

Abstract
Thioredoxin (Trx) and GSH are the major thiol antioxidants protecting cells from oxidative stress-induced cytotoxicity. Redox states of Trx and GSH have been used as indicators of oxidative stress. Accumulating studies suggest that Trx and GSH redox systems regulate cell signaling and metabolic pathways differently and independently during diverse stressful conditions. In the current study, we used a mass spectrometry-based redox proteomics approach to test responses of the cysteine (Cys) proteome to selective disruption of the Trx- and GSH-dependent systems. Auranofin (ARF) was used to inhibit Trx reductase without detectable oxidation of the GSH/GSSG couple, and buthionine sulfoximine (BSO) was used to deplete GSH without detectable oxidation of Trx1. Results for 606 Cys-containing peptides (peptidyl Cys) showed that 36% were oxidized more than 1.3-fold by ARF, whereas BSO-induced oxidation of peptidyl Cys was only 10%. Mean fold oxidation of these peptides was also higher by ARF than BSO treatment. Analysis of potential functional pathways showed that ARF oxidized peptides associated with glycolysis, cytoskeleton remodeling, translation and cell adhesion. Of 60 peptidyl Cys oxidized due to depletion of GSH, 41 were also oxidized by ARF and included proteins of translation and cell adhesion but not glycolysis or cytoskeletal remodeling. Studies to test functional correlates showed that pyruvate kinase activity and lactate levels were decreased with ARF but not BSO, confirming the effects on glycolysis-associated proteins are sensitive to oxidation by ARF. These data show that the Trx system regulates a broader range of proteins than the GSH system, support distinct function of Trx and GSH in cellular redox control, and show for the first time in mammalian cells selective targeting peptidyl Cys and biological pathways due to deficient function of the Trx system.
Oxidative stress is associated with numerous human diseases and results from alteration in cellular redox systems (1–3). Cellular redox mechanisms and signaling are controlled by two major thiol antioxidants, thioredoxin (Trx)1 and glutathione (GSH). The Trx system, composed of Trx, Trx reductase (TrxR), Trx peroxidase/peroxiredoxin (Prx), and NADPH, has a wide range of cellular activities in redox control, cell proliferation, growth and survival, regulation of transcription, and cell morphology and structure (4–6). The Trx family includes evolutionary conserved proteins that contain two catalytically active cysteine (Cys) residues that can reduce disulfide bonds of numerous target proteins in the Cys proteome, e.g. apoptosis signaling kinase-1 (ASK-1), Trx1-interacting protein (Txnip), transcription factors, and actin. In addition to catalytic activity, Trx binds to target proteins and further controls protein activity and biological function. For example, ASK-1 binds to Trx in physiologic conditions; however, under stressful conditions, it is dissociated from Trx, which then stimulates apoptotic signaling leading to cell death (7). The diverse functions of Trx suggest a nonspecific nature to its function, yet kinetic studies also show differences in activities with different substrates (8–10), implying that an underlying redox organizational structure of the Trx-dependent Cys proteome could exist.
The GSH redox system likewise plays a key role in maintaining reduced cellular redox state and regulates multiple signaling pathways by protecting cells from oxidative stress. The GSH system is dependent on NADPH and consists of GSH, GSSG reductase, and glutaredoxins. This system differs from Trx in that GSH can be present at up to 10 mm whereas Trx is about 1 μm in cells. Genetic studies of yeast show that the GSH system protects against acute but not chronic oxidative stress (11), suggesting that an underlying redox organization of the GSH-dependent Cys proteome could also exist. Studies in mammalian cells show that maintenance of GSH is essential, e.g. for prevention of protein aggregation in the lens, protection against apoptosis, support of cell proliferation and survival signaling, differentiation, and protection against genomic arrangement (12, 13). Redox interactions of GSH with protein Cys are catalyzed by glutaredoxins, and glutathionylation/deglutathionylation of actin is known to occur under physiologic conditions (14). Direct chemical reactions of GSH with some peptidyl Cys may also be important because the concentration of GSH can be four orders of magnitude greater than Trx. Together with previous evidence that the GSH and Trx redox systems are controlled independently in mammalian cells, these lines of evidence suggest that the function of the GSH system could differ from Trx in the redox organizational structure controlling the Cys proteome in mammalian cells.
Previous studies show genetic manipulations of Trx and GSH systems in yeast have distinct functions in controlling the redox proteome (15). Mammalian cell studies show that Trx- and GSH-dependent systems have overlapping antioxidant functions under toxicologic and pathologic conditions (16, 17). During the past several years we have found that under diverse conditions of differentiation, nutritional limitation and metal ion toxicity in mammalian cells, these major thiol and disulfide redox control systems are maintained with distinct redox potentials and respond in a semiautonomous manner (18–21). The latter results are consistent with an accumulating literature that, in addition to functions during toxicologic stresses, the Trx and GSH systems function constitutively in the dynamic regulation of cells (4–6, 12, 13). To test the hypothesis that Trx and GSH systems regulate distinct protein targets in the absence of toxicologic challenge, such as ones commonly used in oxidative stress research, we employed selective inhibition of the Trx and GSH systems in cell culture without added oxidants. We then analyzed effects of the selective inhibition of Trx and GSH systems on oxidation of peptidyl Cys using a mass spectrometry-based redox proteomics method.
We used auranofin (ARF; 2,3,4,6-tetra-O-acetyl-1-thio-β-d-glucopyranosato-S-gold) and BSO (l-buthionine-[S,R]-sulfoximine) to selectively inhibit Trx and GSH systems, respectively. ARF and aurothioglucose [ATG; gold (I)(2S, 3S,4R,5S)-3,4,5-trihydroxy-6-(hydroxymethyl)-oxane-2-thiolate] are gold-containing compounds that inhibit catalytic activity of TrxR, are used to treat rheumatoid arthritis (22), and show potential as anticancer and antiparasitic drugs (23). TrxR maintains reduced Trx and is important in controlling apoptosis (24). Cox et al. showed that ARF-induced apoptosis in Jurkat T-lymphoma cells was associated with oxidation of mitochondrial Prx 3 (25) and a recent study also showed ARF caused Trx oxidation (25), showing that ARF-induced inhibition of TrxR disrupts the Trx/TrxR redox system. Similarly, BSO, a well-known inhibitor of γ-glutamylcysteine synthetase (γ-GCS) is a widely used tool to lower GSH (26) and disrupt the GSH redox system. A recent study using cultured organogenesis-stage rat conceptuses showed that BSO treatment at 1 mm resulted in oxidation of many embryonic protein thiols and affected protein concentrations supporting the observation that altered GSH redox state was of sufficient magnitude to alter the cellular redox proteome (27). In addition, depletion of cellular GSH with BSO pretreatment affects a broad range of cellular mechanisms such as cell proliferation, but most notably sensitizes cells to chemical-induced toxicity.
In the current study, we selected human colon carcinoma HT29 cells because the Trx and GSH systems have been extensively characterized and have different steady states of oxidation under defined cell culture conditions (20, 28, 29). We performed dose-response studies to obtain concentration of ARF that oxidized Trx1 without oxidation of GSH/GSSG and to obtain concentration of BSO that oxidized GSH/GSSG without oxidation of Trx1. We then employed redox ICAT (Isotope Coded Affinity Tag)-combined mass spectrometry to measure percentage of oxidation of Cys in peptides (29–31), and used the associated proteins to map to metabolic pathways using MetaCore (https://portal.genego.com/) pathway analysis tools. The results show that specific peptidyl Cys/proteins and pathways are selectively regulated by the Trx and GSH systems.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments
HT29 cells were purchased from American Tissue Culture Collection (Rockville, MD) and cultured in 10-cm plates in DMEM (37 °C, 5% CO2) with 10% FBS and antibiotics (penicillin/streptomycin) (20, 29, 32). Cells with 80 to 90% confluency were washed with Hanks balanced salt solution (HBSS) and treated with ARF, ATG, BSO purchased from Sigma-Aldrich (St. Louis, MO). After initial screening, ARF was selected for inhibition of TrxR, and the conditions for treating cells with ARF and BSO were optimized from the results of dose-response experiments to obtain conditions where each inhibitor did not have significant effect on the other redox system. Exposure times with these inhibitors were selected wherein treated cells did not show morphological change compared with control cells.
Measurements of Trx1 and GSH/GSSG Redox States
To analyze redox forms of Trx1, HT29 cells treated with BSO, ATG, or ARF were washed three times with ice-cold PBS and lysed in G-lysis buffer (6 m guanidine-HCl, 50 mm Tris, pH 8.3, 3 mm EDTA) including 50 mm iodoacetic acid (IAA) (33). After incubation of lysate at 37 °C for 30 min, excess amounts of IAA were removed from lysate by G-25 spin column (GE Healthcare, UK). Lysates were then analyzed to examine Trx1 redox state by native gel electrophoresis, blotting, and probing with antibody specific to Trx1 (AbFrontier, Korea) (33, 34). Bands corresponding to reduced and oxidized forms of Trx1 were visualized using an Odyssey scanner (LI-COR, Lincoln, NE) and steady-state redox potential (Eh, redox state) was calculated by the Nernst equation (EoTrx1 = −240 mV, pH 7.4) as outlined (33, 35, 36). To examine GSH redox state, control cells and cells treated with BSO, ATG, or ARF were analyzed for GSH and glutathione disulfide (GSSG) using high performance liquid chromatography (HPLC) with fluorescence detection (37). Values of GSH and GSSG concentrations were used to calculate the steady-state redox potential for cellular GSH/GSSG (EhGSSG) using the measured concentrations, the Nernst equation, and corresponding EoGSSG (-264 mV, pH 7.4) (36, 37). Concentrations of GSH and GSSG obtained from HPLC analysis were normalized to the protein concentration of each sample prepared using cultured HT29 cells. Because of the proliferating nature of HT29 cells under normal conditions, GSH (3.8–4.6 mm) and GSSG (41.7–113.8 μm) concentrations and EhGSSG level (-254.0 ∼ −241.2 mV) measured in HT29 cells at control condition varied slightly by experiments, consistent with previous reports (20, 28, 36).
