

Abstract
The propagation of phosphorylation downstream of receptor tyrosine kinases is a key dynamic cellular event involved in signal transduction, which is often deregulated in disease states such as cancer. Probing phosphorylation dynamics is therefore crucial for understanding receptor tyrosine kinases' function and finding ways to inhibit their effects. MS methods combined with metabolic labeling such as stable isotope labeling with amino acids in cell culture (SILAC) have already proven successful in deciphering temporal phosphotyrosine perturbations. However, they are limited in terms of multiplexing, and they also are time consuming, because several experiments need to be performed separately. Here, we introduce an innovative approach based on 5-plex SILAC that allows monitoring of phosphotyrosine signaling perturbations induced by a drug treatment in one single experiment. Using this new labeling strategy specifically tailored for phosphotyrosines, it was possible to generate the time profiles for 318 unique phosphopeptides belonging to 215 proteins from an erlotinib-treated breast cancer cell line model. Hierarchical clustering of the time profiles followed by pathway enrichment analysis highlighted epidermal growth factor receptor (EGFR or ErbB1) and ErbB2 signaling as the major pathways affected by erlotinib, thereby validating the method. Moreover, based on the similarity of its time profile to those of other proteins in the ErbB pathways, the phosphorylation at Tyr453 of protein FAM59A, a recently described adaptor of EGFR, was confirmed as tightly involved in the signaling cascade. The present investigation also demonstrates the remote effect of EGFR inhibition on ErbB3 phosphorylation sites such as Tyr1289 and Tyr1328, as well as a potential feedback effect on Tyr877 of ErbB2. Overall, the 5-plex SILAC is a straightforward approach that extends sample multiplexing and builds up the arsenal of methods for tyrosine phosphorylation dynamics.
The transmission of information in a cell is largely driven by protein phosphorylation (1, 2). It is used to convey a signal within the cell and modulate different biological outcomes such as transcription regulation or cytoskeleton rearrangement. The receptor tyrosine kinases represent key players at the plasma membrane interface because they operate as initiators of phosphorylation cascades (3, 4). Among the 58 receptor tyrosine kinases encoded by the human genome, the ErbB family members—composed of ErbB1 (also called epidermal growth factor receptor (EGFR)),1 ErbB2, ErbB3, and ErbB4—play a pivotal role because they control important processes such as cell differentiation, proliferation, and apoptosis (5). Deregulation of ErbB signaling networks is known to be connected to various diseases such as cancer (6, 7). Thus, the ErbB family members represent an important target class for understanding and curing disease—indeed, both small-molecule tyrosine kinase inhibitors (TKIs) and monoclonal antibodies are under extensive development (8). In order to evaluate the proper effect of drugs on signaling pathways and better recognize their mode of action, one must assess tyrosine phosphorylation changes of ErbBs and their associated downstream proteins.
Recently, MS-based quantitative phosphoproteomics approaches have developed into attractive and powerful tools for studying signaling networks (9–12). Moreover, it has become apparent that investigations based on “static” binary comparisons (control versus treated) need to be replaced by alternative ones in which the dynamics of the networks can be monitored (13–16). Also, peptide-centric strategies are preferred because they permit deeper sampling of the phosphoproteome and discriminate between differentially regulated sites on a single protein (15). Typically, phosphoproteome dynamics screens are performed using one of two approaches: stable isotope labeling with amino acids in cell culture (SILAC) using three different versions of labeled lysine and arginine (3-plex SILAC) (17), or isobaric tags for relative and absolute quantitation (iTRAQ) using four (4-plex iTRAQ) (18) and eight (8-plex iTRAQ) (19) labeling reagents. MS signal quantitation is then performed using the precursor (MS1-based) and reporter ions (MS2-based) for SILAC and iTRAQ, respectively. Although both strategies are highly valuable, they also have some shortcomings. The 3-plex SILAC approach requires the combination of two separate parallel experiments, including a common reference for normalization, to obtain a five-time-point kinetic. This implies that the two samples also need to be processed and measured individually, augmenting the analysis time per condition. The iTRAQ approach constricts the labeling and mixing steps at a late stage of the sample preparation procedure, introducing variability issues. Moreover, during data acquisition, co-fragmentation of interfering peptides present in the isolation window of the precursor ions perturbs the quantitative information encoded in the reporter ion region (20–22). It has also been reported that iTRAQ derivatized phosphopeptides tend to undergo charge enhancement during ionization, leading to a significant drop in identification efficiency (23).
Here, we describe the development of a new 5-plex SILAC-based strategy combining serial peptide immunoaffinity purification and LC-MS/MS detection for the examination of tyrosine phosphorylation dynamics. The approach relies on robust MS1-based quantitation and offers the advantage of increased sample multiplexing. The model breast cancer cell line KPL-4 was differentially labeled into five populations using a mixture of light and heavy isotope-encoded lysines, arginines, and tyrosines. After sample combination, each detected tyrosine-phosphorylated tryptic peptide was present as a quintuplet in MS, which allowed the monitoring of five time points in a single experiment. This method was applied to investigate the effects of the selective EGFR TKI erlotinib (Tarceva) on ErbB signaling over a 1-h treatment. It was possible to identify, quantitate, and generate the temporal drug response profiles of 318 unique tyrosine-phosphorylated peptides belonging to 215 proteins. After hierarchical clustering and pathway enrichment analysis, the data clearly show EGFR and ErbB2 as the main affected signaling networks.
EXPERIMENTAL PROCEDURES
Materials
D4-lysine (Lys4), [13C6,15N2]-lysine (Lys8), 13C6-arginine (Arg6), [13C6,15N4]-arginine (Arg10), 13C6-tyrosine (Tyr6), and [13C9,15N1]-tyrosine (Tyr10) were obtained from Cambridge Isotope Laboratories (Andover, MA), and unlabeled lysine (Lys0), arginine (Arg0), and tyrosine (Tyr0) were obtained from Sigma (Buchs, Switzerland). Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/l glucose and deficient in glutamine, lysine, arginine, and tyrosine was prepared in-house. Amino acids were added at 0.798, 0.398, and 0.461 mm final concentration for lysine, arginine, and tyrosine, respectively. To prevent arginine-to-proline conversion, 200 mg/l (1.737 mm) proline was added to the medium (24). The human breast cancer cell line KPL-4 (25) was a kind gift from J. Kurebayashi (Kawasaki Medical School, Okayama, Japan).
