

Abstract
We describe a novel amine-reactive chemical label that exploits differential neutron-binding energy between 13C and 15N isotopes. These neutron-encoded (NeuCode) chemical labels enable up to 12-plex MS1-based protein quantification. Each structurally identical, but isotopically unique, tag is encoded with a 12.6-mDa mass difference—relative to its nearest neighbor—so that peptides bearing these NeuCode signatures do not increase spectral complexity and are detected only upon analysis with very high mass-resolving powers. We demonstrate that the method provides quantitative performance that is comparable to both metabolic labeling and isobaric tagging while combining the benefits of both strategies. Finally, we employ the tags to characterize the proteome of Saccharomyces cerevisiae during the diauxic shift, a metabolic transition from fermentation to aerobic respiration.
Proteome quantification is an increasingly essential component of modern biology and translational medicine (1, 2). Whether targeted or global, stable isotope incorporation with mass spectrometry (MS) analysis is a core technique for protein abundance measurements. There are numerous approaches that can be used to introduce stable isotopes into peptides, the most frequently used being stable isotope labeling with amino acids in cell culture (SILAC)1 and isobaric tagging (tandem mass tags or isobaric tags for relative and absolute quantitation) (3–7). Both of these methods incorporate heavy isotopes to increase mass by at least 1 Da. SILAC is the quantification gold standard for global proteomic analysis. However, the SILAC approach is not easily adapted for tissue sample analysis; SILAC mouse labeling, for example, requires feeding mice a specialized diet for multiple generations (8). Tissue samples can be analyzed if they are mixed with SILAC cell-culture-based labeled standards, but this strategy does not permit multiplexing (9). Isobaric labels, in contrast, are conjugated to the primary amines of peptides following proteolytic digestion and thus have the advantage of being completely compatible with samples from virtually any source (10, 11). That said, isobaric tagging suffers from dynamic range suppression caused by co-isolation of precursor peptides (12). Multiple studies have revealed that this problem greatly erodes quantitative accuracy—for example, 10-fold changes often are detected as much smaller ∼4-fold changes (13, 14). Here we propose a new approach to protein quantification, one that achieves tissue-compatible 4-plexed MS1-based quantification without increasing spectral complexity.
Recently, we described the use of mass defects to expand SILAC quantification from 3-plex to 12-plex and beyond (i.e., NeuCode SILAC) (15). NeuCode SILAC exploits the subtle mass differences that exist in atoms as a result of the varying energies of nuclear binding in common stable isotopes (e.g., carbon, nitrogen, hydrogen, and oxygen) by using the extremely high resolving power of modern Fourier transform mass spectrometer systems (16, 17). For example, the multiplexing capability of tandem mass tagging was increased from 6 to 8 by incorporating a difference in mass of 6.3 mDa in specific reporter ions by swapping 14N for a 15N atom while concomitantly switching a 13C with a 12C atom (18, 19). This method requires only 30,000 resolving power to resolve the reporter ions, but it still suffers from the interference problem described above. Repetition of this process, within the context of an analyte molecule, can generate several chemically identical isotopologues that, when analyzed under normal MS analysis conditions (resolving power R) are indistinguishable (i.e., produce one m/z peak). Analysis of these NeuCode m/z peaks with high resolving power (480,000), however, often reveals distinct m/z peaks whose abundances can be extracted and used to determine analyte quantity across the sundry conditions. This strategy permits very high levels of MS1-based multiplexing (>10), which has several advantages. First, MS1 scans across the entire analyte elution profile can be averaged to increase quantitative accuracy and precision. Second, a tandem mass spectrum is not necessary for quantification. Mann and colleagues showed that once a peptide is identified via tandem MS analysis, it can be confidently identified in other runs with only the exact mass and elution profile matching (20). Third, MS1-based quantification does not suffer from the pervasive problem of precursor interference that cripples the quantitative accuracy of the isobaric tagging strategies (13).
The NeuCode SILAC approach, however, relies on the use of amino acid isotopologues and still requires metabolic incorporation. Chemical labeling strategies for proteome quantification can be convenient and, for certain systems, are requisite. We reasoned that our NeuCode strategy could be extended to create novel chemical reagents for proteome quantification. We describe here the design, synthesis, and use of 12-plex NeuCode amine reactive labels for global proteome quantification.
EXPERIMENTAL PROCEDURES
Theoretical Calculations
First, a library of 65,011 yeast trypsin-derived peptides identified via mass spectrometry was composed. The theoretical full width at 10% maximum peak height (FWTM) for each library peptide across resolving powers (R) from 15,000 to 1,000,000 is calculated by
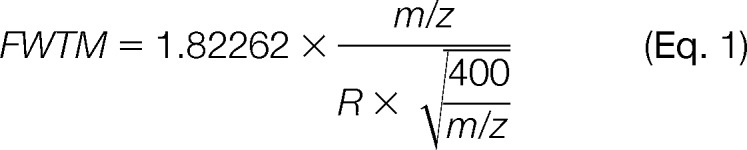 |
where the resolving power is defined as the minimum m/z difference that can be resolved at 400 m/z and the coefficient (1.82262) is derived from Gaussian peak shape modeling. The m/z difference (Δm/z) for each theoretical isotope doublet assuming isotopologue mass differences (ΔI) of 6.3, 12.6, and 25.2 mDa is
 |
where n is the number of primary amines (lysines and N termini) in the peptide sequence and z is the charge of the peptide. An isotopologue pair is considered resolvable at the tested isotopologue mass difference and resolving power only if Δm/z > FWTM.
Label Synthesis
NeuCode labels were synthesized on preloaded Gly-Wang resin (EMD Millipore, Billerica, MA) via typical Fmoc solid-phase synthesis. Mass defects were incorporated by generating tri-peptides with the isotopic distributions described in Fig. 1C using protected amino acids (EMD Millipore) and protected amino acid isotopes (Cambridge Isotopes, Tewksbury, MA). The N termini and lysine side chains were reacted with either 13C4 acetic anhydride (Cambridge Isotopes) or light acetic anhydride (Sigma Aldrich). NHS esters were formed via reaction of the tri-peptide in dimethyl sulfoxide with 10 equivalents of diisopropylcarbodiimide, 1.5 equivalents of N-hydroxysuccinimide, and 10 equivalents of pyridine. NHS esters were precipitated by means of dropwise addition to ice-cold ether. Precipitate was dissolved in dimethyl sulfoxide.
Fig. 1.
