

Abstract
In the past decade, mass-spectrometry-based methods have emerged for the quantitative profiling of dynamic changes in protein phosphorylation, allowing the behavior of thousands of phosphorylation sites to be monitored in a single experiment. However, when one is interested in specific signaling pathways, such shotgun methodologies are not ideal because they lack selectivity and are not cost and time efficient with respect to instrument and data analysis time.
Here we evaluate and explore a peptide-centric antibody generated to selectively enrich peptides containing the cAMP-dependent protein kinase (PKA) consensus motif. This targeted phosphoproteomic strategy is used to profile temporal quantitative changes of potential PKA substrates in Jurkat T lymphocytes upon prostaglandin E2 (PGE2) stimulation, which increases intracellular cAMP, activating PKA. Our method combines ultra-high-specificity motif-based immunoaffinity purification with cost-efficient stable isotope dimethyl labeling. We identified 655 phosphopeptides, of which 642 (i.e. 98%) contained the consensus motif [R/K][R/K/X]X[pS/pT]. When our data were compared with a large-scale Jurkat T-lymphocyte phosphoproteomics dataset containing more than 10,500 phosphosites, a minimal overlap of 0.2% was observed. This stresses the need for such targeted analyses when the interest is in a particular kinase.
Our data provide a resource of likely substrates of PKA, and potentially some substrates of closely related kinases. Network analysis revealed that about half of the observed substrates have been implicated in cAMP-induced signaling. Still, the other half of the here-identified substrates have been less well characterized, representing a valuable resource for future research.
The identification and quantification of protein phosphorylation under system perturbations is an integral part of systems biology (1, 2). The combination of phosphopeptide enrichment (3–6), stable isotope labeling, and high-resolution mass spectrometry (MS) methods (7–9) has become the method of choice for the identification of novel phosphorylation sites and for the quantitation of temporal dynamics within signaling networks (10, 11), allowing the behavior of thousands of phosphorylation sites to be studied in a single experiment (10, 12, 13). Nowadays, one of the most commonly adopted high-throughput phosphoproteomics strategies utilizes two consecutive separation steps: (i) an initial fractionation to reduce the sample complexity, and (ii) a phosphopeptide-specific affinity purification. Such techniques include strong cation exchange fractionation under acidic conditions (3), followed by a chelation-based method with the use of metal ions (i.e. immobilized metal ion affinity chromatography (4), metal oxide affinity chromatography (10, 14), or Ti4+ immobilized metal ion affinity chromatography (6)). Alternatives to strong cation exchange for the first sample fractionation step have also been reported, including the use of electrostatic repulsion liquid chromatography (15, 16), which is well suited for the identification of multiply phosphorylated peptides, or hydrophilic interaction chromatography (17).
Although the number of detected phosphorylated peptides is nowadays impressive, these kinds of methodologies are still inclined to identify/quantify the more abundant phosphoproteins present in a sample. For example, phosphotyrosine peptides are underrepresented because of their relatively lower abundance.
In order to analyze key signaling events that may occur on less abundant phosphoproteins, more targeted approaches, focused on a specific pathway or a specific post-translational modification, are thus still essential. Studies examining post-translational modifications are often based on immunoaffinity purification at the protein or peptide level using dedicated antibodies. Recent examples include the selective enrichment of acetylated lysines (18) and phosphorylated tyrosines (19, 20). More recently, the first specific methods targeting serine/threonine phosphorylation motifs using immune-affinity assays have emerged (21, 22). The advantages of targeted approaches are their potentially higher sensitivity and more specific throughput with, as a consequence, relatively faster and easier data interpretation, which make them attractive for many systems biology applications.
Immunoaffinity enrichment can be applied at both the protein and the peptide level, and both have been explored to study protein tyrosine phosphorylation (23). The first one results mainly in information on total protein phosphorylation levels. The detection of the actual phosphoresidue might be hampered by the high content of unmodified peptides derived from the immune-purified phosphoprotein and its binding partners. Immunoprecipitation at the peptide level (20, 24, 25), in contrast, leads to improved phosphosite characterization, with the identification of hundreds of sites, albeit with the loss (generally) of information regarding total protein expression.
To profile the dynamic regulation of phosphorylation events via mass spectrometry, stable isotope labeling is often implemented, either with the use of amino acids in cell culture (10) or via chemical peptide labeling of the proteolytic digests (26, 27). To identify low-abundant signaling events, phosphoprotein/phosphopeptide immunoprecipitation is typically performed on several milligrams of material because of the substoichiometric abundance of post-translational modifications. This may hamper the use of expensive isotope-labeling reagents such as iTRAQ or tandem mass tag reagents, given the large amount of chemicals needed. Boersema et al. (28) introduced an alternative sensitive and accurate triplex labeling approach using inexpensive reagents (i.e. formaldehyde) that is much less limited in terms of the sample type or amount. We combined this latter stable-isotope dimethyl labeling approach (27–29) with highly specific antibodies raised against a set of cAMP-dependent protein kinase (PKA) phosphorylated substrates as based on the current literature (11, 30–34). It is generally accepted that PKA phosphorylates sites with the reasonably stringent consensus motif [R/K][R/K/X]X[pS/pT]. It should be noted that this consensus motif resembles somewhat the motifs of other AGC kinases (e.g. Akt, PKG, PKC).
The basicity of the PKA motifs may hamper their analysis via MS-based proteomics, especially when trypsin is used as a protease, as the peptides may become too small to be sequenced. The use of trypsin is also unfavorable in the approach presented here when attempting to immunoprecipitate peptides bearing the PKA motif. Therefore, we decided to use Lys-C in order to keep the (dominant (RRX[pS/pT])) phosphorylated motif intact. To enhance identification, we applied decision-tree MS/MS technology (9), which makes use of HCD and ETD for more efficient fragmentation, higher mass accuracy in tandem MS mode, and less background noise (35).
We applied this method to screen the response of Jurkat T cells to prostaglandin E2 (PGE2) treatment. PGE2 is a potent inflammatory mediator that plays an important role in several immune-regulatory actions (36). It is produced by many different cell types, including tumor cells, where carcinogenesis is associated with chronic inflammatory responses (37). PGE2 signaling in T cells is initiated by its binding to the G protein–coupled receptors EP1, -2, -3, and -4. Signaling pathways that are initiated by PGE2 are for the most part under control of the second messenger cyclic adenosine monophosphate (cAMP),1 which is generated from ATP by adenylyl cyclase when PGE2 binds to EP2 or EP4 receptors. One of the primary targets of cAMP is PKA—cAMP binding releases the catalytic subunit activating the kinase. In the current study, we efficiently enriched close to 650 phosphopeptides containing the [R/K][R/K/X]X[pS/pT] consensus motif. Almost all these sites were absent in a recently reported comprehensive phosphoproteomics dataset of Jurkat T cells (12), compiled using shotgun strong cation exchange–immobilized metal ion affinity chromatography analysis and containing ∼10,500 phosphorylation sites, illustrative of the complementarity and selectivity of our approach. The qualitative and quantitative data presented here provide a wide-ranging and credible resource of likely PKA substrates. Network analysis confirmed several established cAMP-dependent signaling nodes in our dataset, although most identified potential PKA substrates are “novel” (i.e. not previously reported and/or linked to PKA). Therefore, the dataset presented here can be considered as a comprehensive and reliable resource for future research into cAMP-related signaling.