5-plex SILAC Cell Culturing, Treatment, and Digest Preparation
KPL-4 cells were grown as five different populations for at least six doublings in DMEM supplemented with 10% dialyzed fetal bovine serum and glutamine-containing antibiotics (Pen Strep Glutamine, Invitrogen) and with the following amino acid combinations: SILAC medium 1 (SM1): Lys0, Arg0, Tyr0; SM2: Lys4, Arg6, Tyr0; SM3: Lys8, Arg10, Tyr0; SM4: Lys8, Arg10, Tyr6; SM5: Lys8, Arg10, Tyr10 (see Fig. 1). Plated cells (2 × 107) were serum starved for 24 h, and SM2–5 were treated with erlotinib (5 μM end concentration) for 5, 10, 30, and 60 min, respectively. SM1 was treated with dimethyl sulfoxide to serve as the control. Cells were washed twice with cold PBS before the addition of lysis buffer for 10 min at room temperature. Lysis buffer consisted of 6 m urea, 2 m thiourea, 200 mm ammonium bicarbonate pH 8.5, and phosphatase inhibitors (PhosStop, Roche). Lysed cells were collected by scraping the plates, homogenized for 3 × 10 s on ice using a tip sonifier (Branson), and centrifuged at 12,000 × g for 10 min. A 10-μl aliquot was taken from each sample for protein quantitation using a modified Bradford assay. After mixing in equal portions, 20 mg of total proteins were reduced with 5 mm DTT for 1 h and alkylated with 55 mm iodoacetamide for 1 h at room temperature. The sample was diluted 8-fold with 50 mm ammonium bicarbonate pH 8.5, 200 μg of trypsin (Roche) was added, and proteins were digested overnight at 37 °C. A supplementary 200 μg of trypsin was added, and digestion continued for 4 h at 37 °C. After acidification with formic acid at 5% v/v end concentration, samples were concentrated onto C18 Sep-Pak cartridges (100 mg, Waters, Etten-Leur, The Netherlands), and eluted peptides were dried down using a SpeedVac. Three independent biological replicates were prepared at one-week intervals following the same procedure.
Fig. 1.
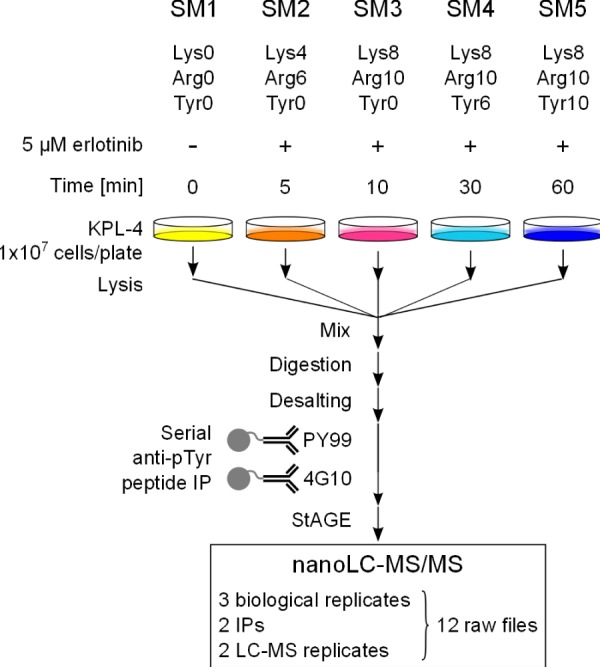
Experimental workflow for the analysis of tyrosine phosphorylated peptides via 5-plex SILAC. Breast cancer KPL-4 cells were grown in SM1–5 containing different combinations of unlabeled or stable isotopically labeled Lys, Arg, and Tyr. Each population was treated with the selective EGFR inhibitor erlotinib for the indicated times. SM1–5 cell lysates were combined and proteins were enzymatically digested. Tyrosine phosphorylated peptides were selectively enriched via two successive IPs with PY99- and 4G10-agarose conjugates. Eluted peptides were analyzed via nano-LC-MS/MS on an Orbitrap Velos.
Immunoprecipitation
Tyrosine phosphorylated peptides were sequentially immunoprecipitated with two antibodies following the procedure described by Boersema et al. (26), with some modifications. Briefly, samples were re-suspended in immunoprecipitation (IP) buffer (50 mm Tris-HCl pH 7.2, 150 mm NaCl, and 1 mm PMSF) and incubated with pre-equilibrated PY99-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4 °C. Following the first PY99 IP, the same samples were further incubated with pre-equilibrated 4G10-agarose beads (Millipore, Billerica, MA) for 4 h at 4 °C. Both types of beads were washed three times with IP buffer and twice with water before elution with 0.15% TFA for 15 min at room temperature. Eluted peptide fractions from PY99 and 4G10 IPs were concentrated on stop-and-go extraction tips, dried down, and stored at −20 °C. Separate PY99 and 4G10 samples were re-suspended in 5% formic acid/2% acetonitrile and analyzed in duplicate via LC-MS.
Online Nanoflow Liquid Chromatography Mass Spectrometry
Nanoflow LC-MS/MS was performed by coupling an Easy-nLC system (Proxeon, Odensa, Denmark) to an LTQ Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany) equipped with a nanoelectrospray source (Proxeon) and an active background-ion reduction device (ABIRD, ESI Source Solutions, Woburn, MA). Peptides were loaded onto an Aqua C18 (Phenomenex, Torrance, CA) trapping column (packed in-house, 10 mm length × 100 μm inner diameter, 5 μm particle size) and separated on a Reprosil-Pur C18-AQ (Dr. Maisch GmbH, Ammerbuch, Germany) analytical column (200 mm length × 75 μm inner diameter, 3 μm particle size, 120 Å). Trapping of the peptides was performed using the intelligent flow control at a maximal backpressure of 250 bar with 100% solvent A (0.6% acetic acid), and elution was carried out at a flow rate of 250 nl/min using the following gradient of solvent B (acetonitrile and 0.6% acetic acid): 0%–2% B in 2 min, 2%–35% B in 106 min, 35%–80% in 2 min, and 80% B for 10 min. The eluent was electrosprayed via a normal coated emitter tip butt-connected to the analytical column using a PicoClear union (New Objective, Cambridge, MA). The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS. Survey full-scan MS spectra were acquired from m/z 350 to 1700 in the Orbitrap with a resolution of r = 60,000 (at m/z 400) after accumulation to a target value of 1 × 106 in the linear ion trap for a maximum time of 500 ms. The detected ions were recalibrated on the fly using the ambient air polysiloxane at m/z 445.120024 as lock-mass (27). The 10 most intense ions with a charge state >1+ and a threshold above 1000 were selected for collision-induced dissociation in the linear ion trap. Ions were accumulated to a target value of 10,000 using the predictive ion trap fill time mode, isolated with a width of 2 amu, and collision-induced dissociation was performed at a normalized collision energy of 35% with an activation Q of 0.25 for 10 ms and wideband activation mode enabled. Fragmented ions with an m/z width of ±10 ppm were placed on an exclusion list for 30 s and a maximal size of 500.