NeuCode amine-reactive label development and design. A, theoretical calculation depicting the percentage of peptides that are resolved at full width at 10% maximum peak height when spaced at 6.3, 12.6, or 25.2 mDa for resolving powers of 15,000 to 1,000,000 on an Orbitrap (n = 65,011). B, acetylarginine-acetyllysine-glycine-NHS structure and isotopic composition. The asterisks denote atoms in which a 13C or 15N isotope can be incorporated. C, the specific isotopic compositions that encode the 12.6-mDa mass differences for each of the 4-plex labels are listed in the table. D, spectra from synthesized acetylarginine-acetyllysine-glycine tripeptide encoding 12.6-mDa mass differences.
Yeast Cultures and Lysis
The wild-type W303 Saccharomyces cerevisae strain was obtained from the laboratory of Jarred Rutter. In biological triplicate, a single colony of yeast was used to inoculate a starter culture of yeast extract peptone dextrose media and incubated (30 °C, 230 rpm) for 12 h. Yeast extract peptone dextrose media (1 l) was inoculated with yeast cells from the starter culture and incubated (30 °C, 230 rpm). Yeast cell pellets were isolated at four points during the culture (1–4; Fig. 3A) via centrifugation and immediately frozen in N2(l). The concentration of glucose in the media was determined with a Glucose (HK) Assay Kit (Sigma). The entirety of the following yeast lysis procedure was performed at 4 °C. SEH lysis buffer (0.6 m sorbitol, 20 mm HEPES, pH 7.4, 2 mm MgCl2, 1 mm EGTA, protease inhibitors) and glass beads (BioSpec, Bartlesville, OK) were added to the yeast cell pellet. The cells were lysed by bead beating. The beads were allowed to settle to the bottom of the tube, and the supernatant (lysate) was frozen in N2(l) and stored at −80 °C.
Fig. 3.
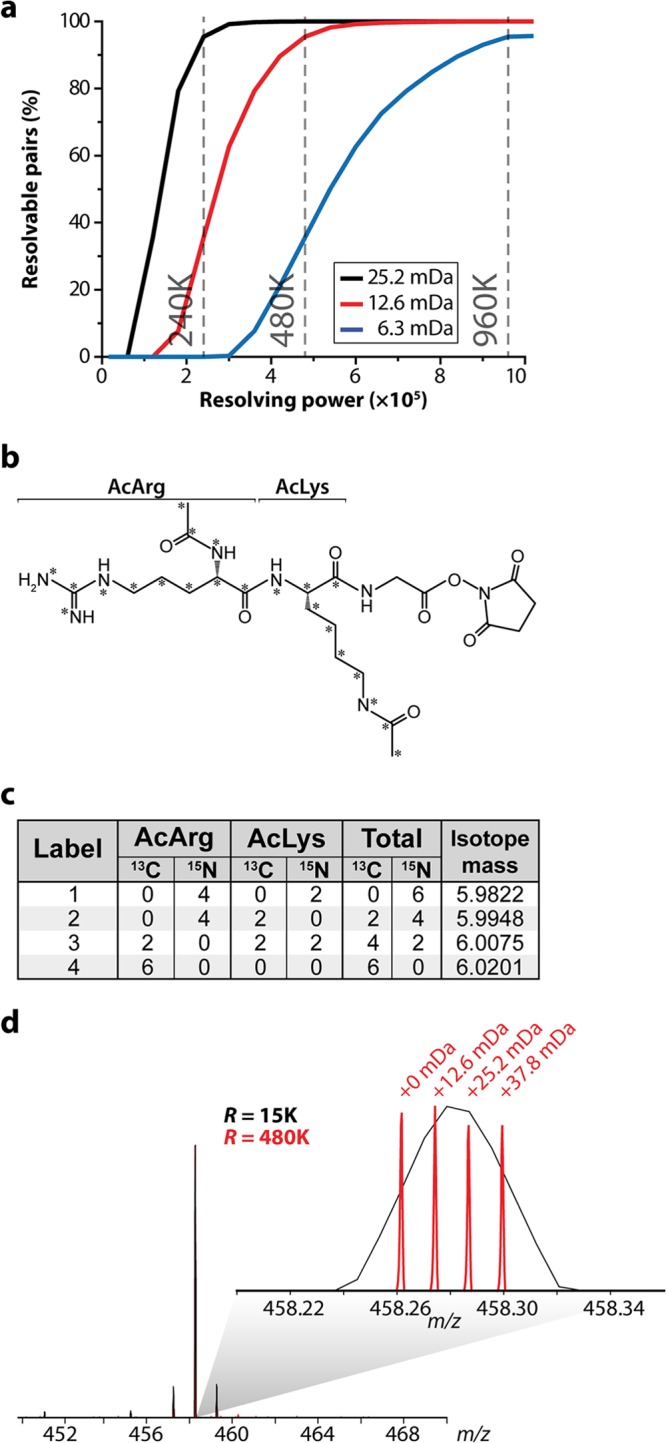
Comparison between the widths of single peaks and 4-plex clusters. A, single peak widths (red) were calculated as the FWTM when analyzed at R = 30,000. The width of a 4-plex cluster (blue) is calculated as the difference between the largest and smallest m/z that corresponds to 10% maximum peak height when analyzed at R = 480,000. B, Boxplot of calculated widths for single peaks and for 4-plex clusters from a library of 65,011 identified peptides.
Nano-HPLC-MS/MS Analysis
The samples were reduced and alkylated, digested overnight with trypsin (Promega, Madison, WI), desalted, and dried. Peptides were resuspended in dimethyl sulfoxide and triethylamine. Labels were added to individual samples, incubated at ambient temperature for 2 h, and combined. The 4- and 12-plex samples were fractionated via high-pH reversed-phase chromatography (pH 10). Each sample was analyzed via nano-reversed-phase liquid chromatography coupled to an Orbitrap Elite mass spectrometer (Thermo Scientific). The MS acquisition method is described in the “Results” section.
Data Analysis
Raw files were converted to searchable text files using COMPASS 1.2.2.3. Data were searched using semitryptic enzyme specificity against a target-decoy database containing 13,306 entries (Saccharomyces cerevisiae, downloaded from UniProt on January 20, 2013) using the Open Source Mass Spectrometry Search Algorithm (OMSSA 2.1.8), allowing for up to three missed cleavages. For all samples, methionine oxidation and cysteine carbamidomethylation were searched as variable and fixed modifications, respectively. 4-plex and known-ratio samples were searched with the average 4-plex NeuCode label mass (+431.2399) as a fixed modification on peptide N termini and lysines and as a variable modification on tyrosines. 12-plex was searched exactly like the 4-plex, except the data were searched independently for NeuCode light (+431.2399), medium (+435.2533), and heavy (+439.2667). Survey scan mass tolerances were set to 3.5 Da. Fragment mass tolerances were 0.1 Da for Orbitrap higher energy collision-activated dissociation spectra and 0.35 for ion-trap collision-activated dissociation spectra. Little difference in the number of identifications was observed between spectra cleared of tag ions and those not; this step was not included in the analyses. Search results were filtered to a 1% false discovery rate based on E-values and mass error (21). The maximum mass error for all identified peptides was 15 ppm. Peptides were grouped into proteins and filtered to a 1% false discovery rate according to rules previously described (22). Tagging efficiency was calculated from peptide identification counts. The number of labels on primary amines was divided by the total number of amines.