EXPERIMENTAL PROCEDURES
Cell Culture, Stimulation, and Digest Preparation
Jurkat T lymphoma cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (Lonza, Basel, Switzerland). For PGE2 stimulation, cells were centrifuged for 2 min at 1500 × g. The growth medium was removed, and the cells were resuspended at a final concentration of 1 × 106 to 2 × 106 cells/ml in RPMI, supplemented with 0 (control) or 10 μm PGE2, and incubated for 1 or 60 min. After treatment, Jurkat cells were harvested and lysed on ice via sonication in 20 mm HEPES pH 8.0, 8 m urea, 1 mm sodium vanadate supplemented with 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, and an EDTA-free protease inhibitor mixture (Roche). The total protein concentration was determined using a Bradford assay (Bio-Rad). The total protein lysate from each condition (6 mg) was reduced with DTT at a final concentration of 4 mm at 56 °C for 30 min; subsequently, samples were alkylated with iodoacetamide at a final concentration of 8 mm at room temperature for 30 min in the dark. For digestion, the protease Lys-C (1:50; Wako, Richmond, VA) was added. Digestion was performed for 4 h at 37 °C.
Stable Isotope Labeling by Reductive Amination of Lys-C Peptides
Lys-C peptides were desalted and stable-isotope labeled using a Sep-Pak C18 column (Waters, Etten-Leur, The Netherlands) as described previously (28). Equal amounts of protein sample were labeled on-column using “light,” “intermediate,” and “heavy” dimethyl labeling reagents. The light label (L) was used for the control sample and the intermediate (I) and heavy (H) dimethyl labels were used for the PGE2-stimulated cells for 1 and 60 min, respectively. The resulting solution was then dried in vacuo and stored at −80 °C. The differentially dimethyl-labeled samples were mixed in a 1:1:1 ratio.
Immunoprecipitation
The immunoprecipitation (IP) was performed according to the manufacturer's protocol. Briefly, the mixed, labeled peptides from the three experiments were desalted, dried down, and resuspended in 1.4 ml of IP buffer (50 mm MOPS, pH 7.2, 10 mm sodium phosphate, and 50 mm NaCl). The labeled peptide mixture was added to 80 μl of the phospho-PKA substrate antibody beads (Cell Signaling Technology, Danvers, MA), and incubation was performed for 2 h at 4 °C with gentle shaking. Beads were washed two times with 1 ml IP buffer and four times with 1 ml MQ-water, all at 4 °C. Peptides were eluted by the addition of 0.15% TFA for 20 min at room temperature. Eluted peptides were desalted and concentrated on stop-and-go extraction tips.
On-line Nanoflow LC-MS/MS
The analysis of the enriched phosphopeptides and of the pre-IP cell lysate digest (MIX) was performed on a reversed-phase nano-LC coupled to an LTQ Orbitrap Velos mass spectrometer equipped with an ETD source (Thermo Fisher Scientific). An EASY-nLC 1000 (Thermo Fisher Scientific) was equipped with a 20-mm Aqua C18 (Phenomenex, Utrecht, The Netherlands) trapping column (packed in-house; 100-μm inner diameter, 5-μm particle size) and a 400-mm Zorbax SB-C18 (Agilent, Amstelveen, The Netherlands) analytical column (packed in-house; either 50-μm or 75-μm inner diameter; 1.8-μm particle size). Trapping and washing were performed at 10 μl/min for 4 min with solvent A (0.1 m acetic acid in water). Subsequently, peptides were transferred to the analytical column at about 150 nl/min in a total analysis time of 180 min with a gradient of 3%–40% (v/v) solvent B (0.1 m acetic acid in 80% acetonitrile) in 150 min.
The eluent was sprayed by a distal coated fused silica emitter (360-μm outer diameter, 20-μm inner diameter, 10-μm tip inner diameter; constructed in-house) butt-connected to the analytical columns. The ion spray voltage was set to 1.7 kV. The mass spectrometer was operated in a data-dependent mode to automatically switch between MS and MS/MS. Briefly, survey full-scan MS spectra were acquired after accumulation to a target value of 500,000 in the linear ion trap from m/z 350 to m/z 1500 in the Orbitrap with a resolution of 60,000 at m/z 400. For internal mass calibration, the 445.120025 ion was used as a lock mass with a target lock mass abundance of 0%. Charge state screening was enabled, and precursors with an unknown charge state or a charge state of 1 were excluded. After the survey scans, the 10 most intense precursors were subjected to HCD, ETD with ion trap detection, or ETD–Fourier transform fragmentation. A programmed data-dependent decision tree determined the choice of the most appropriate technique for a selected precursor (9). In essence, doubly charged peptides were subjected to HCD fragmentation, and more highly charged peptides were fragmented using ETD. The normalized collision energy for HCD was set to 40%. Supplemental activation was enabled for ETD. Dynamic exclusion was enabled (exclusion size list = 500, exclusion duration = 60 s).
Data Analysis
Each raw data file recorded by the mass spectrometer was processed and quantified with Proteome Discoverer (version 1.3, Thermo Scientific). Peak lists containing HCD and ETD fragmentation were generated with Proteome Discoverer with a signal-to-noise threshold of 1.5. The ETD non-fragment filter was also taken into account with the following settings: the precursor peak was removed within a 4-Da window, charged reduced precursors were removed within a 2-Da window, neutral losses from charged reduced precursors were removed within a 2-Da window, and the maximum neutral loss mass was set to 120 Da. All generated peak lists of the IP were searched against a concatenated forward-decoy Swissprot human database (version 2012 09, 40,992 sequences) and the MIX file was searched against Swissprot database version 2012 09 with taxonomy Homo sapiens (20,235 sequences) using Mascot software (version 2.3.02, Matrix Science, London, UK). The database search was performed with the following parameters: a mass tolerance of ±50 ppm for precursor masses, ±0.6 Da for ETD-ion trap fragment ions, and ±0.05 Da for HCD and ETD-Orbitrap fragment ions; two missed cleavages allowed; and cysteine carbamidomethylation as a fixed modification. Light, intermediate, and heavy dimethylation of peptide N termini and lysine residues; methionine oxidation; and phosphorylation on serine and threonine (only for the IP) were set as variable modifications. The enzyme was specified as Lys-C, and the fragment ion type was specified as electrospray ionization Fourier transform MS electron-capture dissociation, ETD–ion trap, or electrospray ionization quadrupole time-of-flight for the corresponding mass spectra.