LC-MS Data Processing
Raw files belonging to a biological replicate were combined for multi-dimensional protein identification technology and processed using Proteome Discoverer version 1.2 (Thermo Fisher Scientific), comprising a self-designed peptide/protein identification and quantitation workflow (all details can be found in supplemental Fig. S1). Data were searched using Mascot version 2.3.2 (Matrix Science, London, UK) against the human UniProt/SwissProt database (release 2011 04, 20,232 sequences) with trypsin as an enzyme, allowing a maximum of two miscleavages and 20 ppm and 0.5 Da as precursor and fragment mass tolerances, respectively. Carbamidomethylated cysteine was selected as a static modification, and the following were set as dynamic modifications: oxidized methionine; phosphorylated serine, threonine, and tyrosine, with the latter additionally in the Tyr6 labeled (+85.986460 Da and +85.9358 Da as monoisotopic and average mass shifts, respectively) and Tyr10 labeled (89.993559 and 89.9072 as monoisotopic and average mass, respectively) forms; Lys4 and Lys8 labeled lysine; and Arg6 and Arg10 labeled arginine. Protein grouping was enabled, and leucine and isoleucine were considered as equal. Individual top-ranked peptide-to-spectrum matches (PSMs) were accepted for a Mascot score of ≥20 (26). PSMs were filtered by keeping only sequences containing a tyrosine residue and a phosphorylation, and the resulting initial dataset was used to address the identification efficiency of the method (see “Evaluation of the 5-plex SILAC Dataset”). The false discovery rate at this score was experimentally tested via a concatenated target-decoy database search that yielded <1% for phosphotyrosine containing peptides for the three biological replicates (supplemental Fig. S2). A common number of natural isotopes for all members of a quintuplet were used to create extracted ion chromatograms, compute the peak areas, and determine relative peptide ratios. To generate the extracted ion chromatograms, the precursor ion mass precision between successive scans was set to 2 ppm, the minimal signal-to-noise value was set at 1, and quintuplets were assembled allowing a maximum retention time deviation of 0.2 min. PSMs with no quantitation value (approximately 34% to 40% of PSMs) were removed. Quintuplet ratios from tyrosine containing unphosphorylated peptides nonspecifically co-immunoprecipitated in the experiment were used for data normalization with each separate biological replicate. Phosphopeptide quintuplet ratios originating from different biological replicates were log2-averaged, standard deviations and statistical significance using a paired two-tailed Student's t test were calculated in Excel, and only the peptide with the highest Mascot score when multiple isotopologues were identified was reported. Peptides bearing the same phosphorylation site and showing identical time profiles but with additional oxidized methionine and/or miscleavages were considered as redundant and were excluded. The false localization rate was tested using the Mascot delta score between two successive identical peptide sequences differing only in phosphorylation site assignment and accepted for values greater than or equal to 6 (see “Discussion”) (28).
Data Clustering and Analysis
The phosphotyrosine time profiles were clustered hierarchically using Euclidean distance and Ward linkage. Applying a distance threshold of 0.32 resulted in nine time profile clusters determined to be optimal for subsequent pathway enrichment analysis. The correlation between a cluster's proteins and signaling pathways was assessed as follows: enrichment statistical significance of proteins belonging to each individual cluster (and combinations of adjacent clusters) in gene sets available from the Reactome pathway database (version 40, March 2012) was calculated via Fisher's exact test with the significance threshold set to 0.01. Gene sets were pre-filtered to consider only candidate proteins (N) complemented with the rest of the dataset (215-N “background” proteins), and at least three proteins per enriched pathway were required.
RESULTS
Overview of the 5-plex SILAC Approach
Focusing on the phosphotyrosine fraction of the phosphoproteome enabled us to utilize Tyr as an additional amino acid for protein labeling (29), along with the more conventional Lys and Arg. By selecting a proper combination of light and heavy labeled Lys, Arg, and Tyr, we designed a new 5-plex SILAC strategy to distinguish tyrosine-containing peptides that originate from five different cell populations. The cell culture media were prepared with a mixture of Lys0/Arg0/Tyr0, Lys4/Arg6/Tyr0, Lys8/Arg10/Tyr0, Lys8/Arg10/Tyr6, or Lys8/Arg10/Tyr10 and numbered SM1 to SM5, respectively (Fig. 1). As a consequence, after digestion of the proteins with trypsin, every tyrosine-containing peptide holding a C-terminal Lys was detected in the MS with mass shifts of 4, 8, 14, and 18 Da (supplemental Fig. S3). When mixed in equal amounts, peptides of interest were present as SILAC quintuplets. The selection of the heavy labeled amino acids was done in such a way that a minimal mass shift of 4 Da between two peptide isotopologues was guaranteed. This constraint was of importance for minimizing the overlap between the isotope clusters within a quintuplet. A similar picture was observed when the peptide C terminus was an Arg, with mass shifts of 6, 10, 16, and 20 Da, respectively.
Four of the five KPL-4 cell populations were subjected to the selective EGFR TKI erlotinib for 5, 10, 30, and 60 min, and the last one was treated with dimethyl sulfoxide to serve as a control. In the present study, SM1—containing only light amino acids—served as the control, and SM2–5 were successively used for the longer treatment times, so that the “heaviest medium” represented the end point of the kinetic. After the lysates had been mixed in equal proportions and the proteins had been trypsinized, tyrosine phosphorylated peptides were enriched from the crude digest via the application of a serial anti-pTyr IP procedure. That is, peptides were initially incubated with anti-phosphotyrosine (PY99) affinity resin, and the unbound peptides were then additionally treated with the second anti-phosphotyrosine (4G10) affinity resin. Finally, both phosphotyrosine enriched peptide samples were analyzed separately via nano-LC-MS/MS on an Orbitrap Velos.