Enzyme Activity Assays
TrxR1 activity was measured using purified rat TrxR1 (Cayman Chemical, MI) treated with ARF by following the procedures as described previously (38). Glycolysis assays were performed by measuring cellular and extracellular pyruvate kinase enzyme activity and lactate levels after treating cells with BSO (100 μm, 15 h), ARF (20 μm, 2 h), or with no treatment following the protocol provided by manufacturer (Abcam, Cambridge, MA).
Quantification of Cys-containing Protein Peptides (peptidyl Cys) by Redox Proteomic Analysis
Redox ICAT was performed using ICAT-based mass spectrometry (29, 32). This method uses an iodoacetamide-containing reagent that traps thiols under the conditions used in the absence of exogenous oxidants. The reaction is dependent on the efficiency of alkylation relative to oxidation (See Hansen and Winther (39) for a detailed discussion). We have studied this issue extensively for Trxs, Prxs, GSH and other thiol/disulfide couples in different cell types, subcellular compartments and in vivo (see (40–43) and references therein) and used the following precautions to minimize artifacts: (1) rapid processing, (2) extraction at 0°, (3) storage of samples for mass spectrometry at −80°, with effort to minimize time to analysis. Importantly, these experiments do not include addition of exogenous oxidants and therefore do not have the same risk of artifact during processing that is present in many studies of oxidative stress. Briefly, cells treated with BSO (100 μm, 15 h), ARF (20 μm, 2 h), or with no treatment were washed with ice-cold PBS 3 times to remove excess BSO and ARF. Cells were lysed by ice-cold 10% trichloroacetic acid (TCA), and protein precipitate (120 μg) was washed with ice-cold acetone, resuspended in 80 μl denaturing buffer (50 mm Tris, 0.1% SDS, pH8.5) provided by the manufacturer (AB Sciex, CA), and treated with the biotin-conjugated thiol reagent (Heavy isotopic (h-ICAT)) for 1 h at 37°C. Protein was then precipitated by 10% TCA for 30 min on ice, pelleted, washed with acetone, and resuspended in 80 μl denaturing buffer. Unlabeled disulfides and sulfenic acids in the proteins were then reduced by 1 mm TCEP (tris-(2-carboxyethyl phosphine)) and labeled with another biotin-conjugated thiol reagent (Light isotopic (l-ICAT)) at 37°C for 1 h. Samples containing both h- and l-ICAT-labeled proteins were digested with trypsin for 18 h, fractionated by cationic exchange using the ICAT cation-exchange cartridge and elution buffer (10 mm potassium phosphate/25% acetonitrile/350 mm KCl, pH 3.0) following the procedures provided by the manufacturer (AB Sciex). To purify biotinylated l- and h-ICAT-labeled peptides, ICAT-avidin cartridge (AB Sciex) and affinity buffer (30% acetonitrile, 0.4% trifluoroacetic acid) were used. Samples including purified h- and l-ICAT-labeled peptides were analyzed by mass spectrometry as described below. ICAT-labeled peptidyl Cys were identified with an h to l ratio as a measure of the reduced/oxidized state of the protein, expressed as percentage values, and labeled as “% oxidized state.” Identified peptides were individually processed to eliminate redundancies, matched to proteins based on amino acid sequences and assigned to be compared between each treatment (control, CR; ARF; BSO). Fold oxidation was calculated by dividing % oxidized state [l-ICAT/(l-ICAT + h-ICAT)] of individual peptidyl Cys from ARF or BSO treatment into % oxidized state of the identical peptidyl Cys from CR. BSO and ARF-increased fold oxidation compared with no treatment (CR) were obtained from three biological replicates for each BSO, ARF, and CR redox ICAT analysis.
Mass Spectrometry
ICAT-labeled Cys peptides were analyzed by reverse-phase liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) (44). Peptide eluents were monitored in an MS survey scan followed by ten data-dependent MS/MS scans on an LTQ-Orbitrap ion trap mass spectrometer (Thermo Finnigan, San Jose, CA). The LTQ was used to acquire MS/MS spectra (2 m/z isolation width, 35% collision energy, 5000 AGC target, 200 ms maximum ion time). The Orbitrap was used to collect MS scans (300–1600 m/z, 1,000,000 AGC target, 500 ms maximum ion time, resolution 30,000). All data were converted from .raw files to the .dta format using ExtractMS version 2.0 (Thermo Finnigan, San Jose, CA). The acquired MS/MS spectra were searched against a concatenated target-decoy human RefSeq (release 37 - September 2009 - 38108 target proteins) database of the National Center for Biotechnology Information using the SEQUEST Sorcerer algorithm (version 3.11, SAGE-N) (45). Searching parameters included partially tryptic restriction, parent ion mass tolerance (± 20 ppm), dynamic modifications of oxidized Met (+15.9949 Da), differential ICAT-modified Cys (+9.0302 Da), and static ICAT modification of Cys (+227.1270 Da). The peptides were classified by charge state and tryptic state (fully and partial) and first filtered by mass accuracy (10 ppm for high-resolution MS), and then dynamically by increasing XCorr and ΔCn values to reduce protein false discovery rate to less than 1%, according to the target-decoy strategy (46, 47). Parameter settings allowed detection of 100% oxidation and 100% reduction as well as H:L ratios for partially oxidized peptidyl Cys. Protein quantification was performed using an in-house program that quantified paired light and heavy ICAT peptide peaks (48, 49). Peptide ion peaks from survey scans were first defined by m/z, retention time, peak intensity, and signal-to-noise. Ion peak pairs were matched by predicted mass accuracy (10 ppm) and retention time (50). The raw data including all peptides are presented in supplemental File S1. Peptide spectra and raw data is available in the Proteomics Identification Database (PRIDE, http://www.ebi.ac.uk/pride/) (51).
Determination of ARF in Protein Extracts
To determine whether residual ARF was present in protein pellets, posing a potential interference with H- and l-ICAT labeling efficiency, we analyzed residual ARF in protein extracts by LC/MS. Sample was prepared as above; protein pellets were washed with acetone and resuspended with denaturing buffer (AB Sciex) including proteinase K (Sigma-Aldrich). Quantification of ARF was obtained using HPLC (Thermo Fisher UltiMate 3000, San Jose, CA) coupled to Fourier transform-ion cyclotron MS (Thermo Fisher LTQ-FT). Briefly, extracts and standard solutions of ARF were analyzed via anion exchange chromatography (Hamilton Analytical Inc., Reno, NV; PRP-X110S, 2.1 × 10 cm, 7 μm diameter particle size) using a formic acid gradient (0–0.1% formic acid: acetonitrile, 1:1). The MS was operated in selective ion monitoring (SIM) within the linear ion trap using positive electrospray ionization and the following tune parameters (52): spray voltage of 6 kV, sheath gas of 60, capillary temperature of 275°C, capillary voltage of 44 V and tube lens of 120 V. Quantification was completed on the most abundant ion specific to ARF, which was determined via full scan monitoring of 85–2000 m/z during direct infusion of 20 μm ARF prepared in 1:1 0.1% formic acid/acetonitrile and found to be 993.21 m/z, the parent compound coordinated by two gold atoms and two triethylphosphines and one thioglucopyranose as described by Albert et al. (53). Absolute quantification of residual ARF was accomplished by comparison of a five-point external calibration curve prepared by serial dilution of ARF (Sigma-Aldrich). No ARF was detectable in the protein pellets with a detection limit of 20 pmol/120 μg protein extract based on a signal-to-noise ratio of 3.
Biological Network Analysis by MetaCore
Functional genomic and proteomic networks affected by protein redox state were analyzed using MetaCore (Thomson Reuters; https://portal.genego.com/). Protein peptide redox data of three biological replicates for each treatment with BSO and ARF were converted from H:L ratio to fold oxidation compared with that with no treatment (CR). Peptidyl Cys with fold oxidation value (≥1.3) were analyzed for pathway maps by MetaCore. Protein/gene ID and fold oxidation value of each peptidyl Cys were loaded into MetaCore (Version 6.13, build 43450). Ranking of relevant integrated pathways was based on hypergeometric p values (54). Pathway map analysis was specified with a significance level for the false discovery rate (FDR) equal to 0.05.
