

Abstract
MicroRNAs (miRNAs) regulate gene expression through translational repression and RNA degradation. Recently developed high-throughput proteomic methods measure gene expression changes at protein level and therefore can reveal the direct effects of miRNAs' translational repression. Here, we present a web server, ProteoMirExpress, that integrates proteomic and mRNA expression data together to infer miRNA-centered regulatory networks. With both types of high-throughput data from the users, ProteoMirExpress is able to discover not only miRNA targets that have decreased mRNA, but also subgroups of targets with suppressed proteins whose mRNAs are not significantly changed or with decreased mRNA whose proteins are not significantly changed, which are usually ignored by most current methods. Furthermore, both direct and indirect targets of miRNAs can be detected. Therefore, ProteoMirExpress provides more comprehensive miRNA-centered regulatory networks. We used several published data to assess the quality of our inferred networks and prove the value of our server. ProteoMirExpress is available online, with free access to academic users.
MicroRNAs (miRNAs)1 are small noncoding RNAs that regulate gene expression by causing either translation inhibition or mRNA decay (1). Posttranscriptional regulation by miRNA is an important level of the complex gene regulatory network, and it controls a wide range of biological processes. Deregulation of miRNA expression can lead to various diseases, including many human cancers (2). Therefore, understanding the regulatory networks of miRNAs in different biological processes is crucial to unraveling their functional importance and to providing a pool of targets for medical therapies.
Several approaches have been proposed to predict miRNA's targets and to construct miRNA-centered regulatory networks. Computational approaches include miRNA target prediction based on the binding energy of miRNA–mRNA interactions (3, 4) and on the evolutionary conservation of the seed regions (5–7). Experimental approaches include the identification of destabilized mRNAs in the presence of an miRNA or high-throughput methods to detect mRNAs bound by argonaute proteins and miRNA cleavage sites (8). Databases such as MiRecords (9) and TarBase (10) collect experimentally validated miRNA targets, and starBase (8) collects miRNA–mRNA interaction maps from argonaute CLIP-Seq and Degradome-Seq data.
The anticorrelation between miRNAs and their targets has been widely used to infer miRNA–target relationships. Several web servers have been developed to infer miRNA targets based on the expression profiles of miRNAs and mRNAs from the same set of biological samples (11–16). For example, the generative model for miRNA regulation (GenMir++) uses a Bayesian model to predict miRNA targets based on both target genes' 3′ UTR region sequence features and the correlation between expressions of miRNA and its targets (15). When miRNA expression data are not available, active miRNA and its targets can be inferred from the enrichment of its recognized motifs in the 3′ UTRs of suppressed genes in a biological state or process (17–20). Moreover, the condition-specific mRNA–miRNA network integrator (mirConnX) uses transcription factor (TF) binding in the promoter region of miRNAs, as well as mRNA, to construct a transcriptional-posttranscriptional regulatory network (21). In addition, miRNA function can also be annotated by its target genes' enrichments in biological pathways, gene ontology, or diseases (11, 22–24).
Despite the great success of these methods, none of them consider the effects of an miRNA on a target gene's output at both mRNA and protein levels. In addition to destabilization of the mRNA product, translational repression has been proposed to be another major mechanism of miRNA regulation. Many examples have shown that miRNA is able to decrease protein levels without changing mRNA abundance (25–27). It is considered that when the mature miRNA pairs with its target perfectly, the argonaute protein's endonucleolytic active site will cleave the target mRNA's nucleotides that pair with bases 10 and 11 of the miRNA guide strand (1). In cases of imperfect pairing or of argonaute proteins lacking endonucleolytic activity, miRNAs regulate genes through translation repression (1, 28). However, a recent study by Brodersen et al. found that translational repression happened irrespective of the degree of complementarity or location of target sites within mRNAs (29). Complete complementary pairing miRNA may engage in both mRNA cleavage and translational repression, and consequentially lead to decreased protein product. Several models have been suggested to describe the translation repression mechanism, including competition between miRNA-induced silencing complexes and the essential translation initiation factor elF4E for binding to the mRNA 5′ cap structure, deadenylation of the mRNA tail, ribosome drop-off, and reduced translation elongation (1). Two recent papers reported that miRNA first inhibited translation initiation and then induced mRNA deadenylation and decay (30, 31). Even though most of the miRNA targets undergo translational repression followed by decay, there is a subgroup of targets primarily regulated by translation repression without significant mRNA decay (30). It is still unclear why some target mRNAs are degraded and others are not.