Quantification
The general algorithm employed for the quantification of NeuCode peptide isotopologues is described elsewhere (15). This approach was modified in the following ways to accommodate sets of four isotopologues: sets of peaks were extracted from an m/z range extending from 10 ppm below the adjusted m/z for the lightest isotopologue to 10 ppm above the adjusted m/z for the heaviest isotopologue; to be eligible for quantification, a set had to contain at least two peaks that were properly spaced (i.e., within the spacing tolerance of the theoretical spacing ±3.5 mDa); and a noise-based value was added to missing isotopologue channels for peak sets meeting the abovementioned requirements. This algorithm was applied independently to isotopologue sets in different isotope clusters (e.g., for the 12-plex experiments). For protein quantification, the intensities for each isotopologue channel were summed for all a protein's observed peptides, and the resulting protein ratios were normalized such that the distribution median was centered at a ratio of 1. Resolution was not used as criterion for the acceptance of quantitative data. Known contaminants were not quantified.
RESULTS
Theoretical Considerations
NeuCode SILAC requires that the mDa mass signatures be embedded into individual amino acids. Amino acids, of course, offer a limited palette—that is, there are only so many atoms to adjust (23). For lysine, C6H14N2O2, up to 18 atoms can be adjusted—C6H9N2O1—after excluding exchangeable atoms and losses during incorporation. Achieving the highest plexing with NeuCode SILAC will require stable isotope labeled isotopologues that make use of all these atoms (i.e., C, H, N, and O). Some elements, however, have a greater mass defect than others, such as 12C/13C (+3.3 mDa), 1H/2H (+6.3 mDa), 16O/18O (+4.2 mDa), and 14N/15N (−4.2 mDa). Deuterium offers the largest mass defect; however, it also can induce subtle chromatographic peak shifts (24). We reasoned that deuterium should be avoided. Thus, we restricted our tag design to only use the differential incorporation of C and N isotopes. In this scenario, each concomitant exchange of a 13C atom for a 12C and a 14N for a 15N imparts a 6.3-mDa mass defect.
To evaluate the feasibility and practicality of this approach, we constructed a library containing 65,011 tryptic yeast peptides and generated (in silico) versions of each library entry that were spaced by multiples of the 6.3-mDa mass defect (6.3, 12.6, and 25.2 mDa). Next, we plotted the percentage of the proteome that was quantifiable (i.e., resolved, full-width at 10% maximum, FWTM) at resolving powers ranging from 15,000 to 106. The underlying calculation accounts for the decreased resolution at higher m/z, as well as the mass (m), charge (z), label mass, and number of primary amines of each precursor (Fig. 1A) (25). In this calculation, m/z peaks overlapping by up to 3.2% peak area (FWTM) are considered resolvable and, thus, quantifiable. Partially resolved peaks overlapping by >10% to 100% full width of maximum do not meet this criteria. The optimal mass defect produces the highest percentage of quantifiable identifications at the lowest resolving power and is also capable of the highest degree of multiplexing (i.e., smallest mass spacing). Although Orbitrap mass analyzers can achieve resolutions of over 106, current commercial implementations can routinely achieve up to 480,000 resolution (available when the Thermo Scientific Developer's software is installed) (26, 27). From this analysis we conclude that 12.6 mDa is the current optimal mass defect for incorporation into NeuCode tags. Although a mass defect of 6.3 mDa, the exchanging of a heavy N for a heavy C, would offer the highest plexing capacity, ∼35% of the peptides carrying this signature would be quantifiable. Doubling this spacing to 12.6 mDa will render 95% of the proteome quantifiable at the standard commercial resolution of 480,000. We note the 6.3-mDa spaced tags will be more accessible in the coming years, as the resolution of MS instruments is expected to continually improve. Fourier transform–ion cyclotron resonance–MS instruments offer the highest resolution and can analyze samples labeled with the NeuCode tags described here (28, 29). The resolution modeling for this instrument type is displayed in supplemental Fig. S1.
Label Characteristics
With these theoretical calculations in hand, we next sought to develop a proof-of-concept NeuCode tag with the following objectives: (1) demonstrate compatibility of the NeuCode method with chemical labeling, (2) deliver high plexing capacity, and (3) confirm that the quantitative accuracy and precision are comparable to those of other MS1-based quantification methods. Design of the tag structure must balance a number of considerations. The tag should have a limited effect on nano-HPLC-MS/MS parameters, such as peptide chromatographic retention, ionization, and fragmentation. Thus, the tag should be reasonably hydrophilic (hydrophilic amino acids) and contain a basic moiety that cannot be a primary amine. Further, the number of nitrogen atoms should be maximized while limiting label size (30). Incorporation of a large quantity of 15N into a tag is more challenging than the incorporation of 13C, simply because most molecules have fewer N atoms than C atoms.
We reasoned that amino acids present somewhat flexible building blocks from which to construct the NeuCode tagging reagents. Specifically, many isotope varieties of amino acids are commercially available and are readily combined by means of straightforward Fmoc solid-phase synthesis (31, 32). After considering a number of properties, we decided on the sequence acetylArg-acetylLys-Gly with an NHS ester at the C terminus (Fig. 1B). Arg and Lys were selected for their relatively high N content. In fact, Arg has the highest ratio of N to mass (4 N:175 Da) of any amino acid. The presence of Arg also ensures that the tag contains a positive charge. Gly was present on the solid-phase synthesis resin to simplify the initial coupling step. The acetyl groups block the primary amines and prevent the tag from polymerizing during NHS ester activation. The NHS ester moiety is reactive toward primary amines and is commonly used for conjugating labels to peptides prior to MS analysis (6, 7). Asp and Glu were avoided because of their potential to deamidate. Fig. 1C presents the isotope incorporation plan to generate a 4-plex NeuCode tag using the acetylArg-acetylLys-Gly tag scaffold. Our plan incorporates six heavy atoms in four combinations, 13C0 15N6, 13C2 15N4, 13C4 15N2, and 13C6 15N0, spanning 37.9 mDa. Using commercial components and standard Fmoc chemistry, we synthesized all four of these NeuCode tags and then examined a 1:1:1:1 mixture of them using a linear ion trap–Orbitrap hybrid mass spectrometer (Fig. 1D). At 15,000 resolving power, only a single isotopic cluster was observed, and the presence of the four isotopologues was cloaked; collection of the same MS1 scan at 480,000 resolution, however, revealed the four reagents, spaced by precisely 12.6 mDa. We note that, given its size, this label is intended not as a long-term strategy for the incorporation of mass defect signatures, but as a rather convenient scaffold to demonstrate the concept.