An illustration of the approach is shown at the peptide level with the quintuplet matched to the doubly charged peptide VTIADDpYSDPFDAK from the SH2 domain-containing adapter protein B (SHB) and phosphorylated on Tyr246 (Fig. 2). The corresponding ion was recorded at m/z 818.8421 for the isotopologue originating from SM1. Along with the unlabeled form, the co-eluting SM2–5 isotopologues at tR = 53.6 min were found at m/z 820.8543 (+4 Da), 822.8491 (+8 Da), 825.8594 (+14 Da), and 827.8630 (+18 Da), respectively (Fig. 2A). Because of the mass shifts observed within the quintuplet, the candidate phosphopeptide could be identified as containing a C-terminal lysine. Despite the complex nature of the sample, all five isotopologue precursor ions were selected for fragmentation, which demonstrates that the MS duty cycle was short enough to handle 5-plex SILAC labeled samples. Moreover, for this particular candidate, all five MS/MS events resulted in successful peptide identification when submitted to a database search (Fig. 2C). Relative quantitation was achieved by comparing quintuplets' precursor ion intensities after extracted ion chromatograms, followed by data normalization and averaging across biological replicates. For Tyr246 of SHB, erlotinib showed only a moderate down-regulative effect, with the minimum of the time profile observed after 30 min (approximately a 2-fold decrease) followed by a slow return to the basal level after 60 min (Fig. 2D).
Fig. 2.

Overview of the MS data produced via the 5-plex SILAC method. A, full-scan MS quintuplet detected for the doubly charged (z = 2) phosphopeptide VTIADDpYSDPFDAK from SH2 domain-containing adapter protein B (SHB). The SM1–5 isotopologues are found at m/z 818.84, 820.85 (+4 Da), 822.85 (+8 Da), 825.86 (+14 Da), and 827.86 (+18 Da), respectively. B, extracted ion chromatograms (EICs) of the five co-eluting isotopologues at tR = 53.6–53.7 min. C, annotated MS/MS for the SM1–5 isotopologues as identified by a Mascot database search. The phosphorylation site is unambiguously assigned to Tyr246. The y and b fragment ion series, the Mascot ion score, and the measured mass error (ΔM) in ppm are indicated. D, corresponding Tyr phosphorylation time profile. Data are normalized, and the average values (n = 3) are log2-transformed; bars corresponding to the standard deviations and statistical significance (paired two-tailed Student's t test; *p < 0.05; **p < 0.01) are indicated.
Evaluation of the 5-plex SILAC Dataset
Three independent biological replicates, each including two IPs that were both analyzed in duplicate via LC-MS, were performed (see Fig. 1). Searching the 12 raw files against a human protein database (see “Experimental Procedures” for the details) resulted in 6163 PSMs passing the ≥20 Mascot score criterion for phosphotyrosine-immunoprecipitated peptides (26). In order to get an idea of the number of identified isotopologues per quintuplet and how deep the peptide sequencing for a 5-plex SILAC experiment is, the PSMs were combined, and distinct phosphopeptides (i.e. same phosphorylation site with different versions of light or heavy Lys, Arg, and Tyr) were counted (supplemental Table S1). A total number of 846 phosphopeptides that mapped to 340 proteins was obtained. It is of importance to emphasize that this preliminary “uncurated” dataset was intended for method evaluation only, as it still contained redundancy. A consistent fraction (412 peptides representing 49% of the total) was obtained from the fragmentation of a single isotopologue (Fig. 3). Focusing on this subset, 260 (63%) were “unlabeled” SM1, whereas 37 (9%), 80 (19%), 12 (3%), and 23 (6%) were from labeled SM2–5 precursor ions, respectively. Also, examination of precursor ion masses indicated that the heavier the peptide labels were, the greater the bias toward the identification of low-m/z candidates was (supplemental Fig. S4). A possible explanation for this is the unavoidable increased overlap between isotopologues for large peptides leading to mixed (or “chimeric”) MS/MS (30). Overall, about half of the identified phosphopeptides were related to a single isotopologue form, whereas as few as 7% had all five SM1–5 “versions” resulting in peptide identification.
Fig. 3.
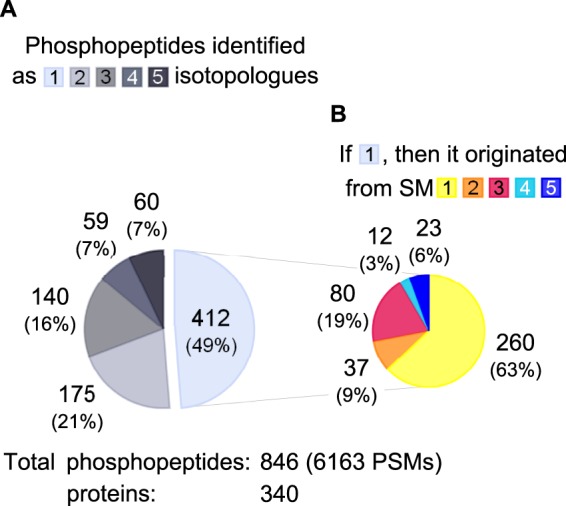
Phosphopeptide identification frequency from 5-plex SILAC data. A, distribution of the number of identified isotopologues per phosphopeptide. For instance, 59 peptides (7% of total) were identified as four (out of the five possible) different isotopologue versions. B, SM origin of single isotopologue identifications. In total, 6163 PSMs from three biological replicates resulted in 846 different phosphopeptides grouped into 340 proteins.
In order to reduce the PSMs down to a list of unique phosphotyrosine peptides, the aforementioned candidate list was manually curated. For instance, entries containing no quantitation value, additional methionine oxidation, or miscleavage were removed. However, a phosphopeptide presenting an additional phosphorylation site was considered as non-redundant, and only the candidate with the highest Mascot score was kept when found in several biological replicates. Furthermore, phosphorylation site localization was tested using the Mascot delta score and considered confident for values greater than or equal to 6, based on the fact that the antibodies used are specific for phosphorylated tyrosine, rather than serine/threonine (28). This resulted in a collection of 318 unique identified and quantified phosphotyrosine peptides grouped into 215 proteins (Fig. 4, supplemental Table S2). All annotated MS/MS together with their time profiles are provided in supplemental Fig. S5. When the reproducibility of 5-plex SILAC was taken into account, 98 peptides (31%) were identified in all biological replicates, and 187 peptides (59%) were present in at least two replicates (Fig. 4A). It has to be mentioned that the 318 peptides in this final dataset were detected only as quintuplets in the MS—that is, each time point had a quantifiable signal. Peptides initially detected as a quadruplet or triplet (representing 121 out of the 6163 PSMs) were removed during the manual curation process. The overlap between the three biological replicates was 31% (98 peptides in common), and around 42% to 50% between two of them, similar to what has been reported by others (12, 13). Because two antibodies were used for serial IPs, data were examined to highlight whether a difference in IP efficiency was observed. Substantially more unique peptides (229 versus 89) originated from the 4G10 than from the PY99 agarose-conjugate fractions (see Fig. 4B). This further emphasizes that combining several pTyr peptide affinity enrichment steps allows deeper sample coverage (12, 31).