The phosphorylation site localization of the identified phosphopeptides was performed by phosphoRS algorithm 2.0 (38), implemented in Proteome Discoverer. A site localization probability of at least 0.75 was used as the threshold for the phosphoresidue localization.
The dimethyl-based quantitation method was chosen in Proteome Discoverer, with a mass precision requirement of 2 ppm for consecutive precursor mass measurements. We applied 0.5 min of retention time tolerance for isotope pattern multiplets and allowed spectra with a maximum of 1 missing channel to be quantified. On/off situations were manually quantified, giving an arbitrary value of 100 or 0.01 for extreme up- or down-regulation, which corresponds to the maximum allowed fold change in the used Proteome Discoverer settings.
After identification and quantification, we combined all results originating from the same biological replica and filtered them with the following criteria: (i) mass deviations of ±10 ppm, (ii) Mascot Ion Score of at least 20, (iii) a minimum of six amino acid residues per peptide, and (iv) position rank 1. As a result, we obtained a peptide false discovery rate of ∼2%.
We also analyzed the previously mentioned 1:1:1 peptide mixture (MIX) prior to the IP, to check for proper mixing and evaluate global protein levels for normalization. Mascot results of the MIX analysis were filtered with the same parameters and with the integrated Percolator-based filter using a false discovery rate of <1% (based on peptide spectrum matches). Finally, all phosphopeptide ratios in the IP were normalized against the median of all quantified non-phosphopeptides in the MIX sample. The average log2 ratio and standard deviation of all quantified non-phosphopeptides identified in the pre-IP cell lysate digest (MIX) and in the eluate were used to determine significant regulation. For significant up-regulation or down-regulation, everything outside the 95% confidence interval was considered to be up-regulated or down-regulated—that is, up-regulation for log2 ratios greater than 0.622 (I/L) or 0.669 (H/L) and down-regulation for log2 ratios less than −0.580 (I/L) or −0.634 (H/L).
All identified phosphopeptides were searched against a recent release of PhosphositePlus.
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (39) with the dataset identifier PXD000148.
Interaction Network Analysis
The proteins identified in the study, together with the three PKA catalytic subunits and the alternative kinases Akt, PKC, and PKG, were used to generate interaction networks. Interactions were mapped from STRING v.9.0.5 using experimental and database lines of evidence with the defined high confidence cutoff (0.700). String-derived interactions were imported into Cytoscape v.2.8.1, where additional interactions were retrieved using the PhosphoSitePlus plugin. Individual protein kinase isoforms added to the protein list were then concatenated into a single node. Unconnected nodes were organized by protein type as derived from the PhosphoSitePlus database. The “All Other Protein Classes” category represents many different protein types assigned to only a few proteins in the list.
For functional annotation analysis of phosphoprotein dataset, we used the annotation tool Ingenuity Pathways Analysis. Canonical pathways analysis was performed with p value of 0.01 (Fisher's exact test).
RESULTS
Qualitative Analysis of PGE2-related Phosphorylation in Jurkat T Cells
Here we evaluated and applied a kinase-directed targeted immune-affinity-based proteomics approach to investigate the role of cAMP/PKA signaling downstream of PGE2 in Jurkat T cells. We first performed an initial kinetic analysis of Jurkat PGE2 stimulation (1 to 60 min) by immunoblotting using the antiphospho-PKA substrate antibody (Fig. 1A). From the blots obtained, we concluded that PGE2 stimulus (10 μm) induced a rapid, maximal increase in the phosphorylation of serine/threonine residues with arginine at the -3 position after only 1 min. Phosphorylation remained high up to 10 min before a substantial number of substrates returned to near-basal conditions after 60 min of PGE2 stimulation. On the basis of this result, we selected for our analysis time points 0 (control), 1, and 60 min. Cells were lysed, and proteins were digested with Lys-C, instead of trypsin, to keep the expected arginine-rich PKA motif intact. To allow quantitation of the phosphorylated peptides originating from the three different time points, we made use of stable-isotope dimethyl labeling (28). Control peptides from nonstimulated cells were labeled with the light dimethyl label, and peptides originating from the cells stimulated for 1 and 60 min with PGE2 were labeled with the intermediate and heavy dimethyl labels, respectively. Labeled samples were mixed in a 1:1:1 ratio, and from this mixture, phosphorylated substrates were enriched via targeted immunoprecipitation with the [R/K][R/K/X]X[pS/pT] consensus motif antibody and LC-MS/MS analysis (Fig. 1B).
Fig. 1.
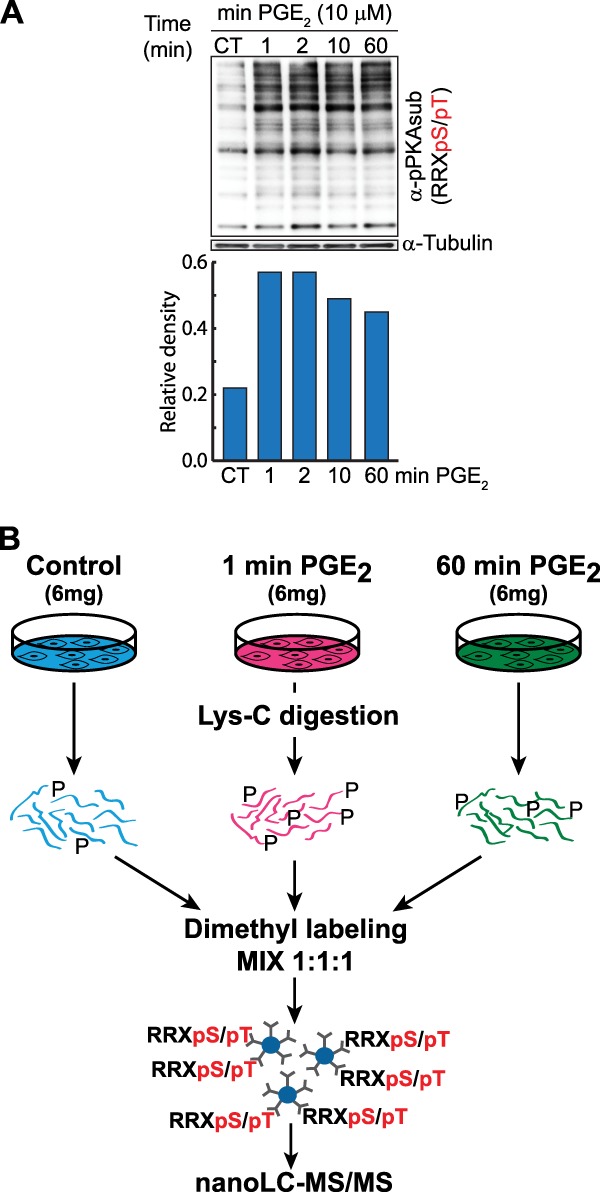
Experimental workflow used for the targeted and quantitative analysis of PKA substrates in Jurkat cells. A, Western blots of Jurkat clone E6.1 cells stimulated with PGE2 (10 μm) over a 60-min time course using the phospho-PKA substrate antibody (top) and a tubulin antibody (bottom) as a control, supplemented by a densitometric analysis of the Western blots. B, quantitative proteomics. Jurkat cells were either left unstimulated (control) or activated with PGE2 (10 μm) over two different stimulation times (1 min and 60 min). After lysis and enzymatic digestion with Lys-C, peptides were differentially labeled with three stable-isotope dimethyl labels and subsequently combined. Next, immunoprecipitation with the immobilized phospho-PKA substrate specific antibody was performed. After stringent washing, the eluate was analyzed via nano-LC-MS. Peptides and proteins were identified through a database search, and a functional analysis was performed using motif search algorithms, as well as Ingenuity and String.