Because of the different mechanisms of miRNA regulation, its targets can be translationally repressed without a significant decrease in mRNA abundance or with a concordant decrease in mRNA abundance, or mRNA abundance might be significantly decreased with few protein changes at a certain time point (28, 32, 33). Current methods that use only mRNA abundance to study miRNA regulatory effects might miss many targets that are suppressed at the protein level without detectable mRNA changes, or they might overestimate miRNA's effect on targets that do not have detectable changes at the protein level but do demonstrate decreases in mRNA. These kinds of false negative and/or false positive links between an miRNA and its targets may lead to misunderstanding of an miRNA's regulatory network, for example, when the miRNA's target is a TF.
Recently, the development of high-throughput quantitative proteomic methods has provided the opportunity to study the effect of miRNA on targets' protein outputs (32, 34, 35). However, most of these studies are restricted to one or a few miRNAs or proteins. Furthermore, there is no published tool for inferring miRNA-centered regulatory networks from high-throughput proteomics data. To fill the gap, we present here a new web server, ProteoMirExpress, which integrates mRNA and protein expression data to infer miRNAs' activities on their direct and indirect targets in the absence or presence of miRNA expression data and construct the regulatory networks controlled by miRNAs. We further use several published data to assess the quality of our inferred networks. The assessment shows that ProteoMirExpress is able to effectively infer a miRNA-centered regulatory network and identify subgroups of miRNA targets, which are usually ignored by the currently available tools. The web server is freely available at http://jjwanglab.org/ProteoMirExpress.
EXPERIMENTAL PROCEDURES
Web Server Integration and Implementation
ProteoMirExpress integrates mRNA and protein expression data together to infer miRNA-centered regulatory networks for a specific biological stage or process. The workflow of ProteoMirExpress is briefly described in Fig. 1. It accepts both high-throughput transcriptomic and proteomic profiling data, and optionally the expression of miRNAs, preferably generated under the same experimental conditions (Fig. 1A). The server has the following functions: (i) In the absence of miRNA expression information, ProteoMirExpress will classify the input genes into different groups according to their mRNA and protein levels and infer miRNA networks by calculating the overlap between the potentially suppressed gene sets and the miRNA's target genes (Fig. 1B). (ii) In the presence of miRNA expression information, the targets of active miRNA will be inferred by the anticorrelation between miRNAs and their potential targets at either the mRNA or the protein level, or both (Fig. 1B). The miRNA target genes are collected from multiple sources, including computational predictions, CLIP/Degradome-Seq, and experimental verifications (Fig. 1C). The inferred active miRNAs and their targets will be ranked according to their p values (Fig. 1D). (iii) Indirect targets of miRNAs are also predicted by scanning suppressed mRNAs' promoters with the binding site information of miRNA-targeted TFs. (iv) The miRNA-centered regulatory network will be visualized on the web page (Fig. 1E). Users can click on each miRNA to inspect all of its targets, as well as miRNA–target interactions.
Fig. 1.

Overview of ProteoMirExpress workflow. A, input of high-throughput transcriptomic mRNA and protein profiling data; input of miRNA expression is optional. B, data analysis of ProteoMirExpress. C, the information sources of miRNA target. D, tabulated outputs. E, visualization of miRNA-centered posttranscriptional regulatory networks and miRNA–target interactions.
Inputs
ProteoMirExpress takes inputs in tab-delimited format containing high-throughput protein and mRNA expressions. With data obtained from the same biological condition, ProteoMirExpress accepts mRNA and protein expression levels from one biological stage or expression changes from two biological stages. For data from one biological stage in one file, the input file should contain three columns: the identifier of the genes, the corresponding expression values of mRNAs, and the expression values of the proteins. Protein and mRNA expression data can also be inputted in two files, one for protein and the other for mRNA. Each file should contain two columns: the first one is the identifier of the protein or mRNA, and the second is the expression value. ProteoMirExpress will match the protein and mRNA from the same gene for the users if the input identifiers are in different types. ProteoMirExpress can recognize protein and mRNA identifiers from various databases, such as RefSeq, Ensemble, UCSC, Uniprot, PDB, etc. With samples from two biological stages, such as before and after a certain biological treatment, ProteoMirExpress accepts the expression fold change (or log2(fold change)) of the mRNA and protein in one file or two files with the same format described above. ProteoMirExpress also considers the expression change p value from two-stage data if the user inputs the p value in the column next to the expression change. Then the input files mentioned above should contain five columns and three columns, respectively. The expression data of miRNAs can also be optionally inputted with the same format as used for mRNA and protein. Furthermore, to serve more users, ProteoMirExpress also accepts data from only mRNA or protein for analysis.