Fig. 4.
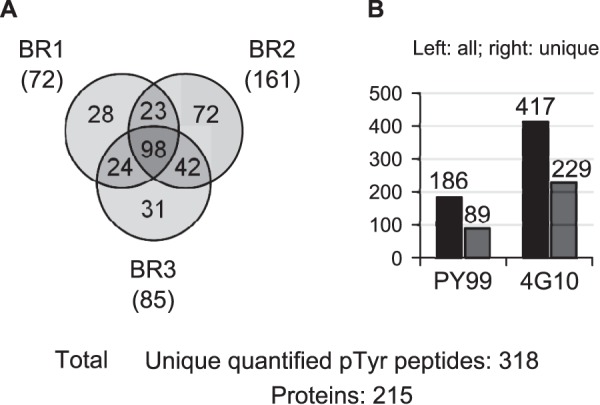
Identified and quantitated Tyr-phosphorylated peptides. A, overlap between the three biological replicates. Peptides unique to a biological replicate (keeping the highest Mascot score candidate) are indicated between brackets. B, distribution among agarose-conjugate resins used for peptide pTyr IPs. Left-hand and right-hand bars represent all and unique peptides, respectively.
Tyr-phosphorylated Sites Modulated by Erlotinib
Erlotinib is a reversible inhibitor of the EGFR kinase that competes with ATP for its binding site. Therefore, the phosphorylation of EGFR, its substrates, and downstream molecules is expected to be down-regulated (32–34). For the present experiment, an erlotinib concentration of 5 μM was selected because it corresponds approximately to the measured plasma level in humans (35). Also, the last point of the investigated time window was selected as 60 min because limited EGFR degradation should be observed within this specific period of time (36). Hierarchical clustering of the 318 pTyr peptide time profiles revealed nine groups and readily allowed the recognition of the most affected phosphosites (Fig. 5, supplemental Table S2). For instance, clusters 1 and 2, containing three and nine time profiles, respectively, exhibited an immediate, strong, and sustained decrease of phosphorylation. In the case of cluster 1 members, the reduction was on the order of 10-fold after only 10 min of treatment and concerned Tyr187 of MAPK1 and Tyr204 of MAPK3 (also known as ERK2 and ERK1, respectively), two markers of the Ras-Raf-Mek pathway connecting EGFR to MAPK/Erk signaling. A similar effect was reported for erlotinib-treated HN5 squamous carcinoma cells (32). Furthermore, a third member belonging to cluster 1, namely, Tyr659 of the GRB2-associated binding protein 1 (GAB1), was found among the most affected phosphorylation sites. The latter is a known substrate of EGFR (37) and is involved in EGF-mediated activation of ERK2 via binding to the protein-tyrosine phosphatase PTPN11 (alternatively named SHP2) (38). Also, a strong down-regulation of GAB1 Tyr659 phosphorylation was observed in H3255 non-small cell lung cancer cells treated with gefitinib, another selective EGFR TKI (39).
Fig. 5.

Hierarchical clustering of phosphotyrosine time profiles. A, dendogram of the 318 time profiles obtained using Euclidean distance and Ward's linkage criterion. The distance threshold was set at 0.32, resulting in nine clusters. B, phosphorylation time profile distribution of the nine individual clusters. The numbers of profiles belonging to each cluster and selected phosphorylation sites are indicated.
Time profiles found in cluster 2 displayed a similar rapid decrease after 10 min, but with a down-regulation magnitude of about 6-fold (Fig. 5B). Two EGFR autophosphorylation sites, Tyr1110 and Tyr1172, were present in this cluster and indicated a reduction of receptor kinase activity. These two EGFR tyrosine residues, along with Tyr1092 and Tyr1197 (found in cluster 4 in the present study), were inhibited by erlotinib in a concentration-dependent manner (33). Other highly down-regulated phosphosites were Tyr771 of phospholipase C-gamma-1 (PLCG1) and Tyr427 of SHC-transforming protein 1 (SHC1). The latter two phosphosites have been described as affected by TKI treatment (32, 39) and are known to specifically bind to phosphorylated Tyr1172 and Tyr1197 of EGFR via their SH2 binding domain (40–42). Another interesting phosphorylation site located in cluster 2 appeared to be Tyr453 of protein FAM59A. This protein (also called GAREM) is a recently discovered EGFR adaptor that interacts with GRB2 and SHP2 and participates in ERK activation (43).
At the other end of the dendogram, a large proportion of the time profiles (68% (215 out of 318)) populate clusters 7–9 (Fig. 5A). These particular phosphorylation sites showed little inhibitory effect, while other sites appeared to increase upon treatment with erlotinib. In particular, phosphorylation of Tyr877 of ErbB2 (member of cluster 9) became gradually up-regulated and showed a 2-fold increase in signal after 60 min. This particular site, located in the activation loop of ErbB2, is known to be a substrate of the Src family kinases (c-Src, Fyn, and Yes) and to modulate the receptor's catalytic activity (44–46). Up-regulated Tyr-phosphorylation sites for c-Src (Tyr530) and Fyn (Tyr185, Tyr213, Tyr420, and Tyr440) were highlighted in the present experiment, and their time profiles were found in clusters 8 and 9 (see also Fig. 6).
Fig. 6.

Representative Tyr phosphorylation sites affected by erlotinib in the EGFR and ErbB2 canonical pathways. The proteins are colored according to their association with the different pathways. Other known undetected members are indicated to complement the pathways. Even though ErbB3 is reported as an ErbB2 signaling pathway member, it is colored gray to avoid confusion. Only EGFR-ErbB2 and EGFR-ErbB3 heterodimers are represented for clarity. Arrows representing selected known interactions between proteins (black) and ErbB's phosphosites and adapter (blue) are indicated.
