinase. Virology 374, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Purves F. C., Deana A. D., Marchiori F., Leader D. P., Pinna L. A. (1986) The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates [corrected] indicate structural requirements distinct from other protein kinases. Biochim. Biophys. Acta 889, 208–215 [DOI] [PubMed] [Google Scholar]
- 55. Leader D. P., Deana A. D., Marchiori F., Purves F. C., Pinna L. A. (1991) Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim. Biophys. Acta 1091, 426–431 [DOI] [PubMed] [Google Scholar]
- 56. Kato A., Yamamoto M., Ohno T., Kodaira H., Nishiyama Y., Kawaguchi Y. (2005) Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79, 9325–9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morrison E. E., Wang Y. F., Meredith D. M. (1998) Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J. Virol. 72, 7108–7114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 [DOI] [PubMed] [Google Scholar]
- 59. Fu H., Subramanian R. R., Masters S. C. (2000) 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 60. van Hemert M. J., Steensma H. Y., van Heusden G. P. (2001) 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays 23, 936–946 [DOI] [PubMed] [Google Scholar]
- 61. Everett R. D., Rechter S., Papior P., Tavalai N., Stamminger T., Orr A. (2006) PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80, 7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gu H., Roizman B. (2003) The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. U.S.A. 100, 8963–8968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boutell C., Canning M., Orr A., Everett R. D. (2005) Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 79, 12342–12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Orzalli M. H., DeLuca N. A., Knipe D. M. (2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U.S.A. 109, E3008–E3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lees-Miller S. P., Long M. C., Kilvert M. A., Lam V., Rice S. A., Spencer C. A. (1996) Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70, 7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Delboy M. G., Nicola A. V. (2011) A pre-immediate-early role for tegument ICP0 in the proteasome-dependent entry of herpes simplex virus. J. Virol. 85, 5910–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Delboy M. G., Roller D. G., Nicola A. V. (2008) Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 82, 3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kobiler O., Brodersen P., Taylor M. P., Ludmir E. B., Enquist L. W. (2011) Herpesvirus replication compartments originate with single incoming viral genomes. MBio 2(6), 00278–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Banfield B. W., Kaufman J. D., Randall J. A., Pickard G. E. (2003) Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. J. Virol. 77, 10106–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Meurens F., Schynts F., Keil G. M., Muylkens B., Vanderplasschen A., Gallego P., Thiry E. (2004) Superinfection prevents recombination of the alphaherpesvirus bovine herpesvirus 1. J. Virol. 78, 3872–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McNabb D. S., Courtney R. J. (1992) Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology 190, 221–232 [DOI] [PubMed] [Google Scholar]
- 72. Meredith D. M., Lindsay J. A., Halliburton I. W., Whittaker G. R. (1991) Post-translational modification of the tegument proteins (VP13 and VP14) of herpes simplex virus type 1 by glycosylation and phosphorylation. J. Gen Virol 72 (Pt 11), 2771–2775 [DOI] [PubMed] [Google Scholar]
- 73. Geiss B. J., Tavis J. E., Metzger L. M., Leib D. A., Morrison L. A. (2001) Temporal regulation of herpes simplex virus type 2 VP22 expression and phosphorylation. J. Virol. 75, 10721–10729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yura Y., Kusaka J., Tsujimoto H., Yoshioka Y., Yoshida H., Sato M. (1997) Effects of protein tyrosine kinase inhibitors on the replication of herpes simplex virus and the phosphorylation of viral proteins. Intervirology 40, 7–14 [DOI] [PubMed] [Google Scholar]
- 75. Sathananthan B., Rødahl E., Ekberg T., Langeland N., Haarr L. (1996) Two-dimensional gel analysis of [35S]methionine labelled and phosphorylated proteins present in virions and light particles of herpes simplex virus type 1, and detection of potentially new structural proteins. Virus Res. 46, 1–18 [DOI] [PubMed] [Google Scholar]
- 76. Bustos D. M., Iglesias A. A. (2006) Intrinsic disorder is a key characteristic in partners that bind 14-3-3 proteins. Proteins 63, 35–42 [DOI] [PubMed] [Google Scholar]
- 77. Pan S., Sehnke P. C., Ferl R. J., Gurley W. B. (1999) Specific interactions with TBP and TFIIB in vitro suggest that 14-3-3 proteins may participate in the regulation of transcription when part of a DNA binding complex. Plant Cell 11, 1591–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xie Y., Avello M., Schirle M., McWhinnie E., Feng Y., Bric-Furlong E., Wilson C., Nathans R., Zhang J., Kirschner M. W., Huang S. M., Cong F. (2013) Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J. Biol. Chem. 288, 2976–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Canning M., Boutell C., Parkinson J., Everett R. D. (2004) A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279, 38160–38168 [DOI] [PubMed] [Google Scholar]
- 80. Godowski P. J., Knipe D. M. (1986) Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc. Natl. Acad. Sci. U.S.A. 83, 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shu M., Taddeo B., Zhang W., Roizman B. (2013) Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc. Natl. Acad. Sci. U.S.A. 110, E1669–E1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Silva L. P., Vanzile M., Bavari S., Aman J. M., Schriemer D. C. (2012) Assembly of Ebola virus matrix protein VP40 is regulated by latch-like properties of N and C terminal tails. PloS One 7, e39978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Timmins J., Schoehn G., Kohlhaas C., Klenk H. D., Ruigrok R. W., Weissenhorn W. (2003) Oligomerization and polymerization of the filovirus matrix protein VP40. Virology 312, 359–368 [DOI] [PubMed] [Google Scholar]
- 84. Hoenen T., Biedenkopf N., Zielecki F., Jung S., Groseth A., Feldmann H., Becker S. (2010) Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol. 84, 7053–7063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hoenen T., Jung S., Herwig A., Groseth A., Becker S. (2010) Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 403, 56–66 [DOI] [PubMed] [Google Scholar]
- 86. Liu Y., Cocka L., Okumura A., Zhang Y. A., Sunyer J. O., Harty R. N. (2010) Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 84, 2294–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.