

Abstract
Determining the half-life of proteins is critical for an understanding of virtually all cellular processes. Current methods for measuring in vivo protein stability, including large-scale approaches, are limited in their throughput or in their ability to discriminate among small differences in stability. We developed a new method, Stable-seq, which uses a simple genetic selection combined with high-throughput DNA sequencing to assess the in vivo stability of a large number of variants of a protein. The variants are fused to a metabolic enzyme, which here is the yeast Leu2 protein. Plasmids encoding these Leu2 fusion proteins are transformed into yeast, with the resultant fusion proteins accumulating to different levels based on their stability and leading to different doubling times when the yeast are grown in the absence of leucine. Sequencing of an input population of variants of a protein and the population of variants after leucine selection allows the stability of tens of thousands of variants to be scored in parallel. By applying the Stable-seq method to variants of the protein degradation signal Deg1 from the yeast Matα2 protein, we generated a high-resolution map that reveals the effect of ∼30,000 mutations on protein stability. We identified mutations that likely affect stability by changing the activity of the degron, by leading to translation from new start codons, or by affecting N-terminal processing. Stable-seq should be applicable to other organisms via the use of suitable reporter proteins, as well as to the analysis of complex mixtures of fusion proteins.
The regulation of protein stability is critical in order for cells to maintain proper functioning of almost every process. Thus, approaches for measuring the in vivo stability of a protein are essential for the identification of components in proteolytic pathways that affect protein turnover and for an understanding of the consequences of their activities. These approaches include traditional biochemical methods such as the Western blot, in which samples taken from time points after the inhibition of protein expression are fractionated via gel electrophoresis and the relevant protein is visualized with the use of an antibody. Another method allows one to track the degradation rate of newly synthesized proteins by metabolically labeling proteins with a radioisotope and following their radioactivity. A third method fuses a protein to a reporter enzyme like β-galactosidase, allowing the steady-state level of a protein to be measured via the enzymatic activity of the reporter enzyme. However, these small-scale methods are limited in the number of samples that they can analyze.
Large-scale methods have been developed that allow the simultaneous quantitation of the in vivo stability of many proteins. For example, Yen et al. fused ∼8,000 human proteins to green fluorescent protein (GFP)1 and followed the amount of each protein over time by using fluorescence-activated cell sorting (FACS) (1). To identify proteins, the plasmids encoding the GFP fusions were isolated and PCR products derived from these plasmids were hybridized to a DNA microarray. This method was applied to identify the substrates of a ubiquitin ligase complex (2). However, this method is limited by the number of bins into which protein fusions can be sorted in the FACS analysis and, consequently, how fine changes in stability can be discriminated. Alternatively, quantitative mass spectrometry has been used to analyze the stability of native proteins (3), but this approach often requires costly labeling and extensive data analysis. Moreover, these large-scale methods generally cannot distinguish differences in in vivo stability, which are sometimes significant, that result from small changes in a protein, such as single amino acid substitutions.
We present a method, Stable-seq, for measuring the in vivo stability of large numbers of variants of a protein that combines a simple genetic selection with high-throughput DNA sequencing. Stable-seq is a form of deep mutational scanning (4, 5), in which a physical association between each protein variant and the DNA that encodes it allows DNA sequencing to score the frequency of each plasmid in a population. Here, our strategy is to fuse protein variants to a stable biosynthetic enzyme, the stability of which becomes dependent on the stability of the attached variant. Plasmids encoding these fusions are transformed into cells, and the activity of the enzyme is then selected for. The selection results in the enrichment or depletion of each plasmid (5) based on the stability of the fusion protein that it encodes, which in turn determines the cell's growth rate. The frequency of each plasmid in the population before and after selection is determined by DNA sequencing. The ratio of the selected frequency to the input frequency, called the enrichment score, serves as a proxy for in vivo stability (Fig. 1A).
Fig. 1.

Overview of Stable-seq. A, variants of a protein are fused to a biosynthetic enzyme that serves as a reporter protein. The variants determine the stability of the reporter, and thereby the growth rate of yeast. A library of plasmids encoding variants fused to such a reporter is constructed, transformed into yeast, and selected for reporter function. Plasmids isolated before and after selection are subjected to high-throughput sequencing. The change in the frequency of each variant is a measure of its stability. B, library design and sequence of Deg1. Residues 3–34 selected for doping to generate a Deg1 mutant library are highlighted in yellow.