Signaling Pathways Affected by Erlotinib
To better understand whether there was a connection between proteins found in a specific cluster and the erlotinib effect, we performed pathway enrichment analysis. For this, the Reactome pathway database was used and the over-representation of members of each cluster, as well as of two adjacent clusters, was calculated (see “Experimental Procedures”). As a result, when members of clusters 1 and 2 were combined, corresponding to a total of 13 candidate proteins, 6 proteins—namely, EGFR, GAB1, PLCG1, MAPK1, MAPK3, and SHC1—were mapped to 51 pathways (supplemental Table S3). In particular, EGFR and ErbB2 signaling were found to be the top enriched pathways. Proteins identified in the present experiment, together with their time profiles, are represented schematically for the EGFR and ErbB2 signaling pathways (Fig. 6). Other proteins containing phosphotyrosine sites that were strongly affected by erlotinib, such as Tyr453 FAM59A and Tyr1135 INPPL1, but which were not previously described as members of EGFR/ErbB2 signaling were included. Our data confirm that SHC1, PLCG1, and GAB1 are central molecules involved in transmission of the signal that regulates the MAPK/Erk signaling pathway, and they therefore validate the 5-plex SILAC approach as a valuable method for monitoring phosphotyrosine dynamics.
In addition to the aforementioned proteins, we examined the behavior of other phosphorylation sites in the ErbB family. Because KPL-4 cells highly express ErbB2 and ErbB3 (no ErbB4 expression) (25), we looked at the crosstalk between EGFR and ErbB2-ErbB3, especially knowing that ErbB2 needs no ligand for activation and ErbB3 cannot signal by itself (47). Grouping time profiles for each individual ErbB family member enabled the identification of ErbB3 phosphorylation sites that were systematically affected by erlotinib relative to ErbB2 (supplemental Fig. S6). That is, four ErbB3 phosphotyrosine sites, Tyr1159, Tyr1289, Tyr1307, and Tyr1328, all located in the C-terminal tail, displayed a sustained down-regulated time profile (clusters 4–6). To better visualize the remote effect of erlotinib on ErbB2 and ErbB3, sequences of the C termini were aligned (supplemental Fig. S6C). The same outcome had already been reported for ErbB3 Tyr1159 and Tyr1328 in the case of dual EGFR/ErbB2 TKI lapatinib-treated BT-474 breast cancer cells (46). Because ErbB3 phosphorylation down-regulation is an indirect result of EGFR inhibition, the observed levels were less than that observed for EGFR, which was in the range of 3-fold reduction. Interestingly, detected ErbB3 downstream interacting partners, such as GRB7 (Tyr284, Tyr388) or the phosphatidylinositol 3-kinase (PI3K) members PIK3R2 (Tyr464) and PIK3R3 (Tyr199, Tyr341), also exhibited decreased phosphorylation over time (supplemental Fig. S6B). The latter could point to an inhibitory effect on the PI3K/Akt signaling pathway (32), which was further tested by immunoblot detection of phospho-Akt (Thr308 and Ser473). However, the connections between EGFR inhibition and PI3K/Akt signaling via phospho-ErbB3 need to be investigated in more detail.
DISCUSSION
The examination of phosphorylation perturbations caused by a drug treatment has evolved from single to multiple time experiments. To allow this, recent developments in MS-based quantitation methods have focused on higher multiplexing (48). We have established an innovative 5-plex SILAC approach involving peptide IP to monitor Tyr-phosphorylation dynamics and compare five different cellular conditions within a single experiment. In addition to Lys and Arg, stable isotopically labeled Tyr (13C6 and [13C9,15N1]) was combined to introduce the necessary peptide mass shifts. A similar 5-plex SILAC method was proposed by Molina et al. to monitor protein expression changes during adipocyte differentiation; however, their approach was limited to the detection of Arg-containing peptides (49). Also, the choice of a minimum mass shift of 4 Da in the method described here results in MS-separated isotopologues, which was considered to deliver more reliable quantitation data than strategies involving overlapping isotopologues and necessitating deconvolution. Our protocol has the advantage of combining all five SILAC labeled samples at the beginning of the workflow and thus keeps variability between experimental conditions at low levels. Also, the reduction to a single sample for monitoring five conditions represents a clear advantage in terms of the shorter analysis time. Because MS signal quantitation is performed at the precursor ion level, the method does not suffer from the weaknesses inherent to isobaric labeling, such as reporter ion signal distortion (due to co-eluting peptide fragmentation) (20–22) and peptide charge enhancement (23).
Using three biological replicates and 20 mg (5 × 4 mg) total starting material, it was possible to identify 846 peptides, quantitate 318 unique phosphotyrosine sites found on 215 proteins, and reveal their dynamics over 60 min of erlotinib treatment. These numbers are in good agreement with what has previously been reported in the literature for temporal (15, 50) and dose-response (51) studies involving peptide anti-pTyr IP. As a typical example, Wolf-Yadlin et al. were able to quantify the dynamics of 222 phosphotyrosines found on 146 proteins from EGF-stimulated 184A1 human mammary epithelial cells (52). In another study, Boersema et al. obtained the dynamics of 73 unique phosphotyrosines (52 proteins) from EGF-treated HeLa cells, albeit using as little as 2 mg of starting material (26). Also, the reproducibility of the 5-plex SILAC approach was similar to that typically achieved by other methods (12, 52), as the overlap represented approximately 30% between two replicates (see Fig. 4A).
Despite the a priori rather complex nature of the 5-plex SILAC encoded samples, more than half the peptides (i.e. 434 out of 846; see Fig. 3A) were identified as ≥2 isotopologue forms. From a quantitation point of view, it is obvious that the identification of only one member of a quintuplet is sufficient for subsequent generation of its tyrosine phosphorylation time profile. As anticipated, the reduction of complexity owing to the IP fractionation resulted in a sample containing a number of peptide quintuplets that is manageable in MS. A certain bias toward the identification of SM1 peptides was observed, presumably due to the higher abundance of untreated control phosphotyrosines relative to their SM2–5 erlotinib down-regulated isotopologues. To better address and circumvent this effect, SM and treatment times could have been randomized among the replicates, but this was outside the scope of the present study. It has to be mentioned that our final phosphopeptide dataset was composed of quintuplets only; that is, no peptide was observed with less than five isotopologues due to complete signal absence. For instance, Tyr204 of MAPK3, one of the most affected phosphotyrosines, was still recorded with minute signals for the SM3–5 isotopologues used for quantitation (supplemental Fig. S7). It is accepted that anti-pTyr pan-specific antibodies possess an intrinsic specificity (12, 31). We chose to have two serial IP steps in our analytical workflow to maximize phosphotyrosine coverage; that is, first we precipitated the lysates with PY99, and then we followed with an incubation of the unbound peptide fraction with 4G10-agarose conjugates. In the present experiment, 89 of the unique identified peptides were from PY99, and 229 (72%) were related to 4G10 (see Fig. 4B). It should be emphasized that this reflects better performance of the latter antibody, because the PSMs with the highest Mascot scores were kept to populate the final list of 318 unique phosphopeptides. Furthermore, the observation should be taken with care, as the two IPs were performed in serial and not in parallel.