The high sensitivity and specificity of our approach are illustrated by the base peak ion chromatogram of the immunoprecipitated peptide mixture obtained in a single 3-h LC-MS/MS run (Fig. 2A). All major peaks represent phosphopeptides. Additionally, when comparing the signal overall ion intensities of the pre-IP MIX sample (100 ng total protein injected) with the post-IP phosphopeptide sample, we estimate that the antiphospho-potential PKA substrate IP step yielded a ∼20,000-fold enrichment. This enrichment factor is roughly comparable to that achieved with the use of similar tyrosine phosphorylated peptide targeting antibodies (19, 20).
Fig. 2.
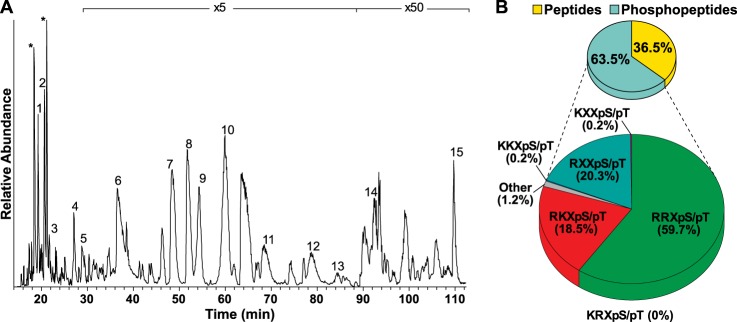
Qualitative evaluation of the phospho-PKA substrate antibody. A, nano-LC-MS base peak chromatogram of the eluate of the immunoprecipitated peptide mixture. The numbered signals represent intense phosphoserine- and phosphothreonine-containing peptides. A numbered list of these abundant phosphopeptides is available as supplemental Table S2. Asterisks (*) represent peaks of ions that could not be identified. B, representation of the PKA consensus motif among all identified phosphopeptides in the eluate displays 98% specificity.
Overall, in the three replicates, we were able to identify 1027 nonredundant peptides, of which 655 (∼64%) were phosphopeptides (supplemental Table S1). An analysis of the detected nonphosphorylated peptides revealed that they originated predominantly from high-abundant proteins, and most of them (∼93%) did not contain the PKA phosphorylation motif. Next we analyzed the enriched phosphopeptides for the presence of a PKA-related phosphorylation motif (11, 30–34) independent from the phosphoresidue localization. Of the 655 phosphopeptide sequences, ∼98% contained a canonical PKA phosphorylation motif (i.e. [R/K][R/K/X]X[pS/pT]), indicative of the extremely high specificity of the antibody (Fig. 2B).
We next probed the amino acid composition surrounding these likely PKA phosphorylation sites with WebLogo (Fig. 3A) (40). This revealed a definite preference for basic residues in P-3 and P-2 positions with the typical distribution of RR ≈ RK ≫ KR ≈ KK, in agreement with what was already known from well-characterized PKA physiological substrates (11, 30–34). We could also confirm the anecdotally reported preference for a small residue (such as serine (11%), proline (15%), or glycine (5%)) at the P-1 position and a hydrophobic residue (i.e. leucine/isoleucine (15%), valine (7%), or phenylalanine (4%)) at the P+1 position (31).
Fig. 3.
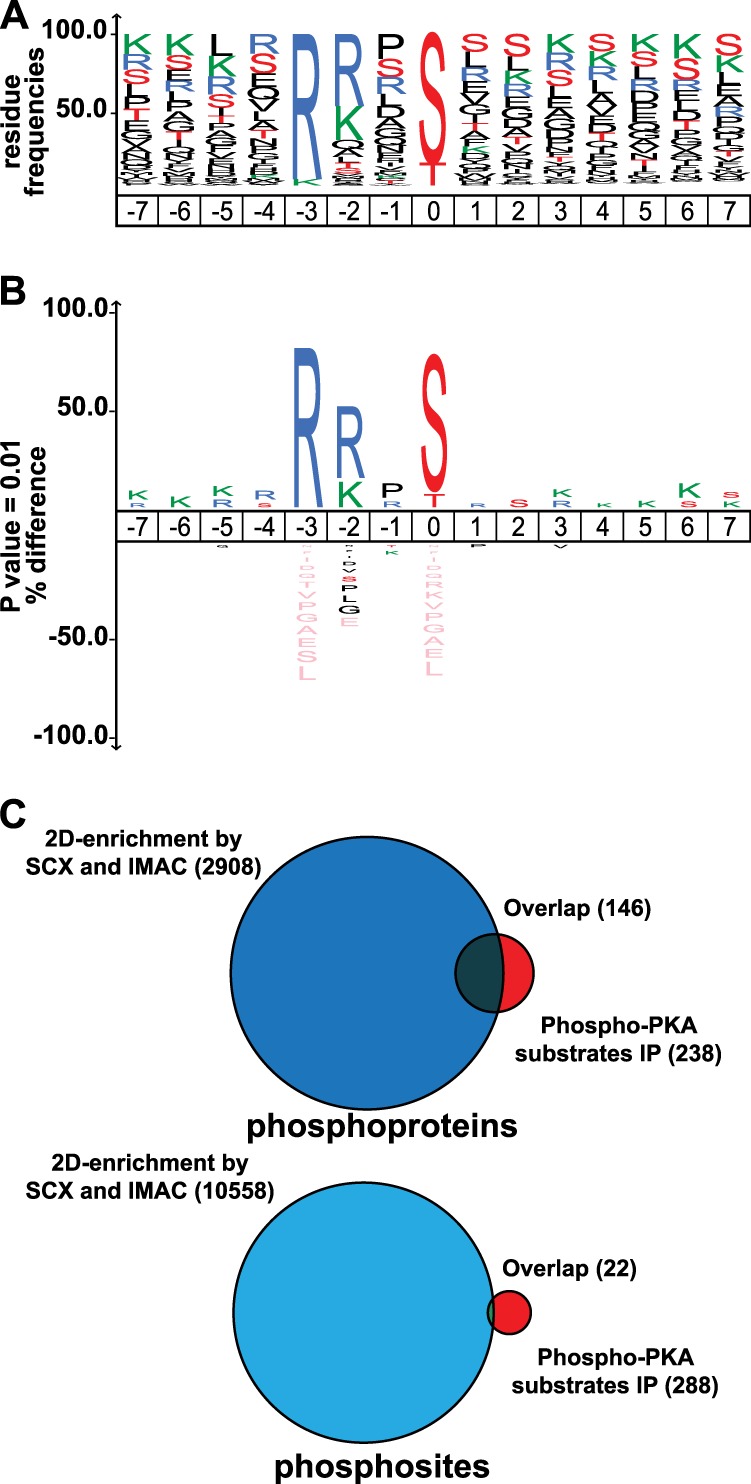
Amino acid distribution (WebLogo) and iceLogo analysis of the potential PKA substrates and their overlap in different phosphorylation datasets. The dataset used to create the frequencies in logo (A) and iceLogo (B) contained phosphopeptides with a PKA phosphosite localized with a probability of >0.75 (pRS probability). C, Venn diagrams representing the overlap between the very large unbiased Jurkat (phospho)proteome dataset reported by Mayya et al. and data from this study with a specific enrichment protocol. The overlap at the phosphoprotein level (top) is still reasonable, but at the phosphopeptide level (bottom) it is extremely low.