We demonstrate the Stable-seq method via the analysis of a degron, which is a protein degradation signal recognized by the proteolytic machinery (6). We fused the well-characterized degron Deg1 from the yeast Matα2 protein (7) to the yeast Leu2 protein, which is necessary for leucine biosynthesis (Fig. 1B). Matα2 and Mata1 are transcriptional repressors required in order to specify mating type in Saccharomyces cerevisiae. When Matα2 forms a heterodimer with Mata1 in diploid a/α cells through its C-terminal tail, it is relatively stable, but it becomes short-lived in haploid α cells through degradation by the ubiquitin proteasome system (8). Deg1, which spans the N-terminal 67 residues of Matα2, is recognized by the E3 enzyme Doa10 (9). In the fusion protein, the stability of Leu2 becomes dependent on Deg1. Thus, any mutation in Deg1 that increases its stability results in the presence of more Deg1–Leu2 fusion protein and increased production of leucine. The additional leucine leads to increased growth of yeast, and thus more copies of the Deg1 sequence containing this mutation. Using this approach, we analyzed the effect of ∼30,000 mutations in Deg1 and identified Deg1 features that affect stability. This approach should work in other organisms with appropriate selections, and it could be scaled up to measure the stability of many different proteins in parallel.
EXPERIMENTAL PROCEDURES
Plasmids and Strains
The p416TEToff-Deg1-LEU2 plasmid was constructed as follows. The SacI site 5′ of the GPD1 promoter in p415GPD was changed to an NheI site to replace the GPD1 promoter with the TEToff promoter cassette from pCM182 (tTA transcriptional activator, CMV promoter, ADH1 terminator, TetO operator, CYC1 promoter, Multiple Cloning Sites, and CYC1 terminator) (10). The LEU2 and Deg1 (Matα21–67) sequences were sequentially cloned into the Multiple Cloning Sites to generate p416TEToff-Deg1-LEU2. A linker (encoding PRRSG) is present between Deg1 and LEU2. Site-directed mutagenesis was carried out for synonymous codon changes in Deg1 to generate a HindIII site at residue 38 and at residues 42–45 to optimize the annealing temperature for an Illumina sequencing primer (Fig. 1B and supplemental Table S1). Deg1-LEU2M1Δ was generated via site-directed mutagenesis to remove the codon for the first methionine in LEU2. FLAG epitope-tagged versions of LEU2, Deg1-LEU2, and Deg1-LEU2M1Δ were cloned into the plasmid with the p416TEToff promoter. Point mutant constructs in Deg1 described in Figs. 3C, 5B, and 6B and in supplemental Figs. S2B and S2C were generated via site-directed mutagenesis. All the clones were confirmed by Sanger sequencing. Detailed plasmid maps and sequences are available upon request. Yeast strain BY4741 and isogenic yeast deletion strains are from Open Biosystems.
Fig. 3.

Selection assay and sequence analysis of Deg1–Leu2M1Δ stability. A, a library of Deg1–Leu2M1Δ variants transformed into yeast and plated without (-Ura) and with (-Leu -Ura) selection for stabilized Leu2M1Δ. 100 times more cells were plated on the selection plate for comparison. B, heat map of enrichment scores of single mutations, with the Deg1 residue numbers along the top (residues in which mutations identified by Johnson et al. (7) were found are shown in boxes) and all possible mutations on the left axis. In the heat map, wild-type Deg1 sequences are shown; mutations identified by Johnson et al. (7) are indicated with black squares, and missing data with gray squares. C, previously identified stabilizing mutations in Deg1. The growth of colonies in the spotting assay and the data from the sequencing are compared to β-galactosidase values of Deg1–β-galactosidase variants identified by Johnson et al (7). The mutations identified by Johnson et al. (7) resulted in increases in stability, measured by β-galactosidase or pulse-chase assays, as shown (β-gal). The log2E values are enrichment scores calculated from DNA sequence data: variant frequencies after leucine selection were divided by frequencies in the input library and then normalized to the wild-type ratio of frequencies.
Fig. 5.

N-terminal processing and its effect on in vivo protein stability. A, enrichment scores of mutations in codon 2. B, spotting assay of the N2 mutants with the highest log2E scores identified in Fig. 5A. Changes to Lys or Arg resulted in good growth on the -Leu -Ura plate. C, effect of the nat3Δ allele on Deg1–Leu2M1Δ production. BY4741 and nat3Δ strains carrying Deg1–Leu2M1Δ variants were spotted on control and selection plates.