RESULTS
Baseline Redox Responses
Dose-dependent redox responses of ARF on TrxR1 activity and redox states of Trx1 and GSH are shown in Fig. 1. As expected, studies with TrxR1 showed that NADPH oxidation to NADP+ in the TrxR1 assay was completely inhibited by preincubating purified TrxR1 protein with ARF at 1:1 ratio (20 nm: 20 nm) for 30 min at RT (Fig 1A top). Addition of ARF to HT29 cells resulted in significant oxidation of Trx1 in a dose-dependent manner at 2 h as shown by redox Western blotting and calculated EhTrx1 measurements (Fig 1A middle, bottom) (33, 34). Analysis of GSH, GSSG, and GSH redox potential (37) under the same condition showed that cellular GSH (0, 4.2 ± 0.2 mm; 10 μm ARF, 4.1 ± 0.2 mm; 20 μm ARF, 3.9 ± 0.2 mm), GSSG (0, 83.8 ± 12.9 μm; 10 μm ARF, 71.4 ± 14.1 μm; 20 μm ARF, 56.2 ± 5.0 μm) and EhGSSG (0, −243.5 ± 2.3 mV; 10 μm ARF, −245.3 ± 2.2 mV; 20 μm ARF, −246.6 ± 0.9 mV) were not significantly altered at 10 and 20 μm ARF (Fig 1B). At 50 μm or higher concentrations of ARF treatment, significant decreases in GSH (0, 4.2 ± 0.2 mm; 50 μm ARF, 3.3 ± 0.2 mm; 100 μm ARF, 2.5 μm ± 0.1 mm) were observed with corresponding oxidation of EhGSSG (0, −243.5 ± 2.3 mV; 50 μm ARF, −237.8 ± 4.2 mV; 100 μm ARF, −237.5 ± 4.7 mV, Fig 1B). These data demonstrate that HT29 cells treated with ARF (20 μm, 2 h) provide a condition that results in oxidation of the Trx1 system with minimal effect on the GSH redox system.
Fig. 1.

Determination of conditions for ARF-induced oxidation of Trx1 with minimal effect on the GSH redox system. TrxR1 activity was measured by monitoring NADPH oxidation rate at absorbance 340 nm after incubating purified TrxR1 protein with ARF as indicated (A top). To examine ARF dose-dependent oxidation in Trx1 redox state, cells treated with indicated amounts of ARF for 2 h were analyzed for EhTrx1 (A, bottom) by using the ratio of oxidized:reduced Trx1 (quantification of two bands' intensity by densitometry) from the redox western assay (A, top), the Eo for the active site dithiol/disulfide of Trx1 and the Nernst equation (33, 34). B, To examine ARF dose-dependent effect on EhGSSG, HT 29 cells treated with ARF as indicated amounts for 2 h were analyzed for GSH and GSSG by HPLC and EhGSSG was calculated using the Nernst equation (Eo = −264, pH 7.4). Bar graphs are shown as mean ± S.E. (n = 3) with significance indicated at p < 0.05.
Dose-response characteristics of BSO were similarly studied using a 15-h incubation period based on previous studies with HT29 cells (28, 55, 56). Cells incubated with BSO (0 – 1000 μm); cellular GSH (0, 4.5 ± 0.1 mm; 50 μm BSO, 2.3 ± 0.1 mm; 100 μm BSO, 1.8 ± 0.1 mm; 200 μm BSO, 1.6 ± 0.1 mm; 500 μm BSO, 1.4 ± 0.1 mm; 1000 μm BSO, 1.2 ± 0.1 mm), GSSG concentrations (0, 96.2 ± 17.6 μm; 50 μm BSO, 56.2 ± 1.0 μm; 100 μm BSO, 54.2 ± 1.0 μm; 200 μm BSO, 55.8 ± 3.3 μm; 500 μm BSO, 48.1 ± 1.0 μm; 1000 μm BSO, 47.9 ± 1.0 μm) and EhGSSG (0, −243.6 ± 2.3 mV; 50 μm BSO, −232.1 ± 0.3 mV; 100 μm BSO, −226.9 ± 0.8 mV; 200 μm BSO, −223.0 ± 1.5 mV; 500 μm BSO, −220.6 ± 1.3 mV; 1000 μm BSO, −217.6 ± 0.8 mV) were significantly affected by BSO as low as 50 μm (Fig 2A). The redox Western analysis showed that Trx1 redox state was not affected by BSO treatment up to 200 μm, but was significantly oxidized at 500 μm (Fig 2B). Cells had a changed appearance at 200 μm compared with lower concentrations, suggesting some initial toxicity (data not shown). Based on these analyses, 100 μm BSO was selected as an appropriate concentration to examine selective effects of perturbing the GSH redox system with minimal alteration in the Trx1 redox system.
Fig. 2.

Determination of conditions for BSO-induced oxidation of GSH with minimal effect on the Trx redox system. A, To examine dose-dependent effect of BSO on GSH redox potential, cells treated with indicated amounts of BSO for 15 h were analyzed for GSH and GSSG, and EhGSSG was calculated using the Nernst equation. B, To examine dose-dependent effect of BSO on EhTrx1, cells treated with BSO, analyzed for reduced and oxidized Trx1 by redox Western blotting, and EhTrx1 was calculated using the Nernst equation. Bar graphs are shown as mean ± S.E. (n = 3) with significance indicated at p < 0.05.
These results were independently verified in conjunction with redox proteomics studies on HT29 cells described below. In these studies, we included examination of the effect of ATG (57) on the Trx1 and GSH redox systems, although we did not study effects of ATG on redox proteomics. The results confirmed effects of ARF on Trx1 and showed that the same concentration of ATG had less of an effect on Trx1 oxidation (EhTrx1, CR, −259 ± 5 mV; ARF, −215 ± 5 mV: ATG, −227 ± 3 mV; Fig 3A). Neither ATG nor ARF affected the cellular GSH redox system, as shown by GSH (None, 4.2 ± 0.4 mm; ATG, 4.0 ± 0.4 mm; ARF, 3.9 ± 0.3 mm) and GSSG amounts (None, 45.4 ± 0.4 μm; ATG, 45.2 ± 2.4 μm; ARF, 45.0 ± 6.3 μm) and the calculated GSH redox state (EhGSSG; None, −251.7 ± 3.8 mV, ATG, −249.9 ± 2.0 mV; ARF, −251.0 ± 5.8 mV) (Fig 3B) (37). Similar to the data shown in Fig. 2, BSO caused significant decrease of both GSH (1.7 ± 0.2 mm) and GSSG (38.8 ± 7.2 μm) Fig 3B), and substantially oxidized EhGSSG (−230.2 ± 2.0 mV). Additionally, BSO had no effect on Trx1 redox state (Fig 3A).
Fig. 3.

Oxidation of Trx1 redox state (EhTrx1) by ARF and oxidation of GSH redox state (EhGSSG) by BSO in HT29 cells under conditions of redox proteomics assay. To verify results from the dose-response studies under conditions of redox proteomics assay, HT29 cells were treated with 100 μm BSO or 20 μm ARF and analyzed by redox Western blotting for EhTrx1 (A) and by HPLC analyses for EhGSSG (B). In A, subsets of cells were treated with DTT (5 mm) and H2O2 (2 mm) as the reduced and oxidized controls, respectively. Quantified intensity of oxidized Trx1 (Trx1Ox1) and reduced Trx1 (TrxRed) bands of each treatment was used for EhTrx1 calculation. Note that a small amount of a hyper-oxidized form of Trx1, termed Trx1Ox2, was detected in some of the experiments with ARF. This has been previously shown to represent oxidation of a C62-C69 dithiol present in a surface α-helix (33). EhGSSG was calculated from Nernst equation with GSH and GSSG concentrations obtained from HPLC analysis (B). Aurothioglucose (ATG) was included for comparison. Bar graphs are shown as mean ± S.E. (n = 3) with significance indicated for p < 0.05.
Effects of Disruption of Trx Redox System and GSH Redox System on the Percentage Oxidation of Peptidyl Cys
The redox ICAT/MS-based redox proteomics methods enable identification of redox-sensitive peptidyl Cys under different conditions and map these to associated proteins and functional pathways (29, 58). We applied this approach to identify proteins regulated by Trx and GSH redox systems from three biological replicates of HT29 cells treated with ARF and BSO under the conditions shown in Fig. 3 and summarized in Fig. 4. Redox ICAT analysis requires that each peptide be detected in both the heavy isotope and light isotope form to allow determination of the percentage oxidation. Consequently, some data were missing for individual experiments and thus criteria were set to include peptidyl Cys detected in at least two out of the three experiments. We filtered peptidyl Cys for fold oxidation compared with control (ARF/CR, BSO/CR), retaining those with values greater than 1.3 (30% more oxidized than CR). The results showed that ARF oxidized 219 peptidyl Cys associated with 181 proteins out of a total of 606 peptidyl Cys associated with 417 proteins (supplemental File S2). BSO oxidized 60 peptidyl Cys associated with 59 proteins out of a total of 594 peptidyl Cys associated with 385 proteins (supplemental File S2). Mean fold oxidation of the 219 peptidyl Cys by ARF and 60 peptidyl Cys by BSO were 3.7 ± 0.4 and 2.3 ± 0.3, respectively (Fig 4A). In addition, distribution data of oxidized peptidyl Cys by ARF and BSO showed that TrxR inhibition and Trx1 oxidation was associated with a greater fold oxidation of proteins compared with that by BSO (Fig 4B).
Fig. 4.