These high-confidence possible PKA phosphorylation sites were further analyzed using iceLogo (41), using the entire human proteome as background. The results, as depicted in Fig. 3B, represent the enrichment of amino acids as positive values, whereas underrepresented amino acids occur as negative values. The dataset generated a unique frequency for amino acids surrounding the phosphorylation site. In accordance with WebLogo, we found an overrepresentation of the basic amino acids R in position P-2 and either R or K in position P-2 (Fig. 3B). The frequency of R at positions P-2 and P-3 was increased by more than 20% and 60% (p = 0.01), respectively, whereas the basic amino acid K was overrepresented mainly at position P-2.
We then qualitatively benchmarked our small but highly defined set of immune-purified phosphosites against several large sources (Fig. 3C). We matched our data against the comprehensive and manually curated database repository PhosphoSitePlus (42) and against a recently reported large-scale phosphoproteomics dataset (∼10,500 phosphorylation sites) acquired in Jurkat T cells (12). Of the most confidently localized phosphorylation sites having the canonical PKA phosphorylation motif (n = 288), a quarter (n = 77) are not yet in PhosphoSitePlus (supplemental Table S3). More remarkable, only 22 out of the 10,500 (0.2%) reported in the large-scale Jurkat phosphoproteome dataset (12) are also in our targeted dataset, indicative of the high selectivity and high orthogonality of our targeted enrichment strategy relative to these shotgun approaches.
Dynamic Phosphorylation in Jurkat Cells upon Stimulation by PGE2
Next, we investigated the quantitative aspect of our approach by evaluating the differential amount of phosphorylation between the control and the two PGE2-stimulated samples (1 and 60 min); this was made possible by our stable-isotope dimethyl labeling ratios. Within the three replicates we were able to differentially quantify 586 (out of 655 (89%)) phosphopeptides (supplemental Table S4). Global evaluation of the quantitative data showed little change and merely 1:1:1 ratios in two control experiments: (i) the pre-IP cell lysate digest earlier referred to as MIX and (ii) the nonphosphorylated peptides that co-purified in the IP (Fig. 4). In contrast, many, but not all, of the detected phosphopeptides bearing a possible PKA motif showed a substantial increase in abundance after stimulation by PGE2 for 1 min and/or 60 min. Interestingly, a small minority of likely PKA phosphopeptides showed down-regulation, mainly after 60 min of stimulation.
Fig. 4.
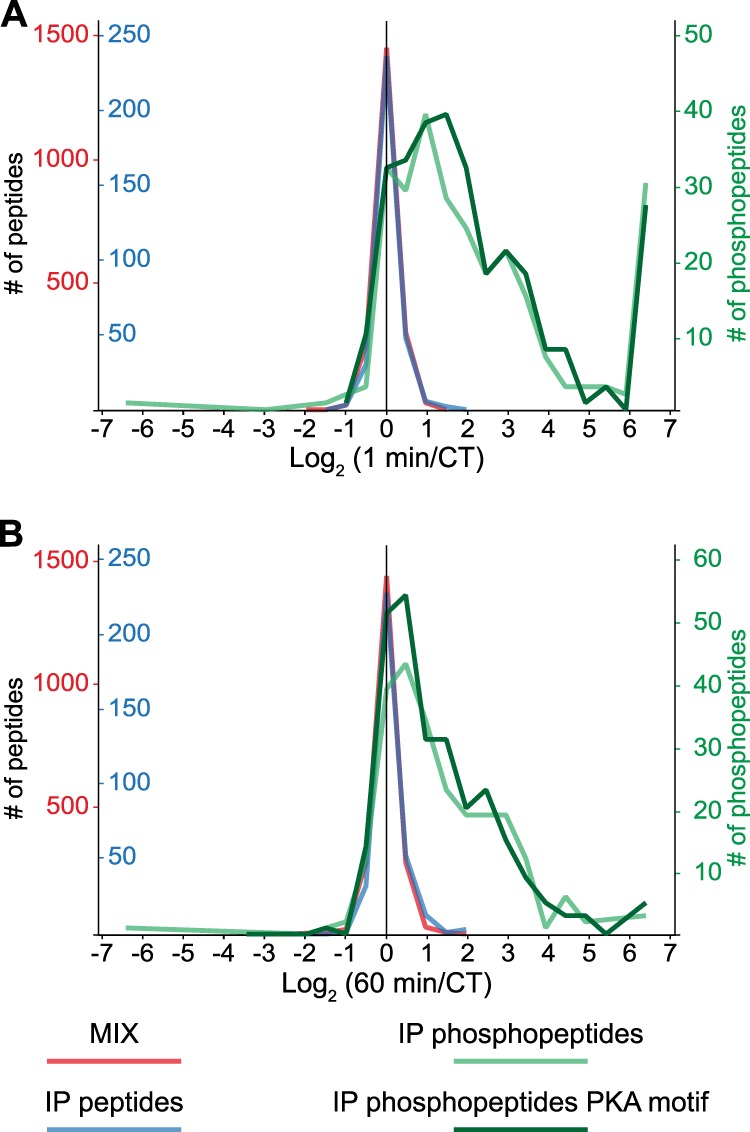
Quantitative analysis of the PGE2-regulated phosphoproteome. Density curves of observed peptide ratios before (MIX) and after (IP) the immunoprecipitation show that the three differentially stable-isotope dimethyl-labeled samples were mixed as 1:1:1, with negligible nonphosphopeptides with a large dimethyl ratio between stimulated and unstimulated cells. Density curves of the quantified phosphopeptides, with or without a localized putative PKA motif, show a general increase in the level of phosphorylation upon PGE2 stimulation after 1 min (A) and 60 min (B).