Fig. 6.

Analysis of epistatic effect of double mutants on protein stability. A, histogram of epistasis scores from 17,196 double mutants. B, spotting assay of double mutants with large positive epistasis, along with the constituent single mutants.
Construction of the Deg1 Mutant Library
An oligonucleotide encoding amino acids 3–34 of Deg1 was synthesized by Trilink Biosciences (Deg13–34 library). In the synthesis, the variable region was doped with 2.088% (0.696% of each non-wild-type nucleotide) to generate on average two nucleotide changes per clone. The oligonucleotide was made double-stranded and then PCR amplified using iProofTM High-Fidelity DNA Polymerase (Bio-Rad) for 15 cycles. Gel-purified PCR fragments were digested with NotI and HindIII and used to replace the wild-type sequence in p416TEToff-Deg1-LEU2M1Δ to generate a library of ∼170,000 variants of the Deg1 sequence (input library). The Deg1N2 library was constructed with an oligonucleotide containing random bases (NNN) at the second codon (Asn) of Deg1, which was cloned into p416TEToff-Deg1-LEU2M1Δ as described for the Deg13–34 library.
Deg1 Library Selection and High-throughput Sequencing
Plasmid DNA of the Deg13–34 library was transformed into yeast strain BY4741 via the lithium acetate method to generate 1.1 × 106 transformants (11, 12). Only moderate transformation efficiency (<0.5%) was achieved in an effort to avoid the co-transformation of two variant plasmids into a single cell, which was determined to occur <2% of the time. Approximately 70,000 yeast transformants were plated onto 20 SC-Leu-Ura (13) 15-cm plates and incubated at 30 °C for 3 days. Colonies were scraped off the plate and used to prepare DNA via the zymolase method. Briefly, yeast cells were lysed with Qiagen Buffer P1 supplemented with 50 mm DTT and 400 μg/ml zymolase 20T at 37 °C for 2 h with occasional shaking, and lysing was followed by incubation at −80 °C for 30 min and thawing at 42 °C for 1 min. The QIAprep Spin Miniprep Kit (Qiagen) protocol was used to elute plasmid DNA contaminated with yeast genomic DNA. Genomic DNA was digested with Exonuclease I (Affymetrix) and lambda exonuclease (New England Biolabs) and removed by a Zymo DNA Clean & ConcentratorTM-5 (Zymo Research) to enrich plasmid DNA in the eluate (selection library). To count the number of plasmids in each pool, we amplified the variable region of Deg1 from input and selection libraries via PCR using iProofTM High-Fidelity DNA Polymerase (Bio-Rad) for 15 cycles, and we sequenced this fragment by MiSeq and HiSeq2000 (Illumina) using the primers listed in supplemental Table S1. The Deg1N2 library was assayed via same procedures as the Deg13–34 library.
Analysis of Sequence Data
The identity of each mutation and its frequency in the input and selected libraries were determined by the Enrich software package (14). We used the E. coli plasmid library of Deg1–LEU2 variants as the input library, because for yeast transformants, even in SC-Ura media (containing 80 mg/l leucine), there was a growth advantage to yeast cells that produced more leucine from a stabilized Deg1. To ensure the quality of sequencing reads, we used paired-end sequencing to read both directions and applied a stringent quality filter. The quality filter included (i) a minimum quality score higher than 20 at every cycle, (ii) fewer than four consecutive mutations, (iii) no ambiguous sequencing bases, and (iv) minimum read counts in the input library of at least 15. The enrichment score (E) of each mutant was calculated as Rm/Rw, with Rm being the frequency of the mutant (m) in the selection library divided by its frequency in the input library, and Rw the frequency of the wild type (w) in the selection library divided by its frequency in the input library. This normalization avoids the bias due to different plasmid frequencies in the input population. Separate aliquots of cells from the selection plates were prepared for sequence analysis by either MiSeq or HiSeq2000. Read counts of each variant from the MiSeq and HiSeq2000 runs were highly correlated (R2 of 0.9999 and 0.9816 for the input and selection libraries, respectively). Therefore, the sets of sequencing data were merged for further analyses. Epistasis scores were calculated as log2Em1,2 − (log2Em1 + log2Em2), where m1,2 represents a double mutant (m1, first single mutant; m2, second single mutant). Epistatic interactions (supplemental Fig. S3) were visualized with a custom script using the D3.js java script library. Other computational and statistical analyses were performed with in-house python scripts and the R statistical package.