4-plex NeuCode Labeling
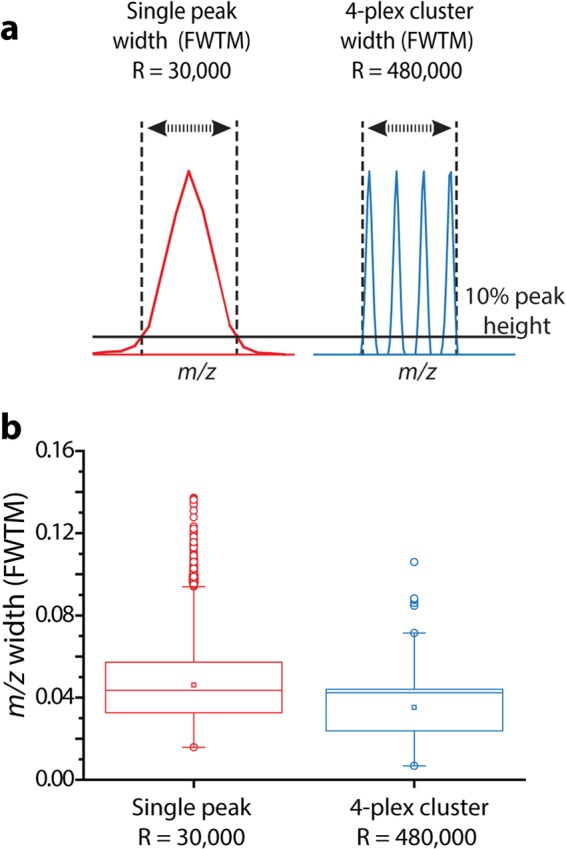
To evaluate the performance of these 4-plex NeuCode tags, we labeled four equivalent aliquots of a complex mixture of yeast tryptic peptides. We confirmed that each sample was >90% labeled, and then we mixed the labeled peptides in various ratios (1:1:2:5) and analyzed the mixture via nano-HPLC-MS/MS. The MS acquisition method included a fast MS1 survey scan (r = 30,000), an MS1 quantitation scan (480,000), and then MS/MS of the 10 most intense precursors (collision-activated dissociation) using the ion trap (note that in some instances we utilized HCD with Orbitrap mass analysis) (33). Previously, we found that the added time required for the high-resolution quantitation scan (∼1.6 s) can be mitigated by the use of the ion trap analyzer for MS/MS scanning (26). We directly compared peptides from a yeast tryptic digest LC-MS/MS analysis with a 60-min gradient elution using a method with a 480,000 resolving power scan and one with only a 30,000 resolving power scan. The 480,000 method resulted in 824 unique protein groups and 4497 unique peptide identifications from 17,150 MS/MS scans, whereas the 30,000 method resulted in 836 unique protein groups and 4725 unique peptides from 19,185 MS/MS scans (supplemental Fig. S2). Fig. 2 presents representative spectra and results from this experiment. Here we consider a precursor at m/z 950 that was selected for MS/MS analysis. The high-resolution quantification scan reveals the embedded NeuCode signature at the appropriate mixing ratios of 1:1:2:5, whereas the MS/MS scan, conducted in the low-resolution ion trap analyzer, shows backbone fragmentation and no evidence of the isotopologue fingerprint. From this experiment, 1738 peptides were confidently identified and 1448 were quantified (83%). We note that the overall MS/MS identification rate was somewhat depressed relative to that of unlabeled peptides, presumably because of the presence of the tag (see discussion below). For peptides that were confidently identified, however, the distribution of experimentally measured ratios was centered within ∼20% of the expected values (median measured peptide ratios = 1.0:1, 1.7:1, 4.0:1; Fig. 2C). The evaluation of median accuracy compared well with reported SILAC and isobaric labeling results. For example, when analyzed in a recent study, the median value for a SILAC 2:1 ratio was 2.9:1, and our own laboratory discovered that 5:1 ratios in isobaric tagging are often detected as much lower (2.5:1 on average) (13, 34). Additionally, calculated standard deviations for each measurement (0.5–0.7, log2) are in line with typical performances for both SILAC (0.35–0.7) and isobaric labeling (0.7–0.8) (13, 14, 34, 35). Precision is improved (0.5 to 0.4 for the 1:1 ratio) when protein values are analyzed, as multiple peptide measurements are often included for calculating a single protein ratio. Results from the 1:1 ratio indicate that 99% of proteins fall within a 100% fold-change (supplemental Fig. S3). An additional 1:2:5:10 known ratio mixture was analyzed (supplemental Fig. S4). Results for the 2:1 and 5:1 were nearly identical to that reported above. We do note that higher ratios exhibit some dynamic range depression and also correlate with decreased precision, just as is often observed in isobaric labeling and traditional SILAC measurements (34, 35). Interestingly, a 4-plex tag with 12.6-mDa spacing analyzed at 480,000 resolving power typically encompasses a similar m/z space as a single peak analyzed at 30,000 resolving power. This suggests that MS1 interference of a 4-plex NeuCode tag will be similar to that of a label-free or SILAC approach (Fig. 3). We conclude that, while there is room for improvement, the overall accuracy, precision, and dynamic range afforded by these NeuCode tags are comparable to what is provided by other current technology, and we anticipate that with continued efforts, improvements in tagging chemistry, instrument control methods, and quantitative software are likely to boost each of these key figures of merit.
Fig. 2.
NeuCode label example and quantitative figures of merit. A, MS1 scan collected from a nano-HPLC-MS/MS analysis of NeuCode 4-plex labeled yeast tryptic peptides mixed in the ratio of 1:1:2:5. The inset displays the 30,000 resolving power survey scan (black) and subsequent 480,000 resolving power quantitation scan (red) for a selected precursor having m/z of 950. B, annotated MS2 spectrum and sequence of the peptide from A, following collision-activated dissociation and ion trap m/z analysis. The inset displays that the SILAC pair is concealed at typical resolution. Fragment ions resulting from tag cleavage are denoted by asterisks. C, boxplots showing the measured (boxes and whiskers) and true (dashed lines) values for mixing ratios of 1:1, 1:2, and 1:5 (n = 1448). Boxplots demarcate the median (stripe), the 25th to 75th percentile (interquartile range, box), 1.5 times the interquartile range (whiskers), and outliers (open circles). The expected mixing ratio, median ratio, and standard deviation of all measurements for each ratio are listed below the boxplots.