Next we clustered the detected temporal profiles of the unique phosphopeptides carrying confidently localized potential PKA phosphosites (306 in total) into five clusters (Fig. 5)—three with an up-regulated pattern, one grouping the PGE2-insensitive sites, and one with a down-regulated profile.
Fig. 5.

Quantitative analysis of the PGE2-regulated possible PKA phosphopeptides. The high-confidence PKA-motif-containing unique phosphopeptides (306 in total) were first grouped into five different clusters. Represented are the ratio profiles (left), the number of members in each cluster, and the sequence and mass spectrum of a representative phosphopeptide. pS, phosphoserine; blue, purple, and green boxes represent the expected regions for the isotopic distribution for the light, intermediate, and heavy peptide isotopomers, respectively. A, cluster 1 (dark green); immediate up-regulation of the phosphorylation site that remains high for up to 60 min. B, cluster 2 (intermediate green); immediate regulation of the phosphosite after 1 min that returned to basal level after 60 min. C, cluster 3 (light green); late-responding phosphorylation sites that were only significantly up-regulated after 60 min. D, cluster 4 (gray); the unaffected sites that presented no change in phosphorylation level at either time point of stimulus. E, cluster 5 (red); sites showing a decrease in their phosphorylation level.
Network analysis was performed using Ingenuity on the dataset containing all the phosphoproteins with an identified PKA-motif-containing peptide in our experiment. This revealed a PKA-focused temporal view of PGE2 stimulation in Jurkat cells. As expected, several of the identified phosphoproteins are involved in the canonical pathway of PKA (Fig. 6), which was also the main enriched pathway (p value < 0.001) if only the sites regulated after 1 min or 60 min of PGE2 treatment were taken into account (supplemental Table S5).
Fig. 6.

Interaction map for regulated PGE2 downstream substrates. Protein–protein interactions were derived from both the STRING database using experimental and database lines of evidence and “high confidence” interactions (score > 0.700), and the substrate lines were derived from the search function in the PhosphoSitePlus database. The interaction map was generated using Cytoscape. Protein class information is from PhosphoSitePlus. Color coding indicates sensitivity to PGE2 stimulation.
Fig. 6 shows an overview of all the proteins detected when using the phosphopeptide targeting antibody. The interaction list used to generate Fig. 6 was created by combining interactions reported in the STRING and PhosphoSitePlus databases. Several of these proteins have been reported to be directly (phosphorylated) or indirectly connected to PKA or to one of the other PGE2-sensible basophilic kinases (supplemental Fig. S1). More interesting, the network analysis revealed the presence of many other proteins not previously linked to cAMP/PGE2 signaling, yet containing a reliable potential PKA consensus motif, becoming phosphorylated upon PGE2 stimulation. These proteins are thus likely substrates and belong to a multitude of protein classes and functionalities (supplemental Table S5), illustrating the widespread role of PKA and the other possible upstream kinases in different biological processes.
DISCUSSION
We describe a highly specific targeted method to quantitatively analyze the interplay between PGE2 and PKA in T-cell activation. We used a new monoclonal antibody able to specifically enrich phosphopeptides containing the [R/K][R/K/X]X[pS/pT] motif, which allowed us to identify a few hundred nonredundant phosphopeptides via LC-MS/MS using a single immunoprecipitation in a single LC-MS/MS run. Moreover, the use of decision-tree MS/MS technology, which takes advantage of ETD fragmentation for the identification of arginine-rich phosphopeptides, permitted the precise localization of phosphorylation for the majority of the identified phosphopeptides (supplemental Fig. S2).
Compared with shotgun phosphoproteomics, our new method proved highly cost and time efficient.
Benchmarking Specificity for Potential PKA Substrate Identification
The overlap of our targeted dataset with the very large unbiased dataset of 10,500 phosphorylation sites of the Jurkat phosphoproteome reported by Mayya et al. (12) comprises merely 22 sites (i.e. 0.2%). The dataset of Mayya et al. was obtained following an extensive phosphopeptide enrichment strategy starting with a similar amount of Jurkat T cell starting material as we used here, albeit using at least 10-fold more instrument and analysis time. Out of the 288 phosphosites identified here, 211 have been reported in the PhosphositePlus database. Closer inspection on where these 211 entries came from revealed that they were predominantly retrieved in experiments using peptide immunopurification with antibodies raised against basophilic kinase motifs (e.g. PKA, Akt, PKC). This clearly indicates that these sites are preferentially observed via immunopurification methods only.
Based on the minimal overlap observed, we conclude that our targeted method is very specific, bringing low-substoichiometric potential PKA phosphorylation sites within reach.
In the shotgun approach, these were likely obscured by the multitude of competing phosphopeptides in the somewhat stochastic process of MS-based peptide identification. The two approaches are complementary and have their own merits. A large-scale, less biased phosphopeptide enrichment provides a more comprehensive cellular phosphoproteome, whereas the targeted approach presented here provides a more detailed view of a specific pathway or kinase, with the possibility of identifying new substrates.
Noncanonical Substrate Motifs
Our data, as depicted in Fig. 3, clearly illustrate that the motif recognized by the antibody used is very well defined, as nearly ∼98% of the identified phosphopeptides contained a potential PKA phosphorylation motif (i.e. [R/K][R/K/X]X[pS/pT]). More in-detail analysis of the remaining phosphopeptides revealed the possible presence of noncanonical motifs. We identified an HRDpS motif on protein DEFI6 and an HSRpS on protein U2AF2, suggesting that histidine instead of R or K can be recognized as a basic residue by PKA. In agreement, it has been reported that PKA can phosphorylate such a histidine-containing motif (HXXpS) (31). Additionally, 16 phosphopeptides in our dataset hold the RQX[pS/pT] motif, with 5 of them having a proline at position P-1. The corroborating RQPpS sequence in the protein KCMA1 is an authentic substrate of PKA (43), suggesting that the proteins in our dataset (PEA15, HD, LMO7, STXB5 and SAMD9) also could be genuine PKA substrates. Therefore, although the sequence specificity of PKA seems to be very constricted, there seems to be room for other noncanonical motifs.