Yeast Cell Growth Assay
To determine the range of in vivo protein stability assayed with Deg1–Leu2 fusions, we monitored the growth rates of yeast cells containing variant fusion constructs in liquid culture over 60 h (Synergy H1, BioTek, Winooski, VT). For the spotting assay, yeast cells containing variant Deg1–LEU2 constructs were grown in SC-Ura media, and equal amounts of cells (OD = 0.5) were spotted onto the plates with 5-fold serial dilutions. The plates were incubated at 30 °C for 1 to 3 days.
Immunoprecipitation and Western Blotting
Equal amounts of yeast cells containing Deg1-LEU2-FLAG constructs were harvested from an exponentially growing culture in a nonselective media (SC-Ura) and lysed with a bead-beater. Cell extracts were equalized using the Bradford assay, and FLAG-tagged Deg1–Leu2 proteins were immunoprecipitated with FLAG-M2 agarose beads (Sigma A2220). The immunoprecipitates were separated via 10% SDS-PAGE, and a Western blot was visualized using anti-FLAG (Sigma F3165), True-Blot® (Rockland Immunochemicals Inc., Gilbertsville, PA) to reduce the signal from the IgG heavy chain (which is similar in size to Deg-Leu2-FLAG), and ECL reagents (GE Healthcare RPN2106).
RESULTS
Strategy for Measuring the Stability of a Protein by Fusing It to a Reporter Protein
We first sought to confirm that fusion of Deg1 to Leu2 generates a fusion protein with a rapid turnover. We tested the growth rate of cells containing Deg1–Leu2 variants via a spotting assay in which 5-fold serial dilutions of a yeast culture were plated on control (-uracil) or selection (-leucine -uracil) plates (Fig. 2A). Yeast expressing Leu2 with no Deg1 sequence grew well under selection, but the expression of Deg1–Leu2 resulted in only moderately reduced growth under selection. We surmised that this modest reduction was due to translation of the fusion downstream from the initiator codon. Although Deg1 contains no methionine except for the initiator, translation could begin from the next in-frame methionine, the start codon of Leu2, especially given the poor context (15) for the Deg1 initiator in this fusion construct. Deletion of the first methionine of Leu2 (Deg1-Leu2M1Δ) nearly eliminated the growth of yeast under selection (Fig. 2A), indicating that Leu2 had been made unstable by its fusion to Deg1.
Fig. 2.
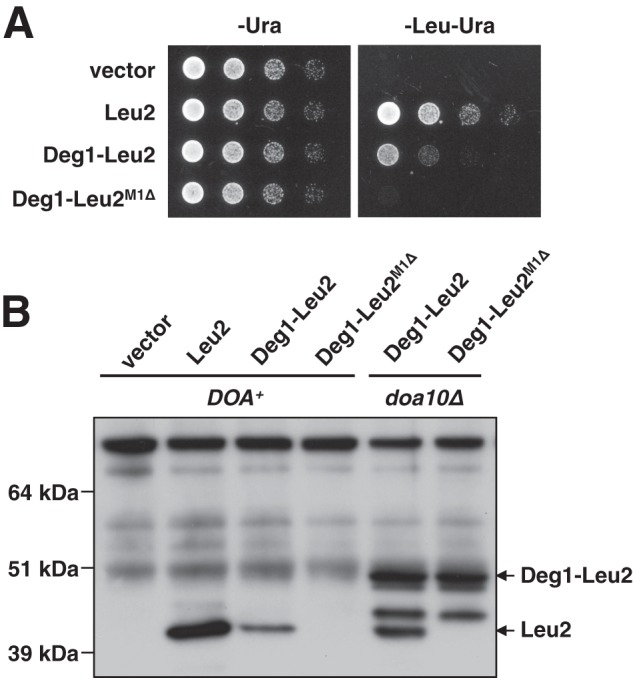
Verification of the Stable-seq assay. A, spotting assay of Deg1–Leu2 variants with 5-fold serial dilutions. Growth on the -Ura plate, which requires only the presence of the URA3 transformation marker, serves as the spotting control, and growth on the -Leu -Ura plate selects for stable versions of Leu2. B, Western blot analysis of C-terminally FLAG-tagged Deg1–Leu2 variants in DOA+ and doa10Δ cells. The full-length Deg1–Leu2 and Deg1–Leu2M1Δ proteins are unstable in DOA+ cells, but Deg1–Leu2 produces a Leu2-sized band. Both Deg1–Leu2 and Deg1–Leu2M1Δ produce a full-length band in doa10Δ cells, as well as a smaller band that runs between Deg1–Leu2 and Leu2 that is likely due to cleavage of the full-length protein.