The decision to apply the newly introduced 5-plex SILAC approach to EGFR signaling was motivated by the fact that the latter is well documented (see Refs. 12, 15, 16, and 40 for a review) and could serve as good model for evaluation. Also, using KPL-4 as the model cell line allowed us to probe the effect of EGFR inhibition on the other two family members ErbB2 and ErbB3. By performing phosphorylation time profile clustering followed by pathway enrichment analysis, we highlighted the involvement of EGFR and ErbB2 signaling as the main pathways affected by erlotinib and thereby confirmed the validity of our approach. The analysis unfortunately did not reveal any additional significant pathways perturbed by erlotinib, as the set of 215 genes was quite small. Despite the fact that many phosphosites identified in the present study were already known interactors of these pathways, additional ones appeared to be also linked, as suggested by their phosphorylation dynamics. For instance, Tyr453 of FAM59A, possessing a time profile similar to those of Tyr1110 and Tyr1172 of EGFR (all three are found in cluster 2; see “Results”), is a confirmed candidate of the EGFR signaling pathway. The effect of erlotinib on other ErbB family members, in particular the inactive kinase ErbB3, was also quantified. Based on the time profiles and their location within the C-terminal tail of ErbB3, Tyr1289, Tyr1307, and Tyr1328 seem to act as alternative docking sites for adaptor proteins. Interestingly, based on the sequence alignments of the three receptor tyrosine kinases, phosphorylated Tyr1307 of ErbB3 might have a distinct role, such as affinity for a specific adaptor, because neither EGFR nor ErbB2 possesses a homologous tyrosine (see supplemental Fig. S6C).
The 5-plex SILAC approach developed here is a valid alternative to existing methods. It has the clear advantage of allowing the analysis of five different conditions within a single sample. It joins the robustness of precursor-ion-based quantitation with higher multiplexing than in conventional iTRAQ- or SILAC-based strategies. Use of the 5-plex SILAC to study EGFR inhibition resulted in the elucidation of phosphotyrosine time profiles specifically affected by a treatment. This information is highly valuable, as it can lead to better understanding of the drug's mode of action and the finding of potential new biomarkers. Future applications of the strategy will be focused on the study of more complex therapies, such as combined small molecule–antibody or antibody–antibody treatments. Finally, the reported 5-plex SILAC approach has the potential to be further improved to increase its overall performance in phosphotyrosine dynamics quantification. For instance, analysis of the samples with a newer generation of MS, such as Q-Exactive, would improve the analytical depth, as thousands of phosphotyrosine sites are now routinely amenable to quantitation. Furthermore, data processing using software tailored for 5-plex SILAC including feature- rather than identification-centric capability would potentially also increase phosphopeptide sampling.
Supplementary Material
Acknowledgments
We thank the following persons: Dr. Georg Schmid and Mr. Jean-Marie Vonach from Roche Discovery Technologies Basel for preparing the SILAC media; Professor Yossi Yarden from the Weizmann Institute of Science Rehovot in Israel and Dr. Max Hasmann from Roche Discovery Oncology Penzberg in Germany for their mentorship during M.T.'s Roche Postdoctoral Fellowship period; and Dr. Tom Dunkley from Translational Technologies and Bioinformatics Basel for comments on the manuscript.
Footnotes
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- EGFR
- epidermal growth factor receptor
- IP
- immunoprecipitation
- iTRAQ
- isobaric tags for relative and absolute quantitation
- PSM
- peptide-to-spectrum match
- pTyr
- phosphotyrosine
- SILAC
- stable isotope labeling with amino acids in cell culture
- SM
- SILAC medium
- TKI
- tyrosine kinase inhibitor
- tR
- retention time.
REFERENCES
- 1. Cohen P. (2000) The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 2. Johnson L. N. (2009) The regulation of protein phosphorylation. Biochem. Soc. Trans. 37, 627–641 [DOI] [PubMed] [Google Scholar]
- 3. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunter T. (2009) Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Citri A., Yarden Y. (2006) EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell. Biol. 7, 505–516 [DOI] [PubMed] [Google Scholar]
- 6. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 7. Hynes N. E., MacDonald G. (2009) ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 21, 177–184 [DOI] [PubMed] [Google Scholar]
- 8. Bublil E. M., Yarden Y. (2007) The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 19, 124–134 [DOI] [PubMed] [Google Scholar]
- 9. Nilsson C. L. (2012) Advances in quantitative phosphoproteomics. Anal. Chem. 84, 735–746 [DOI] [PubMed] [Google Scholar]
- 10. Choudhary C., Mann M. (2010) Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell. Biol. 11, 427–439 [DOI] [PubMed] [Google Scholar]
- 11. Palmisano G., Thingholm T. E. (2010) Strategies for quantitation of phosphoproteomic data. Expert Rev. Proteomics 7, 439–456 [DOI] [PubMed] [Google Scholar]
- 12. Nita-Lazar A., Saito-Benz H., White F. M. (2008) Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics 8, 4433–4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. White F. M. (2008) Quantitative phosphoproteomic analysis of signaling network dynamics. Curr. Opin. Biotechnol. 19, 404–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Del Rosario A. M., White F. M. (2010) Quantifying oncogenic phosphotyrosine signaling networks through systems biology. Curr. Opin. Genet. Dev. 20, 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dengjel J., Kratchmarova I., Blagoev B. (2009) Receptor tyrosine kinase signaling: a view from quantitative proteomics. Mol. Biosyst. 5, 1112–1121 [DOI] [PubMed] [Google Scholar]
- 16. Macek B., Mann M., Olsen J. V. (2009) Global and site-specific quantitative phosphoproteomics: principles and applications. Annu. Rev. Pharmacol. Toxicol. 49, 199–221 [DOI] [PubMed] [Google Scholar]
- 17. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 18. Zhang Y., Wolf-Yadlin A., Ross P. L., Pappin D. J., Rush J., Lauffenburger D. A., White F. M. (2005) Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics 4, 1240–1250 [DOI] [PubMed] [Google Scholar]
- 19. Johnson H., Del Rosario A. M., Bryson B. D., Schroeder M. A., Sarkaria J. N., White F. M. (2012) Molecular characterization of EGFR and EGFRvIII signaling networks in human glioblastoma tumor xenografts. Mol. Cell. Proteomics 11, M112.019984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bantscheff M., Boesche M., Eberhard D., Matthieson T., Sweetman G., Kuster B. (2008) Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol. Cell. Proteomics 7, 1702–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ow S. Y., Salim M., Noirel J., Evans C., Rehman I., Wright P. C. (2009) iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly.” J. Proteome Res. 8, 5347–5355 [DOI] [PubMed] [Google Scholar]