Data Analysis Procedure
The input genes or miRNAs will be classified into different classes according to their expression levels (or changes), as well as their p values, if applicable. Users can input a customized expression level (or change) and a p value cutoff for gene classification. For example, if the expression change cutoff is 1 (using log2, 1 means the fold change cutoff is 2-fold), then genes whose expression decreases to less than 0.5-fold (log2(expression change) < −1) will be classified as decreased (D), whereas genes whose expression change is more than 2-fold (log2(expression change) > 1) will be increased (I), and the rest will be unchanged (U). Genes with low expression or expression suppression at either the protein or the mRNA level will be regarded as potential targets of miRNA for further analysis. If the p value cutoff is set at 0.05, for example, only genes whose p value is lower than 0.05 will be classified as significantly increased or decreased genes.
In the absence of miRNA expression information, ProteoMirExpress will calculate the significance of overlaps between the potential suppressed gene sets and the predicted target genes of each miRNA with a hypergeometric test or permutation test. With N genes in the whole genome, m genes in the potential suppressed gene set (set A), n genes that are the predicted target genes of an miRNA (set B), and k genes in the overlap of gene sets A and B, the hypergeometric p value is calculated by Equation 1.
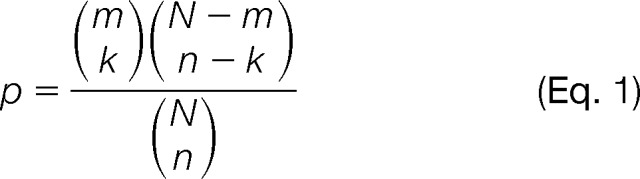 |
The permutation test is performed by randomly selecting m genes from the genome 1000 times. If the selected genes in q of the 1000 times have more than k genes that are the predicted targets of an miRNA, the permutation p value is calculated as p = q/1000 (36). The cutoff for the hypergeometric and permutation p value is set as 0.05 by default, but this can be adjusted by the user.
In the presence of miRNA expression information, the targets of active miRNA will be inferred by the anticorrelation between the miRNA expression levels (or changes) and the mRNA or protein levels (or changes) of the potential targets. The miRNA information is collected from miRBase (37), and information about the target genes is collected from multiple databases. Computational prediction databases include TargetScan (6), miRanda (5), PicTar (7), and PITA (4), and experimental databases include starBase (8), miRecords (9), and TarBase (10). starBase contains miRNA targets identified via the high-throughput methods CLIP/Degradome-Seq, whereas miRecords and TarBase contain experimentally verified targets. To integrate multiple databases from heterogeneous sources, we use an interaction score (IS) to represent the confidence of the link between an miRNA and its target. The IS is calculated as the sum of a weighted proportion of the target databases containing the gene with at least one miRNA target site in the three groups of databases (21). We assign different weights for the three different groups of databases according to reliability, with experimentally validated targets receiving a weight of 4, high-throughput methods a weight of 2, and computational prediction a weight of 1 (Equation 2).
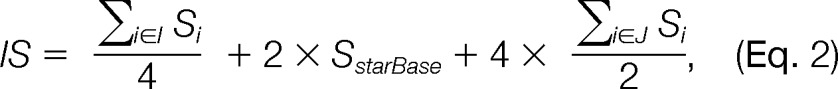 |
where I = (TargetScan, miRanda, PicTar, PITA), J = (miRecords, TarBase), and Si = 1 if the target is present in databases I or J and 0 otherwise. The users can input the cutoff to filter targets with low scores. For example, if the cutoff is 0.5, then only targets reported in at least two of four computational predictions or any one of the CLIP/Degradome-Seq and experiment collection databases will be selected for the analysis.
Besides direct targets, indirect targets of each miRNA are further predicted. When miRNAs suppress the protein abundance of a TF, targets of the TF may also be suppressed indirectly. Thus, after miRNA-targeted TFs are identified, the promoters of genes with suppressed mRNA levels are scanned for putative binding sites of the TFs with the method described by Qin et al. (38). Indirect targets of the miRNAs are defined as genes with decreased mRNA abundance and putative binding sites of miRNA-targeted TFs whose protein abundance is decreased. Users can adjust several parameters for the binding site scanning, including the size of the promoter region, the statistical p value cutoff of binding site significance, the conservation cutoff, and the TF information sources.