Proteome Remodeling During the Diauxic Shift
With these proof-of-concept data in hand, we sought to test the performance of the 4-plex NeuCode tags in quantifying proteome changes during the diauxic shift, a metabolic transition from fermentation to aerobic respiration, in Saccharomyces cerevisiae. This experiment provides an excellent test bed, as the phenomenon has been thoroughly investigated at the genetic and molecular level, but not yet at the global proteome plane (36–38). Briefly, yeast can metabolize organic molecules for energy through two fundamentally different metabolic processes: fermentation and respiration. In the presence of abundant glucose, fermentation predominates. After consuming the available glucose, yeast restructure their metabolic machinery to enable respiration and to use fuel sources other than glucose. Yeast cultures undergo exponential growth in liquid media that contains abundant glucose, temporarily stop proliferating when the glucose is consumed, and then resume growth after a lag period, albeit at a slower rate. The changes in proliferation rate reflect complex changes in gene expression, protein abundance, and post-translational modifications during the diauxic shift (36–40).
Yeast were collected during log phase growth (3.3 h) while glucose was still available and at three time points following glucose exhaustion (5.5, 8.5, and 11.5 h; Fig. 4A). Yeast protein from these four samples was digested with trypsin and independently conjugated to the 4-plex NeuCode tags. Labeled peptides were combined and fractionated via high-pH reversed-phase chromatography and analyzed via nano-HPLC-MS/MS (supplemental Fig. S5). From these data, 1543 proteins were confidently identified (1% false discovery rate), enabling protein fold-change measurement over the time course. As anticipated, major proteomic transformations were observed: between the 3.3-h and 11.5-h time points, 281 proteins had abundances that changed by at least 2-fold (supplemental Table S1). Approximately one-third of the proteins whose abundances decreased (≥2-fold) during the diauxic shift were members of the 40S/60S ribosome, the major yeast cytosolic ribosome. The protein-level data confirm transcriptomic results for these ribosomal genes during the diauxic shift, a response that reflects the decreased availability of nutrients needed to fuel protein synthesis and rapid cell division (36). Alternatively, the abundances of the mitochondrial 37S/54S ribosomal proteins initially decreased but then increased following glucose depletion from the culture media (Fig. 4B) (41). The mitochondrial ribosome is essential for the translation of proteins encoded by mitochondrial DNA, including proteins that are required for oxidative phosphorylation. The observed increase in mitochondrial ribosome abundance may enable an increase in respiratory capacity in response to decreased glucose availability. This result is consistent with previous analyses of the transcriptome and individual proteins of the yeast mitochondrial ribosome, such as MRP20 and MRP49, which are more abundant in media without glucose (42).
Fig. 4.
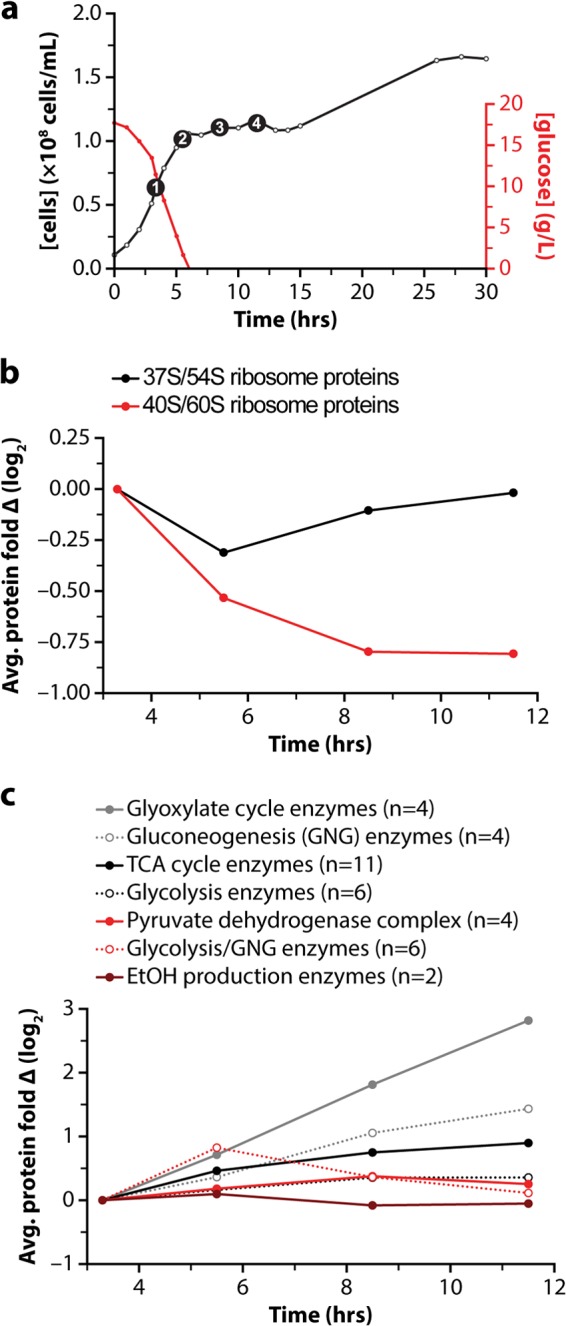
Proteome remodeling during the diauxic shift. A, growth curve for yeast cells (black) and plot of glucose concentration in the media (red) during the diauxic shift experiment. Each of the four selected time points is shown as a black circle. B, average expression change of quantified mitochondrial (red) and cytosolic (black) ribosomes over the time course. C, average expression change of all quantified proteins involved in specific carbon metabolism pathways.
A number of proteins whose abundances increased during the diauxic shift were highly regulated enzymes in carbon metabolism, such as phospho-enolpyruvate carboxykinase, a key enzyme in gluconeogenesis. To further characterize these changes, we compared the average fold changes of enzymes (supplemental Table S1) from multiple pathways of central carbon metabolism (Fig. 4C). As expected, enzymes involved in fermentation and ethanol production did not increase during the diauxic shift; however, enzymes of the tricarboxylic acid cycle and the glyoxylate cycle, metabolic pathways that are tightly linked to respiration, increased during the diauxic shift. Further reflecting the decrease in glucose availability that drives the diauxic shift, an increase in enzymes of gluconeogenesis was observed. These data demonstrate that the NeuCode tagging technology can accurately determine changes in protein abundance from complex biological systems.