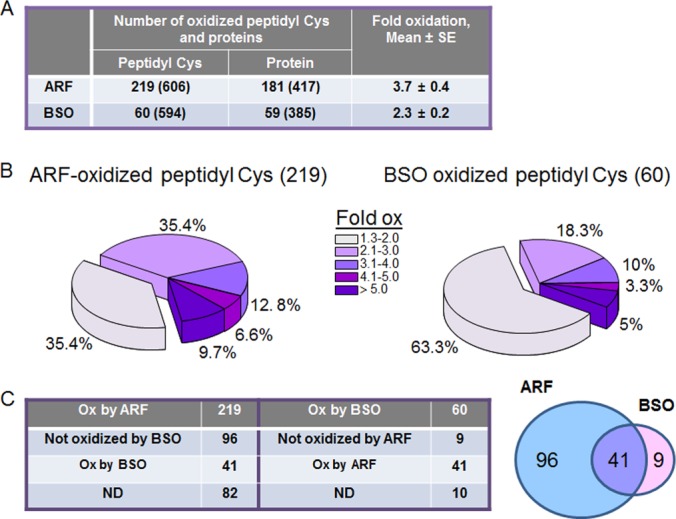
Redox ICAT/MS-based redox proteomics defined peptidyl Cys and proteins regulated by Trx, GSH, and Trx/GSH systems. To determine target proteins regulated by Trx and GSH system, HT29 cells treated with ARF (20 μm, 2 h) and BSO (100 μm, 15 h) were analyzed for redox ICAT-based mass spectrometry as described in the Experimental Procedures. Of the 606 total peptidyl Cys detected in samples treated with ARF, 219 were oxidized 1.3-fold or higher compared with CR (A). On the other hand, of the 594 total peptidyl Cys detected in samples treated with BSO, 60 peptidyl Cys were oxidized 1.3-fold or higher than CR (A). Distribution of ARF and BSO-induced oxidation of peptidyl Cys were visualized by pie charts (B). C, Of the 219 ARF-oxidized peptidyl Cys, 96 were oxidized by only ARF and 41 were by ARF and BSO. Of the 60 BSO-oxidized peptidyl Cys, 9 were oxidized by only BSO and 41 were by BSO and ARF.
To examine whether residual ARF was present in protein extracts used for redox proteomics, cell extracts were treated with proteinase K and analyzed by LC/MS. Results showed that no ARF remained associated with the protein pellet (supplemental File S3, amount of ARF added, 24 nmol/120 μg; limit of detection for ARF remaining in protein pellet, 0.02 nmol/120 μg protein), indicating that oxidation artifacts because of residual ARF were unlikely.
To analyze proteins and associated functional pathways regulated by Trx and GSH redox systems, oxidized peptidyl Cys and proteins were separated into three groups (1) oxidized only by ARF, (2) oxidized by both ARF and BSO, and (3) oxidized only by BSO (Fig 4C). Of the 219 Peptidyl Cys oxidized by ARF, 96 were oxidized only by ARF and 41 were oxidized by both treatments. This grouping excluded 82 peptidyl Cys for further pathway analysis because these had ARF data but lacked consistent data for BSO treatment (see supplemental File S2, worksheet “82 ND”) which precluded categorization into the three groups. Of the 60 oxidized by BSO, nine were oxidized only by BSO, 41 were oxidized by both treatments, and 10 were not categorized because consistent data for ARF treatment were not obtained (see supplemental File S2 worksheet “10 ND”). Because of the relatively large number of peptidyl Cys oxidized by inhibition of the Trx system, we focused on examining the proteins and peptidyl Cys oxidized by ARF. The nine peptidyl Cys oxidized only by BSO treatment were not analyzed further because the number was considered too small for pathway map analysis.
Glycolysis and Gluconeogenesis, and Cytoskeleton Remodeling Pathways are Target Pathways Regulated by the Trx Redox System
To evaluate functional pathway maps associated with these 96 ARF-oxidized peptidyl Cys, we used MetaCore software (https://portal.genego.com/). Thirty-nine pathways (-log p > 2.0, p < 0.01, supplemental File S4) were identified and of the 39 statistically significant pathways, the top ten most significant pathways are shown in Fig. 5. Of these, three were overlapping pathways linked to glycolysis/gluconeogenesis (-log p; 6.4–6.7) and three were overlapping pathways linked to cytoskeleton remodeling (-log p; 4.1–4.9) (Fig. 5). A summary of the peptidyl Cys, protein identifications and fold-oxidation values are shown in Table I. Redox proteomics-identified proteins (ALDOA, ENO3, G3P2, MDH2 PGAM1, PKM2) involved in glycolysis and gluconeogenesis pathways (http://pathwaymaps.com/maps/930/) were identified in MetaCore visualized pathway maps (Fig. 6). To verify ARF-inhibition of glycolysis, we examined HT29 cells for pyruvate kinase (PK) enzyme activity and lactate amount as measures of glycolysis after ARF (20 μm, 2 h) and BSO (100 μm, 15 h) treatments. The result shows both PK activity (CR, 18.4 ± 1.7 mU/ml; ARF, 12.6 ± 1.6 mU/ml) and lactate levels (CR, 100 ± 11.9%; ARF, 52.5 ± 1.1%) were significantly decreased by ARF treatment. In contrast, no significant inhibitory effects were observed by BSO on either PK activity (CR, 18.4 ± 1.7 mU/ml; BSO, 16.1 ± 1.4 mU/ml) or lactate level (CR, 100 ± 11.9%; BSO, 70.0 ± 15.8%) although BSO did appear to exhibit a slight inhibition (Fig. 7).
Fig. 5.

Pathway maps regulated by ARF-oxidized peptidyl Cys. To identify functional pathways affected by disruption in Trx system, 96 peptidyl Cys oxidized by ARF were analyzed by MetaCore Bioinformatics software from Thomson Reuters (https://portal.genego.com/). Of the 39 statistically significant pathways, the top 10 pathways are shown. These include four glycolysis and gluconeogenesis, three cytoskeleton remodeling, oxidative stress by ASK1, CFTR (cystic fibrosis transmembrane conductance regulatory) protein activation, and Parkinson's disease by LRRK2 (Leucine-rich repeat serine/threonine-protein kinase 2).
Table I. Potential target proteins, peptidyl Cys and Cys residues oxidized by ARF-disrupted Trx/TrxR redox system.
| Protein name | Accession | Oxidized Cys residue | Oxidation, Fold ± S.E. |
|---|---|---|---|
| Glycolysis and gluconeogenesis pathway | |||
| Aldolase A (ALDOA) | NP_001121089.1 | YASIC178QQNGIVPIVEPEILPDGDHDLK | 2.5 ± 1.1 |
| Enolase 3 (ENO3) | NP_443739.2 | TGAPC399R | 1.6 ± 0.1 |
| Glyceraldehyde 3-phosphate dehydrogenase (G3P2) | NP_002037.2 | VPTANVSVVDLTC247R | 2.3 ± 0.4 |
| Lactate dehydrogenase A (LDHA) | NP_005557.1 | VIGSGC163NLDSAR | 1.3 ± 0.08 |
| Malate dehydrogenase (MDH2) | NP_005909.2 | GC93DVVVIPAGVPR | 3.5 ± 0.9 |
| Phosphoglycerate mutase1 (PGAM1) | NP_002620.1 | YADLTEDQLPSC153ESLKDTIAR | 2.3 ± 0.8 |
| Pyruvate kinase isozyme (PKM2) | NP_872270.1 | AEGSDVANAVLDGADC358IMLSGETAK | 2.2 ± 0.4 |
| Actin cytoskeleton remodeling | |||
| α-Actinin | NP_004915.2 | AC793LISLGYDVENDR | 7.4 ± 5.9 |
| Cofilin | NP_005498.1 | HELQANCYEEVK | 1.6 ± 0.3 |
| Filamin A | NP_001447.2 | AHVVPC1157FDASK | 2.5 ± 1.2 |
| Myosin light chain (MELC) | NP_066299.2 | ILYSQC32GDVMR | 1.5 ± 0.1 |
| Plectin 1 | NP_000436.2 | C3177RPDQLTGLSLLPLSEK | 1.8 ± 0.5 |
| RhoA | NP_001655.1 | HFC107PNVPIILVGNK | 1.5 ± 0.02 |
| RhoC | NP_001036143.1 | ISAFGYLEC159SAK | 1.4 ± 0.08 |
| Tubulin-α | NP_006000.2 | AYHEQLSVAEITNAC295FEPANQMVK | 1.5 ± 0.06 |
| Oxidative stress/ antioxidants | |||
| HSP70 | NP_004125.3 | MEEFKDQLPADEC608NK | 2.7 ± 1.2 |
| HSP90-α | NP_001017963.2 | HGLEVIYMIEPIDEYC651VQQLK | 3.8 ± 2.3 |
| HSP90-β | NP_031381.2 | GFEVVYMTEPIDEYC521VQQLK | 1.6 ± 0.02 |
| Superoxide dismutase (SOD1) | NP_000445.1 | DGVADVSIEDSVISLSGDHC112IIGR | 5.9 ± 1.8 |
| Thioredoxin | NP_003320.2 | C73MPTFQFFK | 24.9 ± 17.6 |
| Protein synthesis and folding process | |||
| Eukaryotic translation elongation factor 1α (eEF1α) | NP_001393.1 | DGNASGTTLLEALDC234ILPPTRPTDKPLR | 22.5 ± 20.6 |
| Eukaryotic translation elongation factor 1γ (eEF1γ) | NP_001395.1 | WFLTC194INQPQFR | 5.5 ± 3.7 |
| Eukaryotic translation elongation factor 2 (eEF2) | NP_001952.1 | DLEEDHAC567IPIKK | 1.6 ± 0.2 |
| Eukaryotic translation initiation factor 6 (eIF6) | NP_852133.1 | LSALGNVTTC110NDYVALVHPDLDR | 3.7 ± 1.7 |
| Protein disulfide isomerase-associated 3 | NP_005304.3 | FIQENIFGIC244PHMTEDNK | 2.4 ± 0.3 |
| Ribosomal protein L23 | NP_000958.1 | TVFAEHISDEC114K | 3.5 ± 2.0 |
| Ribosomal protein S3a (RPS3A) | NP_000997.1 | NC96LTNFHGMDLTR | 1.5 ± 0.2 |
| Ribosomal protein S5 | NP_001000.2 | KAQC66PIVER | 2.9 ± 0.1 |
| Ribosomal protein S5 | NP_001000.2 | TIAEC172LADELINAAK | 25.8 ± 17.8 |
| RNA-binding protein 3 | NP_909122.1 | C478PSIAAAIAAVNALHGR | 1.9 ± 0.05 |
Fig. 6.