Output
The inferred miRNA-centered regulatory network will be visualized on a Cytoscape page (39). The network contains regulatory relationships between miRNAs and their targets. Each node is an miRNA or a target gene, and each edge is an arrow pointing from an miRNA or a TF to its target, which indicates that the miRNA is directly or indirectly suppressing the target. The weights of the lines are proportional to the IS scores of the miRNA–target pairs. Targets from different expression gene classes are labeled in different colors (Fig. 2). For instance, the class “UD,” which is colored blue, represents genes that have unchanged mRNA levels (U) but decreased protein levels (D), whereas class “ML,” which is colored light purple, represents genes with a medium expression level in mRNA (M) but a low expression level in protein (L).
Fig. 2.

miRNA regulatory network with the top 5 enriched miRNAs in HeLa cells with miR-124 overexpression (p value < 1E-4). Each node is either an miRNA or a gene. Each edge is an arrow pointing from an miRNA to its target, indicating its suppressing role. The weights of the lines are proportional to the IS scores of the miRNA–target pairs. Purple lines: connection between an miRNA from the “Inputted miRNA” list and its targets. Gray lines: connection between other enriched miRNAs and their targets. Nodes in blue: genes in UD class having unchanged mRNA levels (U) but decreased protein levels (D). Nodes in green: genes in ML class having a medium expression level in mRNA (M) but a low expression level in protein (L). Nodes in pink: genes in D class having decreased expression in mRNA or protein when only mRNA or protein data are inputted, but not both.
On the second tap, the inferred active miRNAs are ranked according to their p values in the potential target gene set. When the users input miRNA expression data, the server will report two lists of significant miRNAs; one is listed in the “Inputted miRNA” tap, which contains the miRNAs that have high expression or significant expression changes according to the inputted data, and the other is in the “All enriched miRNA” tap, which also includes other miRNAs that are not inputted miRNAs but have enriched target genes with expression changes. Users can select a subgroup of miRNAs of interest from one or both miRNA lists to redraw the regulatory network. Edges between an miRNA from the “Inputted miRNA” list and its targets will be shown as purple lines, and edges between other enriched miRNAs and their targets will be gray lines. Indirect targets are also shown in the network. Arrows pointing to an indirect target are in sage green. When the user clicks on an miRNA node, a list of its targets will pop up. By hovering over a target node, users can find its inputted identifier and gene symbol. When the target node is clicked on, the user will be taken to its web page from the National Center for Biotechnology Information, where detailed information about the gene will be found.
In addition to targets with suppressed mRNA, ProteoMirExpress also outputs targets with proteins suppressed but mRNAs unchanged. miRNAs enriched in different gene sets classified according to mRNA and protein levels can be viewed by clicking on different “Type” buttons. When an miRNA in the network or tables is clicked on, a list of its targets will be shown in a pop-up window. The miRNA–mRNA interaction site information, the hybridization structure, and their sources can be viewed by clicking on the hyperlinks in the “Interaction” and “RNAHybrid” columns.
RESULTS
To evaluate the performance of ProteoMirExpress, we ran several example tests with data from Baek et al. (35). In the first example, miR-124 was overexpressed in HeLa cells, and the global mRNA and protein expressions were quantified after 24 h and 48 h, respectively. With the inputs of mRNA and protein expression profiles and miR-124 as the known miRNA with an expression change, ProteoMirExpress generated a list of predicted active miRNAs and their targets (Table I). As expected, miR-124 was ranked on the top with the most significant hypergeometric p value (1.19E-15) in the potential target gene set, in which genes were down-regulated in either protein or mRNA level. According to protein and mRNA expression changes, these potential targets were further classified into three gene sets: “DD” containing genes for which both mRNA and protein are down-regulated; “UD” containing genes for which mRNA levels are unchanged but protein levels are decreased; and “DU” containing genes for which mRNA levels are decreased but protein levels are unchanged. In the DD and DU gene sets, the hypergeometric p values of miR-124 also ranked first. In UD, even though miR-124 was not the most significant miRNA, the targets of miR-124 were also significantly enriched. Thus, ProteoMirExpress is able to find not only targets with decreased mRNA abundance, but also those whose protein abundance is decreased.