12-plex NeuCode Labeling
A key benefit of multiplexed analysis is the ability to simultaneously acquire data across multiple time points and biological replicates. By incorporating biological replicate measurements, one can establish a quantitative false discovery rate to distinguish statistically significant biological differences (43, 44). We reasoned that our 4-plex NeuCode tag could be expanded to 12-plex via the superposition of nominal integer (∼4 Da) mass differences onto the 12.6-mDa separated 4-plex. To achieve this, we substituted no, four, or eight additional 13C atoms for 12C atoms in the mass defect labels described above (Fig. 5A). Peptides conjugated to these NeuCode labels will appear as three disparate isotopic distributions when analyzed at 30,000 resolution, similar to triplex SILAC; however, each isotopic cluster peak is resolved into a set of four discrete m/z peaks separated by 12.6 mDa (per tag) when analyzed at higher resolution (∼480,000; Fig. 5B). To test the performance of these 12-plex NeuCode tags, we analyzed the yeast diauxic shift time-course experiment in biological triplicate. Here, every isotopic cluster comprises quantitative data for each of the four time points of a complete biological replicate. From these data, 1056 proteins were detected, 760 of which were quantified across all 12 samples. The increased spectral complexity of the 12-plex experiments (three isotopic clusters) is likely responsible for the decreased proteomic coverage relative to the 4-plex analysis. Fig. 6 demonstrates the reproducibility (r2 values ranging from 0.70 to 0.80) for the NeuCode tagging method between biological replicates. Indeed, recent work with isobaric tagging indicates typical correlation for that chemistry of ∼0.70 (45). Next, we leveraged the replicate measurements to perform rigorous statistical analysis to identify significant biological differences (p < 0.05, Welch's t test with Storey correction). 85, 273, and 270 proteins significantly differed in abundance at 5.5, 8.5, and 11.5 h, as compared with 3.3 h, respectively (2-fold change and p < 0.05; Welch's t test with Storey correction for multiple hypotheses; supplemental Fig. S6). Interestingly, only four proteins were found changing, by the same criteria, between time points three and four, suggesting the yeast proteome had adjusted to the lack of glucose by the third time point, 3 h after glucose was depleted from the media. And, despite the tripled plexing capacity, the average standard deviation between biological replicates was 0.5 (log2 scale), commensurate with the results achieved for the 4-plex NeuCode tag and with both SILAC and isobaric tagging (13, 14, 34, 35). A summary of these proteins and their corresponding quantitative metrics can be found in supplemental Table S2.
Fig. 5.
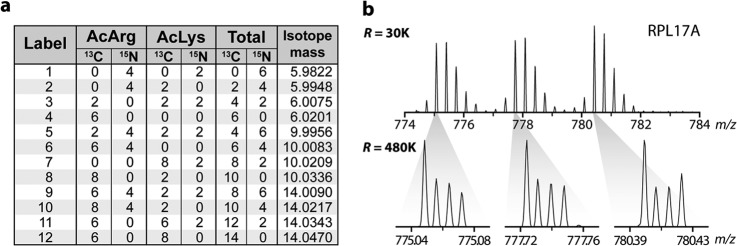
NeuCode 12-plex labels. A, specific isotopic compositions that encode the 12.6-mDa and 4-Da mass differences for each of the 12-plex labels. B, a peptide from RPL17A is observed as three isotopic distributions when analyzed with a 30,000 resolving power scan. A 480,000 resolving power scan (inset) reveals the mass differences imparted by the 12 labels.
Fig. 6.
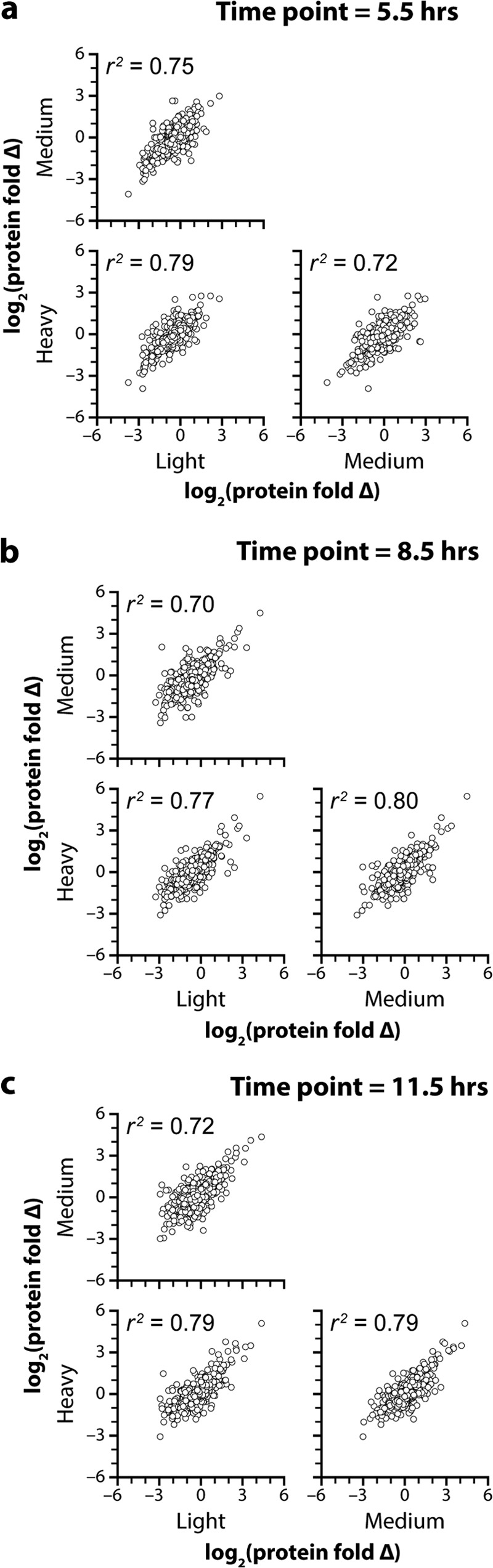
NeuCode 12-plex multi-scatter plots. Multi-scatter plots for each time point compared with the 3.3-h time point. The fold change for each protein with each nominal mass light (+0 Da), medium (+4 Da), and heavy (+8 Da) label is plotted against the fold change for the same protein, measured using a tag with the other two nominal masses. A, 5.5 h. B, 8.5 h. C, 11.5 h. The figure demonstrates that the NeuCode tagging method provides excellent reproducibility between biological replicates, as indicated by r2 values ranging from 0.70 to 0.80.