Inhibition of glycolysis and gluconeogenesis pathway by ARF. Of the top 10 most significant pathways identified by MetaCore analysis (Fig. 5), glycolysis and gluconeogenesis pathway maps are shown. Pathway map details are provided in the link, http://pathwaymaps.com/maps/930/. Of the 66 enzymes and molecules involved in these pathways, seven (indicated with circle, ALDOA, ENO3, G3P2, LDHA, MDH2, PGAM1, PKM2) were significantly oxidized by ARF (see Table I also).
Fig. 7.
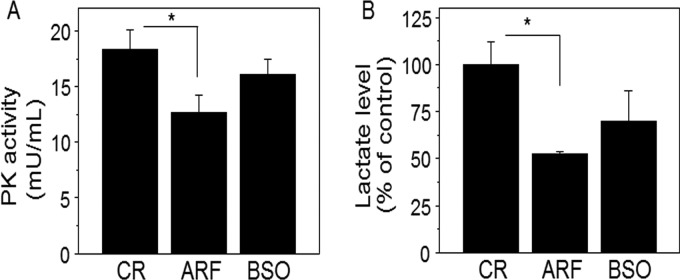
Inhibition of pyruvate kinase (PK) activity and decrease in lactate level by ARF. To verify ARF-inhibited glycolysis pathway identified by redox proteomics, cellular PK activity and lactate levels were measured. HT29 cells treated with ARF (20 μm, 2h) and BSO (100 μm, 15h) or none (CR) were analyzed for PK activity (mU/ml) and l-lactate level (% of control) after preparing standard curves for each PK and l-lactate using fluorometric (Ex/Em = 536/587 nm) assay kits, (Abcam). Bar graphs are shown as mean ± S.E. (n = 3). p < 0.05.
Insulin Regulation of Translation, Lipid Metabolism and Cell Adhesion Pathway Were Affected by both ARF and BSO Treatments
Several peptidyl Cys of proteins involved in translation were oxidized by ARF alone (Table I), but results for proteins oxidized by both ARF and BSO indicated sensitivity to Trx and GSH systems (Table II). For peptidyl Cys oxidized by both ARF and BSO, pathway maps with most significant changes were obtained for insulin regulation of translation, lipid metabolism, and cell adhesion (Fig. 8). Peptidyl Cys and respective proteins, and fold oxidation by ARF and BSO compared with control are shown in the Table II. Information on all 41 peptidyl Cys, associated proteins and fold oxidation is provided in Supplemental data (supplemental File S2).
Table II. Potential target proteins, peptidyl Cys and Cys residues oxidized by both ARF and BSO disrupted Trx1 and GSH redox systems.
| Protein name | Accession | Oxidized Cys residue | Oxidation, Fold ± S.E. |
|
|---|---|---|---|---|
| ARF | BSO | |||
| Insulin regulation of translation and lipid metabolism | ||||
| Eukaryotic translation elongation factor 2 (eEF2) | NP_001952.1 | YVEPIEDVPC466GNIVGLVGVDQFLVK | 4.9 ± 1.6 | 1.7 ± 0.3 |
| Protein phosphatase 1 (PP1) | NP_002699.1 | IC62GDIHGQYYDLLR | 1.5 ± 0.2 | 5.9 ± 2.8 |
| Ribosomal protein S6 (RPS6) | NP_001001.2 | LNISFPATGC12QK | 1.7 ± 0.2 | 1.6 ± 0.1 |
| Cell adhesion and proteolysis | ||||
| Catenin, β1 | NP_001091680.1 | VAAGVLC619ELAQDK | 5.0 ± 2.3 | 2.5 ± 0.8 |
| Plakoglobin | NP_068831.1 | NLALC511PANHAPLQEAAVIPR | 3.3 ± 1.6 | 2.0 ± 0.4 |
| Tubulin-β | NP_821133.1 | LTTPTYGDLNHLVSATMSGVTTC239LR | 1.5 ± 0.1 | 3.3 ± 1.9 |
| Ubiquitin-activating enzyme E1 (UBE1) | NP_003325.2 | YFLVGAGAIGC47ELLK | 3.1 ± 0.7 | 3.1 ± 0.4 |
Fig. 8.

Pathway maps regulated by ARF and BSO-oxidized peptidyl Cys. To identify functional pathway maps affected by disruption in both Trx and GSH system, 41 peptidyl Cys oxidized by both ARF and BSO were analyzed by MetaCore Bioinformatics software from Thomson Reuters (https://portal.genego.com/). Six statistically significant pathways were defined including translation and cell adhesion related pathways (See Table II also).
DISCUSSION
A pharmacological approach based on the use of selective inhibitors together with redox ICAT/MS-based redox proteomics enabled us to test the hypothesis that Trx1 and GSH redox systems selectively control proteins and associated functional pathways in a mammalian cell line. First, we were able to selectively interrupt the Trx and GSH redox systems as evaluated with redox states of Trx1 and GSH in response to concentration-dependent ARF and BSO treatments. Cells exposed to 20 μm ARF for 2 h and 100 μm BSO for 15 h showed selective oxidation in Trx1 and GSH, respectively, without significantly affecting the other redox system. Consequently, we identified potential target proteins and functional pathways responding to altered Trx1 and GSH redox systems using redox proteomics under these conditions.
The differences in time courses of ARF and BSO and variability in detection of the C62-C69 disulfide form of Trx1 with ARF (Trx1Ox2, Fig. 3) leave open the possibility that indirect effects on protein oxidation could be present. The short time frame for ARF treatment suggests that changes in protein abundance are unlikely to impact conclusions. On the other hand, TrxR can reduce proteins other than Trx1 (4, 59, 60), so that the observed effects may not be limited to the Trx system. The longer time frame for BSO treatment could allow changes because of transcriptional activation as shown previously by Harris et al. (27). Although this response was not evaluated in the present study, the redox-ICAT method used to measure percent oxidation is not subject to error because of changes in protein abundance (29).
The present data show that peptidyl Cys associated with proteins of glycolysis and gluconeogenesis pathways are selectively oxidized by ARF. This could be important in conditions of energy limitation, such as can occur during development and tissue repair (61, 62), where oxidation could impair development or tissue repair and recovery. Conversely, enhanced dependence on glycolytic energy metabolism occurs in cancer, a characteristic previously developed as a therapeutic approach for cancer treatment (63). ARF-dependent inhibition of glycolysis in human neutrophils has been previously shown (64), and Le Moan showed that proteins of glycolysis were sensitized to oxidation by H2O2 in yeast with mutations in either the Trx or GSH system (15). Of the 66 proteins and metabolites in the MetaCore human glycolytic/gluconeogenic pathways (http://pathwaymaps.com/maps/930/), 7 were matched in the present study, including aldolase, enolase, glyceraldehyde 3-phosphate dehydrogenase (G3P2), lactate dehydrogenase, malate dehydrogenase, pyruvate kinase isozyme (PKM2), and phosphoglycerate mutase. From our redox ICAT data, we discovered that Cys358 residue of PKM2 was oxidized by ARF treatment (2.2 fold). To confirm a functional relationship, we measured PK enzyme activity and found this to be inhibited by ARF treatment. Oxidation of Cys358 of PKM2 was previously found to result in inhibition of enzymatic activity (65); therefore, our finding of ARF-induced inhibition of PK activity and oxidation of Cys358 of PK are consistent with this previous study. Additionally, our finding of increased oxidation in Cys247 of glyceraldehyde 3-phosphate dehydrogenase (G3P2, 2.3-fold) mediated by ARF is also interesting because modification of this residue is also correlated with decreased enzyme activity (66). Collectively, these findings indicate that the Trx redox system plays an important role in glycolysis by regulating redox states of Cys residues of proteins involved in the glycolytic enzymatic pathways. Interestingly, several other Cys residues of these glycolysis enzymes found to be oxidized by ARF are also S-nitrosylated by a Trx1 transnitrosylation mechanism (67), e.g. Cys247 of G3P2, Cys163 of LDHA, and Cys358 of PKM2 and these Cys modifications appear to be determined by the redox state of Trx1 (68). Taken in concert with the result of studies conducted on yeast and human neutrophils, these data show a central role for redox control in defined peptidyl Cys of multiple proteins in the glycolysis pathway. These results are consistent with previous evidence that the Cys proteome exists in functional redox modules in which multiple proteins within functional networks have evolved similar redox dependences to coordinate activities in response to physiologic and toxicologic challenge (29, 69).