The use of the alternative start codon likely occurred by leaky scanning of the ribosome (15). Initiation codon selection by the eukaryotic ribosome is often determined by the context surrounding the first AUG of the open reading frame. In S. cerevisiae, a 5′-untranslated region rich with A's, especially an A at position -3, is highly favored (15). When this context is not favorable, the next AUG in a better context may be used as an alternative start codon. In the case of Deg1–Leu2, an unfavorable GCGGCCGC precedes the first AUG. The use of the LEU2 AUG was confirmed by Western blot analysis of FLAG epitope-tagged Deg1–Leu2 variants (supplemental Fig. S1A). In DOA+ cells expressing Deg1–Leu2, only a band the size of Leu2 was apparent, whereas these cells expressing Deg1–Leu2M1Δ showed no detectable Leu2 band (Fig. 2B), consistent with the inability of these cells to grow in selection media (supplemental Fig. S1A). In doa10Δ cells, in which the degron is not targeted for degradation, the Deg1–LEU2 plasmid produced a protein consistent with translation starting from the initiator methionine of Deg1 and another protein the size of Leu2; however, the Deg1–LEU2M1Δ plasmid produced the larger species but no Leu2-sized protein (Fig. 2B). The degradation of the Deg1–Leu2 fusion was dependent on both an E3 (Doa10) and an E2 (Ubc7), like other Deg1 fusion proteins (8, 9) (supplemental Fig. S1B and Fig. 2B). Given the failure of Deg1–LEU2M1Δ to provide sufficient Leu2 function, stable variants of Deg1 should result in the production of more leucine, faster growth of yeast on selection media, and thus more copies of the plasmid encoding these variants. We demonstrated that even highly stable variants do not saturate the assay (supplemental Fig. S1C).
Stable-seq Analysis of a Deg1 Mutant Library
To apply the Stable-seq method to many Deg1 variants simultaneously, we used a doped oligonucleotide to mutate residues 3 to 34 of Deg1 fused to Leu2M1Δ (Fig. 1B), generating a library of ∼170,000 Deg1 variant plasmids. Yeast cells transformed with this library formed colonies of similar size if no selection for leucine was imposed, but the transformants produced far fewer colonies, of different sizes, on a selection plate (Fig. 3A). We interpret the leucine selection results as support for the rationale that colony size is dependent on the amount of Deg1–Leu2M1Δ fusion protein, which in turn is determined by the stability of Deg1. We harvested the cells from selection plates, isolated plasmids, and sequenced the DNA encoding Deg1. Comparing the frequency of each variant in the selected yeast to that in the input plasmid library allowed us to assay the effect of mutations on the stability of ∼30,000 variants of Deg1 (supplemental Table S2). Mutations present in the input library but not present after selection might be the result of extreme instability, or they may have been lost due to the limited number of colonies sampled after selection.
Fig. 3B shows the log2 enrichment scores of the single mutations observed, covering 71% of all the possible single mutations. Of the 13 previously identified mutations (7) for which we had DNA sequence data, 10 had positive log2 enrichment scores (>1.5). These scores indicate that the mutations increased in frequency after selection, in accordance with their behavior in the spotting assay (Fig. 3C). The Deg1 residues in which these previously identified mutations occur in several cases could also be mutated to other amino acids with similar or greater enrichment scores (e.g. F18D, F18N, S20N, I22G, L29D, and I32D). In addition to mutations in the previously identified residues, we also found novel mutations with high enrichment scores, including D8R, S21Q, and K27S (Fig. 3B).
In order to compare the scores obtained via Stable-seq with those determined via a previous approach, we examined the growth under leucine selection of cells expressing Deg1–Leu2 that contained one of 13 characterized mutations in Deg1 that lead to stabilization, which had been identified based on the β-galactosidase activity of a Deg1–β-galactosidase fusion (7). These mutations resulted in better growth on the selection plate for cells carrying 10 of these variants (Fig. 3C). The lack of correlation for the other three variants (K19Q, S20P, and I32S) may be due to the different behavior of the Deg1–Leu2 versus the Deg1–β-galactosidase fusion protein in different expression systems and assays.