DISCUSSION
Here we describe a new approach to protein quantification, one that combines the MS1-based quantification of SILAC with the plexing and tissue compatibility of isobaric labeling. The method stems from our recent description of neutron encoded signatures that compress quantitative information into a single isotopic cluster. That work, however, used amino acids and metabolic labeling (NeuCode SILAC) to insert the mDa-spaced labels. Here we expand upon this concept via the design, synthesis, and analysis of NeuCode chemical reagents, permitting NeuCode quantitative analysis of virtually any proteomic sample, including human tissues and fluids. Using amino acid building blocks and standard Fmoc coupling chemistry, we employed a strategy that delivers 12-plex MS1-based quantitation with spectral complexity that is commensurate with triplex SILAC. The specific label structure presented in this work is intended solely as a near-term demonstration for the overall concept of NeuCode labeling and is not intended as a long-term strategy. Using both quantitative models and a biological system of yeast undergoing the diauxic shift, we demonstrated strong performance across all quantitative figures of merit. Briefly, quantitative accuracy, as determined by the fixed ratio experiment, revealed that NeuCode tags provided median measured peptide ratios within ∼20% of known values. Measured standard deviations (i.e., precision) ranged from 0.50 to 0.70, and the r2 correlation of biological replicate measurements (12-plex data) ranged from 0.7 to 0.8. Isobaric tagging, the only method offering comparable plexing capacity, requires some sort of gas-phase purification in order to achieve comparable quantitative accuracy, but even then, at best it offers equivalent precision relative to these NeuCode tags. As expected, given that both are MS1-based quantitative approaches, traditional SILAC and NeuCode tagging offer comparable precision (standard deviations of 0.4 versus 0.5 for a 2:1 mixture, SILAC versus NeuCode tags, respectively) and accuracy. Although beyond the scope of the current work, NeuCode tags should perform comparably for post-translational modification quantitation.
In addition to providing quantitative accuracy and precision comparable to those of SILAC and isobaric tagging, we conclude that our guiding supposition that the NeuCode strategy is indeed compatible with chemical labeling is correct, and that it promises to deliver highly plexed quantitation in an MS1-accessible format. The current tag structure, however, does reduce the overall number of peptide identifications relative to that for peptides having no tag. This result is primarily a function of the tag itself and not of the NeuCode data acquisition method. Supporting this conclusion, no significant loss in peptide identifications was incurred using the same data acquisition strategy for our NeuCode SILAC work (15). Further, ∼5% of tandem mass spectra bearing the NeuCode tags were mapped to sequence, compared with ∼30% for unlabeled peptides. Based on the structure of the NeuCode tag, this reduction in sequencing performance is not entirely unexpected. First, the tag adds five additional amide bonds to each primary amine. Fragmentation of any of these generates sequence-uninformative product ions. Second, the label is relatively large; ∼435 Da is added to each primary amine (i.e., one to two per peptide). Third, Arg is the most basic amino acid and may inhibit random backbone fragmentation by sequestering protons (46, 47). The reduction in proteomic coverage observed in the 12-plex experiments, as compared with the 4-plex data, is likely caused by increased spectral complexity; that is, three isotopic clusters are observed for every unique peptide sequence, so that lower abundance species are less often selected for MS/MS (45, 48). This exact phenomenon is observed during duplex and, especially, triplex SILAC.
Altogether, this study demonstrates that NeuCode chemical reagents will provide a direct route to very highly plexed MS1-based quantification, in a format (MS1) that offers improved accuracy over the current state-of-the-art isobaric tagging chemistry. We posit that future tag architecture should move away from amino acid building blocks and toward small-molecule organic synthesis with tunable 15N and 13C incorporation. Such a strategy will allow for maximal 15N incorporation, so as to increase the plexing capacity while reducing or even eliminating the number of amide linkages of the current tag. And as routine MS resolving power improves to 106, the required NeuCode spacing of 12.6 mDa will be reduced to 6.3 mDa, doubling the plexing capacity for a given tag system.
Supplementary Material
Footnotes
* This work was supported by National Institutes of Health Grant No. R01 GM080148 to J.J.C. A.E.M. gratefully acknowledges support from a National Institutes of Health–funded Genomic Sciences Training Program (5T32HG002760). D.J.P. was supported by a Searle Scholars Award and Shaw Scientist award. J.A.S. was supported by a NIH Ruth L. Kirschstein National Research Service Award (1F30AG043282).
 This article contains supplemental material.
This article contains supplemental material.
Author contributions: A.S.H. designed, performed research, analyzed data, contributed materials, and wrote the paper; A.E.M., contributed analysis tools and analyzed data; J.A.S. designed research, performed research, and analyzed data; D.J.B. contributed analysis tools; C.D.W. contributed analysis tools; M.S.W. analyzed data; D.J.P. designed research; J.J.C. designed research and wrote the paper.
Competing interests statement: A.S.H. and J.J.C. are co-inventors on patent applications (U.S. 13/660677) in part related to the material presented here.
1 The abbreviation used is:
- SILAC
- stable isotope labeling with amino acids in cell culture.