The data presented herein are also consistent with previous evidence that proteins of the actin-cytoskeletal system exist as a functional redox module (29, 58). Substantial evidence indicates that the thiol redox state of the cell, reflected in part by the thiol content of the filamentous actin cytoskeleton, impacts cytoskeletal structure and remodeling (70, 71). In our work, eight peptidyl Cys of actin cytoskeleton-associated proteins (α-actinin, cofilin, filamin A, myosin light chain, plectin1, RhoA, RhoC, tubulin-α) were oxidized by ARF treatment. As shown by previous studies, Trx1 (72, 73) and TrxR1 (4) interact with actin via a thiol redox-dependent mechanism. The interaction between Trx/TrxR and actin cytoskeleton appears to be critical in multiple mammalian cell functions by controlling mechanisms for actin dynamics and cytoskeleton remodeling; in epithelial cells, such as HT29, such activities are important in the cell cycle, responses to stress and maintenance of barrier functions.
The mammalian systems appear to especially differ from yeast in the presence of significant numbers of proteins and pathways sensitive to both Trx and GSH redox systems, as determined by their sensitivity to oxidation by either inhibitor. Based on significance and availability of relevant literature, we briefly discuss pathway results for translation, lipid metabolism, proteolysis, and cell adhesion.
Redox control of translation initiation and elongation has been previously shown in plant and eukaryotic cell studies (74–77). The redox state of translational elongation factor in cyanobacterium Synechocystis, and elongation factor G (EF-G) plays an important role in regulation of translation (74). Oxidation-attributed disulfide bond formation of critical Cys residues results in inactivation of EF-G; however, when the disulfide bond is reduced by Trx, it results in reactivation of translational machinery, suggesting that the translation mechanism is regulated by Trx system by the redox state of EF-G (74). In addition, a number of previous studies have shown that protein synthesis is regulated by redox-dependent activation and inactivation of translational machinery involving interaction of elongation and initiation factors with Trx (75, 76, 78). Consistent with these previous studies, our data show that several translational elongation and initiation factors and other proteins responsible for protein synthesis are oxidized by ARF-induced Trx/TrxR redox disruption (see Table I and II). In addition to Trx redox-dependent control of translation, several studies also show GSH redox-dependent regulation for translation (77, 79, 80). Koonin et al. identified that eEF-1γ contains a GSH transferase (GST) domain (79). Their finding showed the potential GST activity of eEF-1γ and the possibility that GSH redox regulates assembly of multiple EF complex and aminoacyl tRNA synthesis. More evidence for GSH redox system-dependent regulation of translational machinery was shown by glutathionylation of EF, eEF-1α, and eEF2 (35, 81). In the present study, we show that Cys466 of eEF-2, Cys62 of PP1 and Cys12 of RPS6 were oxidized by ARF and BSO treatment. This suggests that redox states of these peptidyl Cys responsible for protein translation are sensitive to general alterations in cellular redox status, regulated by the Trx redox system as well as the GSH redox system. Together, these data provide evidence for a functional redox module associated with translation that has the important character of being dependent on both the Trx and GSH systems and representing a mechanism to integrate biologic systems through redox-sensing cysteine residues (40, 69).
MetaCore analysis identified cell adhesion pathways including plakloglobin, β-catenin, and tubulin as pathways regulated by both the Trx and GSH redox systems. Plakloglobin is highly homologous to β-catenin and is found in adherence junctions (82). Redox regulation of cell surface proteins, including growth factor receptors, transporters, cytokine receptors and integrins, is associated with biologic function (83). Although Trx-redox regulation of junction proteins, e.g. plakloglobin and β-catenin regulating cell-cell adhesion does not appear to be available, GSH- and GSH-redox dependent cell-cell adhesion and cell-matrix adhesion through caveolin/β-catenin pathway has been previously observed (84). Other studies show a link between Trx1 and β-catenin-related to protein expression (85, 86) but do not appear to have established a redox dependence. In conjunction with the present findings, these previous studies support a need for detailed studies to test redox-dependence of cell-cell adhesion pathways as possible redox modules regulated by Trx and GSH systems.
In consideration of global redox control system, the present study is perhaps most informative in comparison to the detailed study of oxidized protein thiols in Saccharomyces cerevisiae with two distinct thiol-labeling approaches by Le Moan et al. (15). They found differences in yeast proteins oxidized by added H2O2 in genetic models with inactivation of Trx and GSH systems, and concluded that Trx and GSH have very distinct thiol redox control systems, with Trx having an exclusive role in H2O2 metabolism and GSH having a presumed thiol redox buffering function. The present study differs from the yeast study in the following ways: (1) using a mammalian cell model, (2) examining redox proteomic changes without exogenously added H2O2, (3) using selective pharmacologic inhibitors, and (4) using a redox-ICAT method to measure peptidyl Cys oxidation. Despite these substantial differences, the central findings are consistent with their findings in yeast, especially showing similarity in sensitivity of glycolysis to genetic inactivation of Trx in yeast and ARF-dependent oxidation of glycolysis/gluconeogenesis proteins in the HT29 cells, and in emphasizing distinct roles for the Trx and GSH systems in other aspects of thiol redox control.
The present findings further indicate that the Trx and GSH system may have different roles in mammalian cells compared with yeast. In particular, the mammalian cell data show that the GSH system is partially redundant to the Trx system in redox control of translation whereas the yeast data showed no effects of either Trx or GSH mutants on translation machinery in the absence of H2O2 challenge. It should be noted, however, that the coverage of proteins involved in translation was sparse in both cases. Additionally, peptidyl Cys of cytoskeletal proteins were highly sensitive to inhibition of TrxR in HT29 cells but such oxidation was not apparent in the reported yeast data. This difference may only reflect life cycle effects or differences in peptides measured and will require additional study. Finally, peptidyl Cys of proteins associated with cell adhesion were sensitive to disruption of either Trx or GSH systems in the mammalian cells. The appearance of this apparent redox module may reflect a change in the Cys proteome with evolution of multicellular complexity (87) and will require additional study across phylogeny.
In summary, the present study provides an evaluation of the differences in oxidation of protein Cys residues resulting from disrupted Trx or GSH redox systems using cultured HT29 cells. HT29 cells are a colon cancer cell line and this makes it likely that some of the current results may differ from what one might find in non-transformed cells. Although further studies are needed to more fully delineate Trx and/or GSH redox-dependent regulation of individual proteins and their associated pathways, the present study supports the hypothesis that the Trx and GSH systems control target proteins and pathways in a selective rather than a general manner. In addition, the data support the interpretation of Le Moan et al. (15) that the Trx system regulates a wider range of proteins and pathways compared with the GSH system. Finally, this study demonstrates that mass spectrometry-based redox proteomics combined with bioinformatics pathway analysis is suitable to identify multiple proteins, pathways and functional networks as components of complex oxidative mechanisms of toxicity and disease.
Supplementary Material
Footnotes
* This work was supported by NIH grants ES009047 and HL113451.
 This article contains supplemental Files S1 to S4.
This article contains supplemental Files S1 to S4.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (88) with the data set identifier ProteomeXchange accession: PXD000353.
1 The abbreviations used are:
- Trx
- thioredoxin
- ARF
- auranofin
- BSO
- buthionine sulfoximine
- CR
- control
- Cys
- cysteine
- Eh
- redox potential
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- H2O2
- hydrogen peroxide
- ICAT
- isotope coded affinity tag
- MS
- mass spectrometry
- PK
- pyruvate kinase
- TrxR
- thioredoxin reductase.