Unlike the previous small-scale study (7), we also identified mutations with negative log2 enrichment scores. These less stable mutants included more than 60% of all the single mutants observed (supplemental Fig. S2A). For example, C33F showed greater instability than wild-type Deg1 (supplemental Fig. S2B), but the degradation was still dependent on Doa10 (supplemental Fig. S2C). These variant Deg1 sequences may be better recognized by the E3 ligase Doa10 than the wild-type Deg1.
Alternative Start Codons and Their Effect on Protein Stability
Changes to methionine at 10 positions, between residues 14 and 32, showed a strong stabilizing effect (Fig. 4A). The sequencing data for several mutants containing a new methionine were also confirmed by a spotting assay (Fig. 4B). It is likely that the new methionines serve as alternative start codons via a leaky scanning mechanism (15). The effect on the stability of truncated Deg1 variants due to alternative start codons correlated well with the results of a previous deletion study (7). Support for this interpretation also comes from 33 double mutants that combine a stop codon with a new methionine C-terminal to this stop (Fig. 4C), which likely initiate or reinitiate translation at the new methionine. Of these double mutants, 21 had a positive log2 enrichment score. The location of the new methionine, or the distance between the stop codon and the new methionine, did not correlate with the enrichment score (data not shown), but the double mutants with a stop codon at residue 17 followed by a methionine strongly stabilized Deg1, suggesting that the underlying mechanism is more complex.
Fig. 4.

Alternative start codon and its effect on in vivo protein stability. A, single mutant changes to methionine at each of residues 3–34 are represented with a heat map and bar plot. B, spotting assay of mutants containing an alternative start codon. These mutants were isolated from the pilot experiments to confirm the quality of the doped oligo library and to verify the assay design, and they include additional mutations. I4M; F30Y, I14M; F18I, I22M; D16A, I25M; S21R, L29R, I32M; D16A. C, heat map of double mutants containing a stop codon followed by a new methionine. Enrichment scores are represented as in Fig. 3B. Missing data are in gray.
N-terminal Processing Effects on in vivo Protein Stability
N-terminal acetylation is the major post-translational modification in eukaryotes, with more than 50% of the proteins in S. cerevisiae undergoing this modification (16). At the N terminus of a protein, the initiator methionine is removed if the second residue is small enough (e.g. Ala, Ser, Thr, Val, or Cys) to be accessed by methionine aminopeptidases, and the exposed second residues are acetylated by the NatA complex (16, 17). Initiator methionines followed by a larger residue are often acetylated by other N-terminal acetyltransferases (e.g. NatB or NatC) depending on the property of the second residue (16, 17). N-terminal acetylation has been proposed as another type of degradation signal based on work using Deg1 fusion proteins as model substrates (18). This concept was also explored with physiological substrates (19, 20), which further expanded the functions of the N-end rule pathway (21, 22).
To determine whether the Stable-seq method could detect changes that affect N-terminal processing, we generated another library that had a random nucleotide sequence specifying only the second residue (Asn) of Deg1 (Deg1N2 library), and we subjected the library to the same assay as the Deg13–34 library. We analyzed all 20 amino acids, as well as the stop codon, and found that the strongest increases on stability were due to the mutations N2K and N2R (Figs. 5A and 5B), consistent with a lack of N-terminal acetylation when the second residue is basic (18).
Acetylation of the wild-type Deg1 occurs via the action of NatB (18). Nat3 is the catalytic subunit of the NatB N-terminal acetyltransferase complex that acts on Matα2. Deletion of the NAT3 gene resulted in stabilization of the Deg1–Leu2M1Δ fusion (Fig. 5C), as observed for another Deg1 fusion (18). This stabilization is consistent with the role of N-terminal acetylation in protein degradation as proposed by Hwang et al. (18).
Epistatic Effects Observed in Double Mutants
In addition to yielding single mutants for analysis, the Stable-seq method also generated data for >17,000 variants (58% of the total variants observed) that contain two mutations. Double mutants can be examined for epistasis, in which the interaction between two mutations causes the double mutant to behave unexpectedly given the behavior of the two constituent single mutations. We used a multiplicative predictive model in which epistasis scores were calculated by subtracting the sum of the log2 enrichment scores of each single mutant (predicted stability) from the log2 enrichment score of the double mutant (observed stability) (Fig. 6A). Based on this model, positive epistasis indicates that the double mutant displayed more stability than predicted, and negative epistasis the opposite. We examined a few double mutants that showed positive epistasis via a spotting assay and validated these unexpected increases in stability, including cases in which neither single mutation alone resulted in an increase in stability (Fig. 6B).