- 22. Karp N. A., Huber W., Sadowski P. G., Charles P. D., Hester S. V., Lilley K. S. (2010) Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 9, 1885–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thingholm T. E., Palmisano G., Kjeldsen F., Larsen M. R. (2010) Undesirable charge-enhancement of isobaric tagged phosphopeptides leads to reduced identification efficiency. J. Proteome Res. 9, 4045–4052 [DOI] [PubMed] [Google Scholar]
- 24. Bendall S. C., Hughes C., Stewart M. H., Doble B., Bhatia M., Lajoie G. A. (2008) Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol. Cell. Proteomics 7, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurebayashi J., Otsuki T., Tang C. K., Kurosumi M., Yamamoto S., Tanaka K., Mochizuki M., Nakamura H., Sonoo H. (1999) Isolation and characterization of a new human breast cancer cell line, KPL-4, expressing the Erb B family receptors and interleukin-6. Br. J. Cancer 79, 707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boersema P. J., Foong L. Y., Ding V. M., Lemeer S., van Breukelen B., Philp R., Boekhorst J., Snel B., den Hertog J., Choo A. B., Heck A. J. (2009) In depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immuno-affinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteomics 9, 84–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 28. Savitski M. M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B. (2011) Confident phosphorylation site localization using the mascot delta score. Mol. Cell. Proteomics 10, M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ibarrola N., Molina H., Iwahori A., Pandey A. (2004) A novel proteomic approach for specific identification of tyrosine kinase substrates using [13C]tyrosine. J. Biol. Chem. 279, 15805–15813 [DOI] [PubMed] [Google Scholar]
- 30. Michalski A., Cox J., Mann M. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 10, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 31. Ding S. J., Qian W. J., Smith R. D. (2007) Quantitative proteomic approaches for studying phosphotyrosine signaling. Expert Rev. Proteomics 4, 13–23 [DOI] [PubMed] [Google Scholar]
- 32. Thelemann A., Petti F., Griffin G., Iwata K., Hunt T., Settinari T., Fenyo D., Gibson N., Haley J. D. (2005) Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol. Cell. Proteomics 4, 356–376 [DOI] [PubMed] [Google Scholar]
- 33. Zhang G., Fang B., Liu R. Z., Lin H., Kinose F., Bai Y., Oguz U., Remily-Wood E. R., Li J., Altiok S., Eschrich S., Koomen J. M., Haura E. B. (2011) Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J. Proteome Res. 10, 305–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weber C., Schreiber T. B., Daub H. (2012) Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J. Proteomics 75, 1343–1356 [DOI] [PubMed] [Google Scholar]
- 35. Ling J., Johnson K. A., Miao Z., Rakhit A., Pantze M. P., Hamilton M., Lum B. L., Prakash C. (2006) Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab. Dispos. 34, 420–426 [DOI] [PubMed] [Google Scholar]
- 36. Marmor M. D., Yarden Y. (2004) Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23, 2057–2070 [DOI] [PubMed] [Google Scholar]
- 37. Rodrigues G. A., Falasca M., Zhang Z., Ong S. H., Schlessinger J. (2000) A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol. Cell. Biol. 20, 1448–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cunnick J. M., Mei L., Doupnik C. A., Wu J. (2001) Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 276, 24380–24387 [DOI] [PubMed] [Google Scholar]
- 39. Moritz A., Li Y., Guo A., Villen J., Wang Y., MacNeill J., Kornhauser J., Sprott K., Zhou J., Possemato A., Ren J. M., Hornbeck P., Cantley L. C., Gygi S. P., Rush J., Comb M. J. (2010) Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morandell S., Stasyk T., Skvortsov S., Ascher S., Huber L. A. (2008) Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics 8, 4383–4401 [DOI] [PubMed] [Google Scholar]
- 41. Schulze W. X., Deng L., Mann M. (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 1, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones R. B., Gordus A., Krall J. A., MacBeath G. (2006) A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439, 168–174 [DOI] [PubMed] [Google Scholar]
- 43. Tashiro K., Tsunematsu T., Okubo H., Ohta T., Sano E., Yamauchi E., Taniguchi H., Konishi H. (2009) GAREM, a novel adaptor protein for growth factor receptor-bound protein 2, contributes to cellular transformation through the activation of extracellular signal-regulated kinase signaling. J. Biol. Chem. 284, 20206–20214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ishizawar R. C., Miyake T., Parsons S. J. (2007) c-Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene 26, 3503–3510 [DOI] [PubMed] [Google Scholar]
- 45. Marcotte R., Zhou L., Kim H., Roskelly C. D., Muller W. J. (2009) c-Src associates with ErbB2 through an interaction between catalytic domains and confers enhanced transforming potential. Mol. Cell. Biol. 29, 5858–5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rexer B. N., Ham A. J., Rinehart C., Hill S., de Matos Granja-Ingram N., Gonzalez-Angulo A. M., Mills G. B., Dave B., Chang J. C., Liebler D. C., Arteaga C. L. (2011) Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene 30, 4163–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 48. Dephoure N., Gygi S. P. (2012) Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signal. 5, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Molina H., Yang Y., Ruch T., Kim J. W., Mortensen P., Otto T., Nalli A., Tang Q. Q., Lane M. D., Chaerkady R., Pandey A. (2009) Temporal profiling of the adipocyte proteome during differentiation using a five-plex SILAC based strategy. J. Proteome Res. 8, 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tedford N. C., Hall A. B., Graham J. R., Murphy C. E., Gordon N. F., Radding J. A. (2009) Quantitative analysis of cell signaling and drug action via mass spectrometry-based systems level phosphoproteomics. Proteomics 9, 1469–1487 [DOI] [PubMed] [Google Scholar]
- 51. Preisinger C., Schwarz J. P., Bleijerveld O. B., Corradini E., Muller P. J., Anderson K. I., Kolch W., Scholten A., Heck A. J. (2013) Imatinib-dependent tyrosine phosphorylation profiling of Bcr-Abl-positive chronic myeloid leukemia cells. Leukemia 27, 743–746 [DOI] [PubMed] [Google Scholar]
- 52. Wolf-Yadlin A., Hautaniemi S., Lauffenburger D. A., White F. M. (2007) Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A. 104, 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.