Response of Jurkat T Cells upon PGE2 Stimulation
With the method described here we were able to monitor in-depth PGE2-induced signaling in Jurkat T cells. The data as presented in Fig. 6 and supplemental Fig. S1 contain multiple PGE2/cAMP benchmarks, including the activation of core pathway components such as PIK3CA/Akt, PKA, and ROCK2 (44) and the well-documented phosphorylation of downstream substrates such as RAF1 (S43), BRAF (S429), and ITGA4 (S1021). Cumulative evidence has revealed that there may be ambiguity and redundancy in kinase–substrate relationships. In particular, kinases in the AGC family have strongly similar substrate specificities. Also, it has been reported that different AGC kinases can phosphorylate the same residues (i.e. BRAF S429 by PKA and Akt). This may be linked to the cell's capability to allow different extracellular stimuli to control the same intracellular signaling pathways, leading to a similar cellular response (45). Evidently, this makes it almost impossible to pinpoint the responsible upstream kinases.
The two kinases RAF1 (46) and BRAF (47) show decreased activity upon PKA phosphorylation, and phosphorylated ITGA4 cannot bind to paxillin (48), an interaction required for the specific promotion of cell migration, enhanced phosphorylation of PTK2, and reduction of cell spreading (49). The network analysis depicted in Fig. 6 shows that many of the identified phosphoproteins indirectly linked to PKA are also putative targets—for instance, the proteins connected to the documented PKA substrates NDE1 (S306) and RPS6 (S235). Interestingly, the quantitative phosphorylation profile of these proteins is similar to that of known, connected PKA targets. This suggests PKA may target multiple binding/interacting partners within a single signaling module. We note that the protein RPS6 can be phosphorylated on S235 by a multitude of other kinases, including Akt and PKC (supplemental Fig. S1), and with the proposed strategy it is not possible to prove that its phosphorylation state is only PKA time-dependent, but our approach provides favorable information for those interested in the PGE2/cAMP pathway.
In-depth Manual Curation Reveals More Links to PKA
The majority of the possible PKA substrates presented here, as depicted in Fig. 6 or supplemental Fig. S1, are currently not connected to PKA or any other phosphoprotein in our analysis. Because of the “incompleteness” of the databases currently available for building signaling networks, manual curation is still valuable for attempting a more in-depth evaluation of the data. Evidently, this is impossible to do for all observed proteins. After performing manual curation on the unconnected proteins, we found two more well-documented PKA substrates, GYS1 phosphorylated on S698 (50) and the transcription factor ATF1 on S63 (51). The latter is a site located in the kinase-inducible domain of ATF1 that, once phosphorylated, enhances the transactivation activity of the protein by promoting recruitment of the co-activator cAMP-responsive element-binding protein (52), which we found phosphorylated on its known PKA phosphorylation site S133 (53) (supplemental Table S4).
Overall, it is clear that the dataset presented here is one of the most extensive and credible datasets of likely PKA substrates, providing a valuable resource for researchers with an interest in cAMP signaling. Obviously, further biological experiments such as site mutations or gene or siRNA knockouts are important in order to further validate putative new PKA substrates. An interesting group of proteins to follow up on might be the observed kinases and phosphatases, as they imply that PKA may have an important role in relaying multiple downstream phosphorylation routes. Also striking is the large set of proteins involved in gene transcription and translation, suggesting that PKA may be a master regulator of Jurkat T-cell function.
Supplementary Material
Acknowledgments
The technical support of Bryon Ricketts in Western blot procedures and Elizabeth A. McClellan in Ingenuity Pathways Analysis is acknowledged.
Footnotes
* The Netherlands Proteomics Centre, a program embedded in the Netherlands Genomics Initiative, is kindly acknowledged for financial support. A.S. acknowledges support from the Utrecht University program “Focus en Massa.” Part of this research was performed within the framework of the PRIME-XS project (Grant No. 262067), funded by the European Union 7th Framework Program and the Netherlands Organization for Scientific Research (NWO)-supported large-scale proteomics facility Proteins@Work (project 184.032.201).
 This article contains supplemental material.
This article contains supplemental material.
Conflicts of interest: M.P.S. and J.C.S. are employees of Cell Signaling Technology, which makes the anti-phospho-PKA antibody commercially available.
1 The abbreviations used are:
- cAMP
- cyclic adenosine monophosphate
- ETD
- electron transfer dissociation
- HCD
- higher energy collision dissociation
- IP
- immunoprecipitation
- MS/MS
- tandem mass spectrometry
- PGE2
- prostaglandin E2
- PKA
- cAMP-dependent protein kinase A.
REFERENCES
- 1. Bensimon A., Heck A. J., Aebersold R. (2012) Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 379–405 [DOI] [PubMed] [Google Scholar]
- 2. Altelaar A. F., Munoz J., Heck A. J. (2012) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48 [DOI] [PubMed] [Google Scholar]
- 3. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villen J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Villen J., Gygi S. P. (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinkse M. W., Mohammed S., Gouw J. W., van Breukelen B., Vos H. R., Heck A. J. (2008) Highly robust, automated, and sensitive online TiO2-based phosphoproteomics applied to study endogenous phosphorylation in Drosophila melanogaster. J. Proteome Res. 7, 687–697 [DOI] [PubMed] [Google Scholar]
- 6. Zhou H., Low T. Y., Hennrich M. L., van der Toorn H., Schwend T., Zou H., Mohammed S., Heck A. J. (2011) Enhancing the identification of phosphopeptides from putative basophilic kinase substrates using Ti (IV) based IMAC enrichment. Mol. Cell. Proteomics 10, M110.006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Good D. M., Wirtala M., McAlister G. C., Coon J. J. (2007) Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 6, 1942–1951 [DOI] [PubMed] [Google Scholar]
- 8. Wiesner J., Premsler T., Sickmann A. (2008) Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics 8, 4466–4483 [DOI] [PubMed] [Google Scholar]
- 9. Frese C. K., Altelaar A. F., Hennrich M. L., Nolting D., Zeller M., Griep-Raming J., Heck A. J., Mohammed S. (2011) Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 10, 2377–2388 [DOI] [PubMed] [Google Scholar]
- 10. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 11. Oberprieler N. G., Lemeer S., Kalland M. E., Torgersen K. M., Heck A. J., Tasken K. (2010) High-resolution mapping of prostaglandin E2-dependent signaling networks identifies a constitutively active PKA signaling node in CD8+CD45RO+ T cells. Blood 116, 2253–2265 [DOI] [PubMed] [Google Scholar]
- 12. Mayya V., Lundgren D. H., Hwang S. I., Rezaul K., Wu L., Eng J. K., Rodionov V., Han D. K. (2009) Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2, ra46. [DOI] [PubMed] [Google Scholar]