REFERENCES
- 1. Grimsrud P. A., Swaney D. L., Wenger C. D., Beauchene N. A., Coon J. J. (2010) Phosphoproteomics for the masses. ACS Chem. Biol. 5, 105–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altelaar A. F. M., Munoz J., Heck A. J. R. (2013) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48 [DOI] [PubMed] [Google Scholar]
- 3. Oda Y., Huang K., Cross F. R., Cowburn D., Chait B. T. (1999) Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 96, 6591–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang H., English A. M. (2002) Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J. Proteome Res. 1, 345–350 [DOI] [PubMed] [Google Scholar]
- 5. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 6. Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 7. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 8. Krüger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fässler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 [DOI] [PubMed] [Google Scholar]
- 9. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 10. Choe L., D'Ascenzo M., Relkin N. R., Pappin D., Ross P., Williamson B., Guertin S., Pribil P., Lee K. H. (2007) 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. Proteomics 7, 3651–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gouw J. W., Krijgsveld J., Heck A. J. R. (2010) Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteomics 9, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ow S. Y., Salim M., Noirel J., Evans C., Rehman I., Wright P. C. (2009) iTRAQ underestimation in simple and complex mixtures: “The Good, the Bad and the Ugly.” J. Proteome Res. 8, 5347–5355 [DOI] [PubMed] [Google Scholar]
- 13. Wenger C. D., Lee M. V., Hebert A. S., McAlister G. C., Phanstiel D. H., Westphall M. S., Coon J. J. (2011) Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 8, 933–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ting L., Rad R., Gygi S. P., Haas W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hebert A. S., Merrill A. E., Bailey D. J., Still A. J., Westphall M. S., Strieter E. R., Pagliarini D. J., Coon J. J. (2013) Neutron-encoded mass signatures for multiplexed proteome quantification. Nat Meth 10, 332–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Atwood J. A., Cheng L., Alvarez-Manilla G., Warren N. L., York W. S., Orlando R. (2007) Quantitation by isobaric labeling: applications to glycomics. J. Proteome Res. 7, 367–374 [DOI] [PubMed] [Google Scholar]
- 17. Sleno L. (2012) The use of mass defect in modern mass spectrometry. J. Mass Spectrom. 47, 226–236 [DOI] [PubMed] [Google Scholar]
- 18. Werner T., Becher I., Sweetman G., Doce C., Savitski M. M., Bantscheff M. (2012) High-resolution enabled TMT 8-plexing. Anal. Chem. 84, 7188–7194 [DOI] [PubMed] [Google Scholar]
- 19. McAlister G. C., Huttlin E. L., Haas W., Ting L., Jedrychowski M. P., Rogers J. C., Kuhn K., Pike I., Grothe R. A., Blethrow J. D., Gygi S. P. (2012) Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 84, 7469–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagaraj N., Alexander Kulak N., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. (2012) System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top orbitrap. Mol. Cell. Proteomics 11, M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., Coon J. J. (2011) COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 23. Molina H., Yang Y., Ruch T., Kim J.-W., Mortensen P., Otto T., Nalli A., Tang Q.-Q., Lane M. D., Chaerkady R., Pandey A. (2008) Temporal profiling of the adipocyte proteome during differentiation using a five-plex SILAC based strategy. J. Proteome Res. 8, 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ong S.-E., Foster L. J., Mann M. (2003) Mass spectrometric-based approaches in quantitative proteomics. Methods 29, 124–130 [DOI] [PubMed] [Google Scholar]
- 25. Makarov A., Denisov E., Lange O. (2009) Performance evaluation of a high-field Orbitrap mass analyzer. J. Am. Soc. Mass Spectrom. 20, 1391–1396 [DOI] [PubMed] [Google Scholar]
- 26. Denisov E., Damoc E., Lange O., Makarov A. (2012) Orbitrap mass spectrometry with resolving powers above 1,000,000. Int. J. Mass Spectrom. 325–327, 80–85 [Google Scholar]
- 27. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Müller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J.-J., Cox J., Horning S., Mann M., Makarov A. (2012) Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11, O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaub T. M., Hendrickson C. L., Horning S., Quinn J. P., Senko M. W., Marshall A. G. (2008) High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 Tesla. Anal. Chem. 80, 3985–3990 [DOI] [PubMed] [Google Scholar]
- 29. Marshall A. G., Hendrickson C. L. (2008) High-resolution mass spectrometers. Annu. Rev. Anal. Chem. 1, 579–599 [DOI] [PubMed] [Google Scholar]
- 30. Pichler P., Köcher T., Holzmann J., Mazanek M., Taus T., Ammerer G., Mechtler K. (2010) Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal. Chem. 82, 6549–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merrifield R. B. (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 [Google Scholar]
- 32. Sieber P. (1987) A new acid-labile anchor group for the solid-phase synthesis of C-terminal peptide amides by the Fmoc method. Tetrahedron Lett. 28, 2107–2110 [Google Scholar]
- 33. McAlister G. C., Phanstiel D., Wenger C. D., Lee M. V., Coon J. J. (2009) Analysis of tandem mass spectra by FTMS for improved large-scale proteomics with superior protein quantification. Anal. Chem. 82, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altelaar A. F. M., Frese C. K., Preisinger C., Hennrich M. L., Schram A. W., Timmers H. T. M., Heck A. J. R., Mohammed S. (2013) Benchmarking stable isotope labeling based quantitative proteomics. Journal of Proteomics 88, 14–26 [DOI] [PubMed] [Google Scholar]
- 35. Bakalarski C. E., Elias J. E., Villén J., Haas W., Gerber S. A., Everley P. A., Gygi S. P. (2008) The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. J. Proteome Res. 7, 4756–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DeRisi J. L., Iyer V. R., Brown P. O. (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686 [DOI] [PubMed] [Google Scholar]
- 37. Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D., Brown P. O. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Picotti P., Bodenmiller B., Mueller L. N., Domon B., Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Futcher B., Latter G. I., Monardo P., McLaughlin C. S., Garrels J. I. (1999) A sampling of the yeast proteome. Mol. Cell. Biol. 19, 7357–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haurie V., Sagliocco F., Boucherie H. (2004) Dissecting regulatory networks by means of two-dimensional gel electrophoresis: application to the study of the diauxic shift in the yeast Saccharomyces cerevisiae. Proteomics 4, 364–373 [DOI] [PubMed] [Google Scholar]
- 41. Graack H. R., Wittmann-Liebold B. (1998) Mitochondrial ribosomal proteins (MRPs) of yeast. Biochem. J. 329, 433–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fearon K., Mason T. L. (1992) Structure and function of MRP20 and MRP49, the nuclear genes for two proteins of the 54S subunit of the yeast mitochondrial ribosome. J. Biol. Chem. 267, 5162–5170 [PubMed] [Google Scholar]
- 43. Storey J. D. (2002) A direct approach to false discovery rates. J. R. Stat. Soc. Series B Stat. Methodol. 64, 479–498 [Google Scholar]
- 44. Phanstiel D. H., Brumbaugh J., Wenger C. D., Tian S., Probasco M. D., Bailey D. J., Swaney D. L., Tervo M. A., Bolin J. M., Ruotti V., Stewart R., Thomson J. A., Coon J. J. (2011) Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 8, 821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mertins P., Udeshi N. D., Clauser K. R., Mani D. R., Patel J., Ong S.-e., Jaffe J. D., Carr S. A. (2011) iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics M111.014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dikler S., Kelly J. W., Russell D. H. (1997) Improving mass spectrometric sequencing of arginine-containing peptides by derivatization with acetylacetone. J. Mass Spectrom. 32, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 47. Fuchs S. W., Sachs C. C., Kegler C., Nollmann F. I., Karas M., Bode H. B. (2012) Neutral loss fragmentation pattern based screening for arginine-rich natural products in Xenorhabdus and Photorhabdus. Anal. Chem. 84, 6948–6955 [DOI] [PubMed] [Google Scholar]
- 48. Boersema P. J., Aye T. T., van Veen T. A. B., Heck A. J. R., Mohammed S. (2008) Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8, 4624–4632 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.