Table I. Active miRNAs inferred by ProteoMirExpress in HeLa cells with miR-124 overexpression (p value < 1E-4).
| miRNA | Number of targets | p value | Number in DDa | p value in DDa | Number in UDa | p value in UDa | Number in DUa | p value in DUa |
|---|---|---|---|---|---|---|---|---|
| hsa-miR-124 | 51 | 1.19E-15 | 12 | 2.04E-08 | 9 | 1.83E-03 | 30 | 2.62E-08 |
| hsa-miR-506 | 46 | 4.96E-13 | 11 | 2.49E-07 | 9 | 1.28E-03 | 26 | 2.03E-06 |
| hsa-miR-760 | 13 | 3.09E-06 | 3 | 5.41E-03 | 3 | 1.44E-02 | 7 | 1.82E-03 |
| hsa-miR-943 | 9 | 1.78E-05 | 3 | 1.01E-03 | 2 | 3.20E-02 | 4 | 1.45E-02 |
| hsa-miR-548m | 16 | 2.19E-05 | 5 | 3.47E-04 | 5 | 1.99E-03 | 6 | 6.31E-02 |
| hsa-miR-545 | 18 | 2.94E-05 | 3 | 4.21E-02 | 5 | 5.24E-03 | 10 | 5.11E-03 |
| hsa-miR-525-3p | 7 | 4.94E-05 | 2 | 7.43E-03 | 0 | – | 5 | 4.21E-04 |
| hsa-miR-802 | 14 | 6.03E-05 | 4 | 1.99E-03 | 3 | 4.03E-02 | 7 | 1.49E-02 |
| hsa-miR-1252 | 16 | 1.15E-04 | 5 | 6.69E-04 | 6 | 5.45E-04 | 5 | 1.50E-01 |
| hsa-miR-432 | 14 | 1.17E-04 | 2 | 1.05E-01 | 5 | 1.38E-03 | 7 | 1.98E-02 |
| hsa-miR-564 | 5 | 1.48E-04 | 1 | 6.58E-02 | 1 | 9.12E-02 | 3 | 4.33E-03 |
| hsa-miR-323b-5p | 10 | 2.50E-04 | 2 | 4.52E-02 | 3 | 1.30E-02 | 5 | 2.27E-02 |
| hsa-miR-513c | 10 | 4.32E-04 | 3 | 5.90E-03 | 2 | 8.99E-02 | 5 | 2.89E-02 |
| hsa-miR-544 | 30 | 4.46E-04 | 6 | 1.50E-02 | 6 | 6.47E-02 | 18 | 1.21E-02 |
| hsa-miR-548p | 18 | 5.42E-04 | 4 | 1.69E-02 | 5 | 1.36E-02 | 9 | 4.25E-02 |
| hsa-miR-922 | 13 | 5.53E-04 | 2 | 1.10E-01 | 2 | 1.73E-01 | 9 | 2.65E-03 |
| hsa-miR-1301 | 10 | 6.76E-04 | 2 | 5.64E-02 | 2 | 9.83E-02 | 6 | 1.11E-02 |
| hsa-miR-518b | 5 | 7.05E-04 | 1 | 9.08E-02 | 2 | 8.42E-03 | 2 | 7.02E-02 |
| hsa-miR-1197 | 8 | 7.33E-04 | 4 | 1.26E-04 | 3 | 6.30E-03 | 1 | 3.55E-01 |
| hsa-miR-518c* | 6 | 7.37E-04 | 0 | – | 2 | 1.85E-02 | 4 | 5.40E-03 |
| hsa-miR-381 | 33 | 7.81E-04 | 6 | 3.07E-02 | 8 | 2.08E-02 | 19 | 2.58E-02 |
| hsa-miR-765 | 11 | 8.46E-04 | 4 | 1.22E-03 | 3 | 2.95E-02 | 4 | 1.31E-01 |
| hsa-miR-1227 | 6 | 8.75E-04 | 1 | 1.37E-01 | 1 | 1.82E-01 | 4 | 6.05E-03 |
| hsa-miR-1266 | 8 | 9.01E-04 | 1 | 2.19E-01 | 2 | 5.50E-02 | 5 | 9.63E-03 |
| hsa-miR-661 | 8 | 9.66E-04 | 3 | 2.57E-03 | 3 | 7.10E-03 | 2 | 2.44E-01 |
a Gene class DD contains genes for which both mRNA and protein are down-regulated. UD contains genes for which mRNA levels are unchanged but protein levels are decreased. DU contains genes for which mRNA levels are decreased but protein levels are unchanged.