- 13. Zhou H., Di Palma S., Preisinger C., Peng M., Polat A. N., Heck A. J., Mohammed S. (2013) Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 12, 260–271 [DOI] [PubMed] [Google Scholar]
- 14. Pinkse M. W., Uitto P. M., Hilhorst M. J., Ooms B., Heck A. J. (2004) Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-nanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 76, 3935–3943 [DOI] [PubMed] [Google Scholar]
- 15. Alpert A. J. (2008) Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem. 80, 62–76 [DOI] [PubMed] [Google Scholar]
- 16. Chien K. Y., Liu H. C., Goshe M. B. (2011) Development and application of a phosphoproteomic method using electrostatic repulsion-hydrophilic interaction chromatography (ERLIC), IMAC, and LC-MS/MS analysis to study Marek's Disease Virus infection. J. Proteome Res. 10, 4041–4053 [DOI] [PubMed] [Google Scholar]
- 17. McNulty D. E., Annan R. S. (2008) Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteomics 7, 971–980 [DOI] [PubMed] [Google Scholar]
- 18. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y., Wolf-Yadlin A., Ross P. L., Pappin D. J., Rush J., Lauffenburger D. A., White F. M. (2005) Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics 4, 1240–1250 [DOI] [PubMed] [Google Scholar]
- 20. Boersema P. J., Foong L. Y., Ding V. M., Lemeer S., van Breukelen B., Philp R., Boekhorst J., Snel B., den Hertog J., Choo A. B., Heck A. J. (2010) In-depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immunoaffinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteomics 9, 84–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 22. Moritz A., Li Y., Guo A., Villen J., Wang Y., MacNeill J., Kornhauser J., Sprott K., Zhou J., Possemato A., Ren J. M., Hornbeck P., Cantley L. C., Gygi S. P., Rush J., Comb M. J. (2010) Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salomon A. R., Ficarro S. B., Brill L. M., Brinker A., Phung Q. T., Ericson C., Sauer K., Brock A., Horn D. M., Schultz P. G., Peters E. C. (2003) Profiling of tyrosine phosphorylation pathways in human cells using mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 100, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X. M., Polakiewicz R. D., Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 25. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., Wetzel R., Macneill J., Ren J. M., Yuan J., Bakalarski C. E., Villen J., Kornhauser J. M., Smith B., Li D., Zhou X., Gygi S. P., Gu T. L., Polakiewicz R. D., Rush J., Comb M. J. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 [DOI] [PubMed] [Google Scholar]
- 26. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 27. Kovanich D., Cappadona S., Raijmakers R., Mohammed S., Scholten A., Heck A. J. (2012) Applications of stable isotope dimethyl labeling in quantitative proteomics. Anal. Bioanal. Chem. 404, 991–1009 [DOI] [PubMed] [Google Scholar]
- 28. Boersema P. J., Raijmakers R., Lemeer S., Mohammed S., Heck A. J. (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 [DOI] [PubMed] [Google Scholar]
- 29. Hsu J. L., Huang S. Y., Chow N. H., Chen S. H. (2003) Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852 [DOI] [PubMed] [Google Scholar]
- 30. Tegge W., Frank R., Hofmann F., Dostmann W. R. (1995) Determination of cyclic nucleotide-dependent protein kinase substrate specificity by the use of peptide libraries on cellulose paper. Biochemistry 34, 10569–10577 [DOI] [PubMed] [Google Scholar]
- 31. Shabb J. B. (2001) Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381–2411 [DOI] [PubMed] [Google Scholar]
- 32. Hutti J. E., Jarrell E. T., Chang J. D., Abbott D. W., Storz P., Toker A., Cantley L. C., Turk B. E. (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29 [DOI] [PubMed] [Google Scholar]
- 33. Kim M., Park Y. S., Shin D. S., Kim J., Kim B. G., Lee Y. S. (2011) Antibody-free peptide substrate screening of serine/threonine kinase (protein kinase A) with a biotinylated detection probe. Anal. Biochem. 413, 30–35 [DOI] [PubMed] [Google Scholar]
- 34. Smith F. D., Samelson B. K., Scott J. D. (2011) Discovery of cellular substrates for protein kinase A using a peptide array screening protocol. Biochem. J. 438, 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swaney D. L., McAlister G. C., Coon J. J. (2008) Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalinski P. (2012) Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chemnitz J. M., Driesen J., Classen S., Riley J. L., Debey S., Beyer M., Popov A., Zander T., Schultze J. L. (2006) Prostaglandin E2 impairs CD4+ T cell activation by inhibition of lck: implications in Hodgkin's lymphoma. Cancer Res. 66, 1114–1122 [DOI] [PubMed] [Google Scholar]
- 38. Taus T., Kocher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 39. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome. Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 42. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu R., Alioua A., Kumar Y., Eghbali M., Stefani E., Toro L. (2006) MaxiK channel partners: physiological impact. J. Physiol. 570, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sreeramkumar V., Fresno M., Cuesta N. (2012) Prostaglandin E2 and T cells: friends or foes? Immunol. Cell Biol. 90, 579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 46. Hafner S., Adler H. S., Mischak H., Janosch P., Heidecker G., Wolfman A., Pippig S., Lohse M., Ueffing M., Kolch W. (1994) Mechanism of inhibition of Raf-1 by protein kinase A. Mol. Cell. Biol. 14, 6696–6703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Konig S., Guibert B., Morice C., Vernier P., Barnier J. V. (2001) Phosphorylation by PKA of a site unique to B-Raf kinase. C. R. Acad. Sci. III 324, 673–681 [DOI] [PubMed] [Google Scholar]
- 48. Han J., Liu S., Rose D. M., Schlaepfer D. D., McDonald H., Ginsberg M. H. (2001) Phosphorylation of the integrin alpha 4 cytoplasmic domain regulates paxillin binding. J. Biol. Chem. 276, 40903–40909 [DOI] [PubMed] [Google Scholar]
- 49. Liu S., Thomas S. M., Woodside D. G., Rose D. M., Kiosses W. B., Pfaff M., Ginsberg M. H. (1999) Binding of paxillin to alpha4 integrins modifies integrin-dependent biological responses. Nature 402, 676–681 [DOI] [PubMed] [Google Scholar]
- 50. Bultot L., Guigas B., Von Wilamowitz-Moellendorff A., Maisin L., Vertommen D., Hussain N., Beullens M., Guinovart J. J., Foretz M., Viollet B., Sakamoto K., Hue L., Rider M. H. (2012) AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem. J. 443, 193–203 [DOI] [PubMed] [Google Scholar]
- 51. Kobayashi M., Shimomura A., Hagiwara M., Kawakami K. (1997) Phosphorylation of ATF-1 enhances its DNA binding and transcription of the Na,K-ATPase alpha 1 subunit gene promoter. Nucleic Acids Res. 25, 877–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng D., Cho Y. Y., Lau A. T., Zhang J., Ma W. Y., Bode A. M., Dong Z. (2008) Cyclin-dependent kinase 3-mediated activating transcription factor 1 phosphorylation enhances cell transformation. Cancer Res. 68, 7650–7660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mosenden R., Tasken K. (2011) Cyclic AMP-mediated immune regulation—overview of mechanisms of action in T cells. Cell. Signal. 23, 1009–1016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.