We further analyzed the double mutants with the most positive epistasis scores (highest 1%) and the most negative epistasis scores (lowest 1%) (supplemental Fig. S3). Positive epistasis in the highest 1% was distributed among many different mutations, with no single mutation accounting for a large fraction of the total number of epistatic interactions. However, much of the negative epistasis in the lowest 1% could be accounted for by a small number of mutations. In most of these cases, the strong stabilizing effect of one mutation in this small grouping was not further increased by the presence of many different second stabilizing mutations. Some of the mutations in the small grouping are changes to a methionine, and others occur in the set of 13 previously identified mutations (7).
Clustering of Read Counts
We analyzed the effect of the proposed mechanisms of stabilization for all the single mutations with positive log2 enrichment scores. These mutations were clustered into five groups, with the median enrichment scores and ranges of each shown by a boxplot (Fig. 7A). Mutations that generate a new methionine or that had been previously identified in the study by Johnson et al. (7) resulted in the highest median enrichment scores. Although there are relatively few mutations in these two groups, they account for more than half of the sequence reads of enriched mutations from the selection library (Fig. 7B).
Fig. 7.
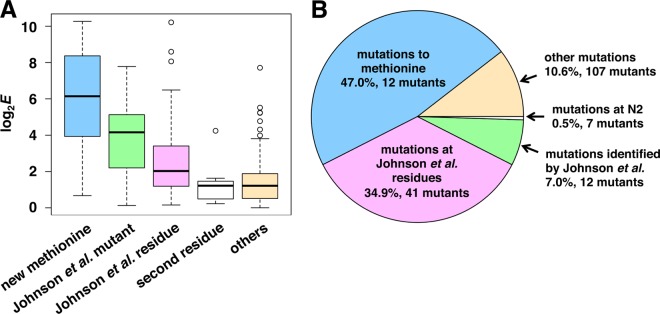
Prevalence and enrichment scores of stabilizing mutations. A, boxplots of groupings of stabilizing single mutations. Median values of log2 enrichment scores are represented with thick black lines. The upper and lower quartiles (interquartile range (IQR)), maximum and minimum values except outliers, and outliers (greater or less than 1.5 times the IQR) are indicated with boxes, whiskers, and circles, respectively. B, fractions of sequence read counts of single mutations that stabilize are represented. Stabilizing mutations are grouped as previously identified by Johnson et al. (7); novel mutations in the same residues in which the mutations identified by Johnson et al. (7) were found; mutations that generate a new methionine, which likely serves as an alternative start codon; mutations at the second residue that may affect N-terminal processing and acetylation; and other stabilizing mutations. Data for codon 2 mutations are from the Deg13–34–Leu2 library.
DISCUSSION
Here, we provide a method, Stable-seq, that uses high-throughput DNA sequencing to assess in vivo protein stability. We show that Stable-seq can identify key features of a protein domain that affect stability, including mutations that affect the full-length domain, alternative start codons that likely truncate the domain, and mutations that appear to affect N-terminal acetylation. The strong correlation between the scores generated via DNA sequencing and the stabilities of Deg1–β-galactosidase variants determined via β-galactosidase assay or pulse-chase analysis (7) indicates that the high-throughput Stable-seq assay is measuring stabilities in a useful range. Moreover, Stable-seq does not require the use of multiple time-points to calculate protein stability as other methods do, and its use of DNA sequencing reactions to compare input and selected populations allows a fine-grained discrimination of protein stability.
Changes of internal residues to a methionine can serve as an alternative start codon. Through this process, the protein becomes truncated at the N terminus, which for Deg1 results in protein stabilization, because the degron is no longer functional. This proposed mechanism is supported by double mutants that contain an upstream termination codon followed by a new methionine. We also found that the identity of the second residue affects stability. This mechanism is likely mediated by whether or not N-terminal acetylation occurs, as the Deg1–Leu2M1Δ fusion is stabilized in the nat3 mutant, which does not carry out NatB-mediated acetylation. Both of these mechanisms may interact together in complex ways, as each new methionine that serves as an alternative start codon is coupled to a new residue in the second position.
We identified double mutants that showed much greater or lesser stability than would be expected based on the behavior of the constituent single mutants. A mutation to a methionine generally showed negative epistasis when it combined with another mutation at an upstream location (supplemental Fig. S3, bottom panel), indicating that the double mutant was less stabilized than expected. These results support the idea that the new methionine serves as an alternative start codon, because the N-terminally truncated Deg1 that initiates from the new methionine would not contain the upstream stabilizing mutation.