REFERENCES
- 1. Shiota M., Yokomizo A., Naito S. (2011) Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic. Biol. Med. 51, 1320–1328 [DOI] [PubMed] [Google Scholar]
- 2. Kondo T., Hirose M., Kageyama K. (2009) Roles of oxidative stress and redox regulation in atherosclerosis. J. Atheroscler. Thromb. 16, 532–538 [DOI] [PubMed] [Google Scholar]
- 3. Zhu H., Jia Z., Misra H., Li Y. R. (2012) Oxidative stress and redox signaling mechanisms of alcoholic liver disease: updated experimental and clinical evidence. J. Dig. Dis. 13, 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thom S. R., Bhopale V. M., Milovanova T. N., Yang M., Bogush M. (2012) Thioredoxin Reductase Linked to Cytoskeleton by Focal Adhesion Kinase Reverses Actin S-Nitrosylation and Restores Neutrophil beta2 Integrin Function. J. Biol. Chem. 287, 30346–30357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biguet C., Wakasugi N., Mishal Z., Holmgren A., Chouaib S., Tursz T., Wakasugi H. (1994) Thioredoxin increases the proliferation of human B-cell lines through a protein kinase C-dependent mechanism. J. Biol. Chem. 269, 28865–28870 [PubMed] [Google Scholar]
- 6. Wei S. J., Botero A., Hirota K., Bradbury C. M., Markovina S., Laszlo A., Spitz D. R., Goswami P. C., Yodoi J., Gius D. (2000) Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 60, 6688–6695 [PubMed] [Google Scholar]
- 7. Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braun H., Lichter A., Haberlein I. (1996) Kinetic evidence for protein complexes between thioredoxin and NADP-malate dehydrogenase and presence of a thioredoxin binding site at the N-terminus of the enzyme. Eur. J. Biochem. 240, 781–788 [DOI] [PubMed] [Google Scholar]
- 9. Matthews J. R., Wakasugi N., Virelizier J. L., Yodoi J., Hay R. T. (1992) Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 20, 3821–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pillay C. S., Hofmeyr J. H., Rohwer J. M. (2011) The logic of kinetic regulation in the thioredoxin system. BMC Syst. Biol. 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spector D., Labarre J., Toledano M. B. (2001) A genetic investigation of the essential role of glutathione: mutations in the proline biosynthesis pathway are the only suppressors of glutathione auxotrophy in yeast. J. Biol. Chem. 276, 7011–7016 [DOI] [PubMed] [Google Scholar]
- 12. Armstrong J. S., Steinauer K. K., Hornung B., Irish J. M., Lecane P., Birrell G. W., Peehl D. M., Knox S. J. (2002) Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 9, 252–263 [DOI] [PubMed] [Google Scholar]
- 13. Reliene R., Schiestl R. H. (2006) Glutathione depletion by buthionine sulfoximine induces DNA deletions in mice. Carcinogenesis 27, 240–244 [DOI] [PubMed] [Google Scholar]
- 14. Wang J., Boja E. S., Tan W., Tekle E., Fales H. M., English S., Mieyal J. J., Chock P. B. (2001) Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 276, 47763–47766 [DOI] [PubMed] [Google Scholar]
- 15. Le Moan N., Clement G., Le Maout S., Tacnet F., Toledano M. B. (2006) The Saccharomyces cerevisiae proteome of oxidized protein thiols: contrasted functions for the thioredoxin and glutathione pathways. J. Biol. Chem. 281, 10420–10430 [DOI] [PubMed] [Google Scholar]
- 16. Reichheld J. P., Khafif M., Riondet C., Droux M., Bonnard G., Meyer Y. (2007) Inactivation of thioredoxin reductases reveals a complex interplay between thioredoxin and glutathione pathways in Arabidopsis development. Plant Cell 19, 1851–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu J., Chew E. H., Holmgren A. (2007) Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U.S.A. 104, 12288–12293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hansen J. M., Zhang H., Jones D. P. (2006) Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic. Biol. Med. 40, 138–145 [DOI] [PubMed] [Google Scholar]
- 19. Zhang H., Go Y. M., Jones D. P. (2007) Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch. Biochem. Biophys. 465, 119–126 [DOI] [PubMed] [Google Scholar]
- 20. Go Y. M., Ziegler T. R., Johnson J. M., Gu L., Hansen J. M., Jones D. P. (2007) Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic. Biol. Med. 42, 363–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Halvey P. J., Hansen J. M., Johnson J. M., Go Y. M., Samali A., Jones D. P. (2007) Selective oxidative stress in cell nuclei by nuclear-targeted D-amino acid oxidase. Antioxid. Redox Signal. 9, 807–816 [DOI] [PubMed] [Google Scholar]
- 22. Finkelstein A. E., Walz D. T., Batista V., Mizraji M., Roisman F., Misher A. (1976) Auranofin. New oral gold compound for treatment of rheumatoid arthritis. Ann. Rheum. Dis. 35, 251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu C., Liu Z., Li M., Li X., Wong Y. S., Ngai S. M., Zheng W., Zhang Y., Chen T. (2013) Enhancement of auranofin-induced apoptosis in MCF-7 human breast cells by selenocystine, a synergistic inhibitor of thioredoxin reductase. PLoS One. 8, e53945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox A. G., Brown K. K., Arner E. S., Hampton M. B. (2008) The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem. Pharmacol. 76, 1097–1109 [DOI] [PubMed] [Google Scholar]
- 25. Cohen-Kutner M., Khomsky L., Trus M., Aisner Y., Niv M. Y., Benhar M., Atlas D. (2013) Thioredoxin-mimetic peptides (TXM) reverse auranofin induced apoptosis and restore insulin secretion in insulinoma cells. Biochem. Pharmacol. 85, 977–990 [DOI] [PubMed] [Google Scholar]
- 26. Drew R., Miners J. O. (1984) The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem. Pharmacol. 33, 2989–2994 [DOI] [PubMed] [Google Scholar]
- 27. Harris C., Shuster D. Z., Roman Gomez R., Sant K. E., Reed M. S., Pohl J., Hansen J. M. (2013) Inhibition of glutathione biosynthesis alters compartmental redox status and the thiol proteome in organogenesis-stage rat conceptuses. Free Radic. Biol. Med. 63, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderson C. L., Iyer S. S., Ziegler T. R., Jones D. P. (2007) Control of extracellular cysteine/cystine redox state by HT-29 cells is independent of cellular glutathione. Am. J. Physiol. Regul Integr Comp Physiol. 293, R1069–R1075 [DOI] [PubMed] [Google Scholar]
- 29. Go Y. M., Duong D. M., Peng J., Jones D. P. (2011) Protein Cysteines Map to Functional Networks According to Steady-state Level of Oxidation. J. Proteomics Bioinform. 4, 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sethuraman M., McComb M. E., Huang H., Huang S., Heibeck T., Costello C. E., Cohen R. A. (2004) Isotope-coded affinity tag (ICAT) approach to redox proteomics: identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J. Proteome Res. 3, 1228–1233 [DOI] [PubMed] [Google Scholar]
- 31. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Go Y. M., Pohl J., Jones D. P. (2009) Quantification of redox conditions in the nucleus. Methods Mol. Biol. 464, 303–317 [DOI] [PubMed] [Google Scholar]
- 33. Watson W. H., Pohl J., Montfort W. R., Stuchlik O., Reed M. S., Powis G., Jones D. P. (2003) Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 278, 33408–33415 [DOI] [PubMed] [Google Scholar]
- 34. Halvey P. J., Watson W. H., Hansen J. M., Go Y. M., Samali A., Jones D. P. (2005) Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem. J. 386, 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamnell-Pamment Y., Lind C., Palmberg C., Bergman T., Cotgreave I. A. (2005) Determination of site-specificity of S-glutathionylated cellular proteins. Biochem. Biophys. Res. Commun. 332, 362–369 [DOI] [PubMed] [Google Scholar]
- 36. Kirlin W. G., Cai J., Thompson S. A., Diaz D., Kavanagh T. J., Jones D. P. (1999) Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic. Biol. Med. 27, 1208–1218 [DOI] [PubMed] [Google Scholar]
- 37. Jones D. P., Liang Y. (2009) Measuring the poise of thiol/disulfide couples in vivo. Free Radic. Biol. Med. 47, 1329–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arner E. S., Holmgren A. (2001) Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. Chapter 7, Unit 74 [DOI] [PubMed] [Google Scholar]
- 39. Hansen R. E., Winther J. R. (2009) An introduction to methods for analyzing thiols and disulfides: Reactions, reagents, and practical considerations. Anal. Biochem. 394, 147–158 [DOI] [PubMed] [Google Scholar]
- 40. Jones D. P. (2010) Redox sensing: orthogonal control in cell cycle and apoptosis signalling. J. Intern. Med. 268, 432–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones D. P. (2008) Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 295, C849–C868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kemp M., Go Y. M., Jones D. P. (2008) Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic. Biol. Med. 44, 921–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones D. P., Go Y. M., Anderson C. L., Ziegler T. R., Kinkade J. M., Jr., Kirlin W. G. (2004) Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. Faseb J. 18, 1246–1248 [DOI] [PubMed] [Google Scholar]
- 44. Xu P., Duong D. M., Peng J. (2009) Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res. 8, 3944–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eng J., McCormack A., Yates J. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 46. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 47. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 48. Seyfried N. T., Gozal Y. M., Donovan L. E., Herskowitz J. H., Dammer E. B., Xia Q., Ku L., Chang J., Duong D. M., Rees H. D., Cooper D. S., Glass J. D., Gearing M., Tansey M. G., Lah J. J., Feng Y., Levey A. I., Peng J. (2012) Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards. J. Proteome Res. 11, 2721–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dammer E. B., Fallini C., Gozal Y. M., Duong D. M., Rossoll W., Xu P., Lah J. J., Levey A. I., Peng J., Bassell G. J., Seyfried N. T. (2012) Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS One. 7, e38658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barsnes H., Vizcaíno J. A., Eidhammer I., Martens L. (2009) PRIDE Converter: making proteomics data-sharing easy. Nat. Biotechnol. 27, 598–599 [DOI] [PubMed] [Google Scholar]
- 52. Soltow Q. A., Strobel F. H., Mansfield K. G., Wachtman L., Park Y., Jones D. P. (2013) High-performance metabolic profiling with dual chromatography-Fourier-transform mass spectrometry (DC-FTMS) for study of the exposome. Metabolomics 9, 132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Albert A., Brauckmann C., Bilaske F., Sperling M., Engelhard C., Karst U. (2012) Speciation analysis of the antirheumatic agent Auranofin and its thiol adducts by LC/ESI-MS and LC/ICP-MS. J. Anal. Atomic Spectrom. 27, 975–981 [Google Scholar]
- 54. Levine D. M., Haynor D. R., Castle J. C., Stepaniants S. B., Pellegrini M., Mao M., Johnson J. M. (2006) Pathway and gene-set activation measurement from mRNA expression data: the tissue distribution of human pathways. Genome Biol. 7, R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knoll N., Ruhe C., Veeriah S., Sauer J., Glei M., Gallagher E. P., Pool-Zobel B. L. (2005) Genotoxicity of 4-hydroxy-2-nonenal in human colon tumor cells is associated with cellular levels of glutathione and the modulation of glutathione S-transferase A4 expression by butyrate. Toxicol. Sci. 86, 27–35 [DOI] [PubMed] [Google Scholar]
- 56. Duyndam M. C., Hulscher T. M., Fontijn D., Pinedo H. M., Boven E. (2001) Induction of vascular endothelial growth factor expression and hypoxia-inducible factor 1alpha protein by the oxidative stressor arsenite. J. Biol. Chem. 276, 48066–48076 [DOI] [PubMed] [Google Scholar]
- 57. Pratesi A., Gabbiani C., Ginanneschi M., Messori L. (2010) Reactions of medicinally relevant gold compounds with the C-terminal motif of thioredoxin reductase elucidated by MS analysis. Chem. Commun. 46, 7001–7003 [DOI] [PubMed] [Google Scholar]
- 58. Go Y. M., Park H., Koval M., Orr M., Reed M., Liang Y., Smith D., Pohl J., Jones D. P. (2010) A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic. Biol. Med. 48, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fang J., Zhong L., Zhao R., Holmgren A. (2005) Ebselen: a thioredoxin reductase-dependent catalyst for alpha-tocopherol quinone reduction. Toxicol. Appl. Pharmacol. 207, 103–109 [DOI] [PubMed] [Google Scholar]
- 60. Mustacich D., Powis G. (2000) Thioredoxin reductase. Biochem. J. 346 Pt 1, 1–8 [PMC free article] [PubMed] [Google Scholar]
- 61. Pavlides S., Whitaker-Menezes D., Castello-Cros R., Flomenberg N., Witkiewicz A. K., Frank P. G., Casimiro M. C., Wang C., Fortina P., Addya S., Pestell R. G., Martinez-Outschoorn U. E., Sotgia F., Lisanti M. P. (2009) The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 8, 3984–4001 [DOI] [PubMed] [Google Scholar]
- 62. Tsuruda T., Hatakeyama K., Nagamachi S., Sekita Y., Sakamoto S., Endo G. J., Nishimura M., Matsuyama M., Yoshimura K., Sato Y., Onitsuka T., Imamura T., Asada Y., Kitamura K. (2012) Inhibition of development of abdominal aortic aneurysm by glycolysis restriction. Arterioscler. Thromb. Vasc. Biol. 32, 1410–1417 [DOI] [PubMed] [Google Scholar]
- 63. Gatenby R. A., Gillies R. J. (2007) Glycolysis in cancer: a potential target for therapy. Int. J. Biochem. Cell Biol. 39, 1358–1366 [DOI] [PubMed] [Google Scholar]
- 64. Anderson R., Van Rensburg C. E., Jooné G. K., Lessing A. (1991) Auranofin inactivates phosphofructokinase in human neutrophils, leading to depletion of intracellular ATP and inhibition of superoxide generation and locomotion. Mol. Pharmacol. 40, 427–434 [PubMed] [Google Scholar]
- 65. Anastasiou D., Poulogiannis G., Asara J. M., Boxer M. B., Jiang J. K., Shen M., Bellinger G., Sasaki A. T., Locasale J. W., Auld D. S., Thomas C. J., Vander Heiden M. G., Cantley L. C. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martyniuk C. J., Fang B., Koomen J. M., Gavin T., Zhang L., Barber D. S., Lopachin R. M. (2011) Molecular mechanism of glyceraldehyde-3-phosphate dehydrogenase inactivation by alpha,beta-unsaturated carbonyl derivatives. Chem. Res. Toxicol. 24, 2302–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu C., Liu T., Chen W., Oka S., Fu C., Jain M. R., Parrott A. M., Baykal A. T., Sadoshima J., Li H. (2010) Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol. Cell. Proteomics. 9, 2262–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sengupta R., Holmgren A. (2013) Thioredoxin and thioredoxin reductase in relation to reversible s-nitrosylation. Antioxid. Redox. Signal. 18, 259–269 [DOI] [PubMed] [Google Scholar]
- 69. Go Y. M., Jones D. P. (2013) Thiol/disulfide redox states in signaling and sensing. Crit. Rev. Biochem. Mol. Biol. 48, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stournaras C. (1990) Exposure of thiol groups and bound nucleotide in G-actin: thiols as an indicator for the native state of actin. Anticancer Res. 10, 1651–1659 [PubMed] [Google Scholar]
- 71. Valentin-Ranc C., Carlier M. F. (1991) Role of ATP-bound divalent metal ion in the conformation and function of actin. Comparison of Mg-ATP, Ca-ATP, and metal ion-free ATP-actin. J. Biol. Chem. 266, 7668–7675 [PubMed] [Google Scholar]
- 72. Zschauer T. C., Kunze K., Jakob S., Haendeler J., Altschmied J. (2011) Oxidative stress-induced degradation of thioredoxin-1 and apoptosis is inhibited by thioredoxin-1-actin interaction in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 31, 650–656 [DOI] [PubMed] [Google Scholar]
- 73. Wang X., Ling S., Zhao D., Sun Q., Li Q., Wu F., Nie J., Qu L., Wang B., Shen X., Bai Y., Li Y., Li Y. (2010) Redox regulation of actin by thioredoxin-1 is mediated by the interaction of the proteins via cysteine 62. Antioxid. Redox. Signal. 13, 565–573 [DOI] [PubMed] [Google Scholar]
- 74. Kojima K., Motohashi K., Morota T., Oshita M., Hisabori T., Hayashi H., Nishiyama Y. (2009) Regulation of translation by the redox state of elongation factor G in the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem. 284, 18685–18691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lemaire S. D., Guillon B., Le Maréchal P., Keryer E., Miginiac-Maslow M., Decottignies P. (2004) New thioredoxin targets in the unicellular photosynthetic eukaryote Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. U.S.A. 101, 7475–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yamazaki D., Motohashi K., Kasama T., Hara Y., Hisabori T. (2004) Target proteins of the cytosolic thioredoxins in Arabidopsis thaliana. Plant Cell Physiol. 45, 18–27 [DOI] [PubMed] [Google Scholar]
- 77. Barnes D., Mayfield S. P. (2003) Redox control of posttranscriptional processes in the chloroplast. Antioxid. Redox. Signal. 5, 89–94 [DOI] [PubMed] [Google Scholar]
- 78. Jun K. O., Song C. H., Kim Y. B., An J., Oh J. H., Choi S. K. (2009) Activation of translation via reduction by thioredoxin-thioredoxin reductase in Saccharomyces cerevisiae. FEBS Lett. 583, 2804–2810 [DOI] [PubMed] [Google Scholar]
- 79. Koonin E. V., Mushegian A. R., Tatusov R. L., Altschul S. F., Bryant S. H., Bork P., Valencia A. (1994) Eukaryotic translation elongation factor 1 gamma contains a glutathione transferase domain–study of a diverse, ancient protein superfamily using motif search and structural modeling. Protein Sci. 3, 2045–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Irihimovitch V., Shapira M. (2000) Glutathione redox potential modulated by reactive oxygen species regulates translation of Rubisco large subunit in the chloroplast. J. Biol. Chem. 275, 16289–16295 [DOI] [PubMed] [Google Scholar]
- 81. Townsend D. M., Findlay V. J., Fazilev F., Ogle M., Fraser J., Saavedra J. E., Ji X., Keefer L. K., Tew K. D. (2006) A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol. Pharmacol. 69, 501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fukunaga Y., Liu H., Shimizu M., Komiya S., Kawasuji M., Nagafuchi A. (2005) Defining the roles of beta-catenin and plakoglobin in cell-cell adhesion: isolation of beta-catenin/plakoglobin-deficient F9 cells. Cell Struct. Funct. 30, 25–34 [DOI] [PubMed] [Google Scholar]
- 83. Metcalfe C., Cresswell P., Ciaccia L., Thomas B., Barclay A. N. (2011) Labile disulfide bonds are common at the leucocyte cell surface. Open Biol. 1, 110010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen R. S., Song Y. M., Zhou Z. Y., Tong T., Li Y., Fu M., Guo X. L., Dong L. J., He X., Qiao H. X., Zhan Q. M., Li W. (2009) Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene 28, 599–609 [DOI] [PubMed] [Google Scholar]
- 85. Kipp A. P., Muller M. F., Göken E. M., Deubel S., Brigelius-Flohe R. (2012) The selenoproteins GPx2, TrxR2 and TrxR3 are regulated by Wnt signalling in the intestinal epithelium. Biochim. Biophys. Acta 1820, 1588–1596 [DOI] [PubMed] [Google Scholar]
- 86. Adluri R. S., Thirunavukkarasu M., Zhan L., Akita Y., Samuel S. M., Otani H., Ho Y. S., Maulik G., Maulik N. (2011) Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: a study using thioredoxin 1 transgenic mice. J. Mol. Cell Cardiol. 50, 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Miseta A., Csutora P. (2000) Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 17, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 88. Vizcaíno J. A., Côté R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Pérez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}