Stable-seq is based on the assumption that the transcription and translation of variants are the same, and thus the level of the nutritional marker is dependent solely on the stability of the variants. However, it is possible that other factors will influence the function of the metabolic enzyme (e.g. Leu2); for example, mutations could change the folding of the enzyme or protein–protein interaction. When the method is applied to assay diverse proteins simultaneously, the presence of factors such as different protein localization signals will make additional controls necessary in order for the resultant stabilities to be validated.
Stable-seq could be adapted to analyze the stability of other degrons and other proteins. The method could be scaled up to handle large complements of proteins simultaneously if libraries of Leu2 fusions with random genomic or cDNA inserts or a collection of defined open reading frames were assayed. Stable-seq should also be amenable to other model organisms or to tissue culture cells if appropriate selection markers such as proteins that confer drug resistance are used.
Supplementary Material
Acknowledgments
We thank members of the Fields lab for help with the computational analyses and experimental procedures. We thank M. Hochstrasser, M. Dunham, and R. Gardner for yeast strains and Douglas Fowler, Christine Queitsch, James Bruce, and Hai Rao for critical reading of the manuscript. S.F. is an investigator of the Howard Hughes Medical Institute.
Footnotes
* This work was supported in part by Grant No. P41 GM103533 from the NIGMS, National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- E2
- ubiquitin-conjugation enzyme
- E3
- ubiquitin-protein ligase
- FACS
- fluorescence-activated cell sorting
- GFP
- green fluorescent protein
- log2E
- log2 enrichment score
- RX
- ratio of the frequency of the x sequence in the selection library to its frequency in the input library.
REFERENCES
- 1. Yen H. C., Xu Q., Chou D. M., Zhao Z., Elledge S. J. (2008) Global protein stability profiling in mammalian cells. Science 322, 918–923 [DOI] [PubMed] [Google Scholar]
- 2. Yen H. C., Elledge S. J. (2008) Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science 322, 923–929 [DOI] [PubMed] [Google Scholar]
- 3. Doherty M. K., Hammond D. E., Clague M. J., Gaskell S. J., Beynon R. J. (2009) Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J. Proteome Res. 8, 104–112 [DOI] [PubMed] [Google Scholar]
- 4. Araya C. L., Fowler D. M. (2011) Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 29, 435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fowler D. M., Araya C. L., Fleishman S. J., Kellogg E. H., Stephany J. J., Baker D., Fields S. (2010) High-resolution mapping of protein sequence-function relationships. Nat. Methods 7, 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ravid T., Hochstrasser M. (2008) Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell. Biol. 9, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson P. R., Swanson R., Rakhilina L., Hochstrasser M. (1998) Degradation signal masking by heterodimerization of MATalpha2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell 94, 217–227 [DOI] [PubMed] [Google Scholar]
- 8. Chen P., Johnson P., Sommer T., Jentsch S., Hochstrasser M. (1993) Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell 74, 357–369 [DOI] [PubMed] [Google Scholar]
- 9. Swanson R., Locher M., Hochstrasser M. (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 15, 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gari E., Piedrafita L., Aldea M., Herrero E. (1997) A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13, 837–848 [DOI] [PubMed] [Google Scholar]
- 11. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 12. Gietz R. D., Schiestl R. H. (2007) Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 38–41 [DOI] [PubMed] [Google Scholar]
- 13. Amberg D. C., Burke D., Strathern J. N., and Cold Spring Harbor Laboratory (2005) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 14. Fowler D. M., Araya C. L., Gerard W., Fields S. (2011) Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27, 3430–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinnebusch A. G. (2011) Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 75, 434–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Starheim K. K., Gevaert K., Arnesen T. (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 17. Arnesen T. (2011) Towards a functional understanding of protein N-terminal acetylation. PLoS Biol. 9, e1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hwang C. S., Shemorry A., Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shemorry A., Hwang C. S., Varshavsky A. (2013) Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell 50, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zattas D., Adle D. J., Rubenstein E. M., Hochstrasser M. (2013) N-terminal acetylation of the yeast Derlin Der1 is essential for Hrd1 ubiquitin-ligase activity toward luminal ER substrates. Mol. Biol. Cell 24, 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Varshavsky A. (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tasaki T., Sriram S. M., Park K. S., Kwon Y. T. (2012) The N-end rule pathway. Annu. Rev. Biochem. 81, 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.