Three miR-124-targeted TFs, SP1 (NM_138473), TFAP4 (NM_003223), and TEAD1 (NM_021961), were found to have predicted targets whose mRNA expressions are suppressed in the presence of miR-124 (Fig. 2). Because only TFs whose protein abundance is decreased will be analyzed in order to predict indirect targets, all three of these TFs have reduced protein levels. However, only for TEAD1 is mRNA significantly decreased as well. The mRNA abundance of SP1 and TFAP4 is not significantly changed (92% and 89% of control, respectively), even though their protein abundance is less than 50% of the control's (29% and 43%, respectively). SP1 and TFAP4 have nine and seven targets in the miR-124-controlled network, respectively, which indicates that they may be important downstream regulators for the function of miR-124. However, these TFs may not be reported as miR-124's targets by other tools that use only mRNA expression data for the analysis.
Other miRNAs with significantly enriched targets in the result list are possibly functionally related to or co-expressed with miR-124 (Table I). For example, hsa-miR-506, belonging to the same miRNA family, has expression profiles that are very similar to those of miR-124 in lung carcinogenesis (40, 41) and in breast cancer samples (42). miR-124 is known as a neural-specific miRNA and is suppressed in Huntington disease, and hsa-miR-760, hsa-miR-432, and hsa-miR-1301 are also found to be down-regulated in that disease (43). The miR-Ontology Database miRò also reports that hsa-miR-124, hsa-miR-760, and hsa-miR-432 are all associated with brain tissues, and other enriched miRNAs, such as hsa-miR-943, hsa-miR-548m, hsa-miR-1301, etc., are associated with Alzheimer disease (44). The regulatory network controlled by the top five enriched miRNAs is shown in Fig. 2. More enriched miRNAs are listed in supplementary Table S1. Both the hypergeometric p value and the permutation p value of each miRNA are shown, and the Pearson's correlation coefficient between two p values is as high as 0.89.
To further test the performance of ProteoMirExpress in inferring co-expressed miRNA, we ran a second example in which miR-223 was knocked out in mouse neutrophils and the mRNA and protein expression were quantified on day 8 from progenitor differentiation (35). Again, miR-223 was reported as the most significantly active miRNA by ProteoMirExpress with the inputs of mRNA and protein expression changes (log2(wild type/knockout)) (supplemental Table S2). Of 57 miRNAs in 20 families that were co-expressed with miR-223, 44 miRNAs in 15 families were shown in the significant miRNA list generated by ProteoMirExpress (supplemental Table S2). The co-expression of miRNAs and the co-occurrence of targeting sites on 3′-UTR are highly correlated (p value < 2.2E-16 in Fisher's exact test). The regulatory network with all co-expressed miRNAs gives an overview of the collaborative regulation of these miRNAs on their targets (Fig. 3). It can be seen that ProteoMirExpress is able to infer active miRNAs in a biological process successfully. In both cases, ProteoMirExpress finishes the analyses within minutes. By integrating protein expression data, ProteoMirExpress takes three subgroups of targets into consideration to construct the miRNA-centered regulatory networks, which provides a more comprehensive understanding of the targets and regulatory functions of miRNAs.
Fig. 3.

miRNA regulatory network with the co-expressed miRNAs in miR-223 expressed mouse neutrophils. Refer to Fig. 2 for a detailed description of the graph.
DISCUSSION
The identification of miRNA targets and the construction of an miRNA regulatory network are two major steps in studying the function of miRNA in the complex gene regulatory system. Perturbation of the expression of an miRNA is commonly used to infer the targets of an miRNA. Genes with significant changes in mRNA or protein levels after the perturbation are usually considered as the targets of the miRNA. However, in our first case study, miR-124 overexpression caused 105 genes to show significantly down-regulated expression, but only 56 of them were direct or indirect targets of miR-124. This left 46% of the down-regulated genes unexplained. Thus, we also reported other miRNAs whose targets are enriched in the differentially expressed gene set, which provides the user with hints about other miRNAs that may be co-expressed with or regulated by miR-124. These regulatory links are shown as gray lines to distinguish them from those of miR-124 (Fig. 2). With both analyses on inputted miRNAs and other enriched miRNAs, as well as the prediction of indirect targets, ProteoMirExpress provides a more complete view of miRNA's effects on the suppressed genes, which covers 92% of the down-regulated genes. Besides perturbation experiments with miRNAs, our web server can also be applied to other biological studies, such as comparisons between different developmental stages or disease status, inferring an miRNA-centered regulatory network that controls the gene expression changes between different biological conditions.
With the recent development of high-throughput proteomic methods such as stable isotope labeling by amino acids in cell culture (45, 46), the proteomic investigation strategy for mammals (47), and label-free quantitative methods (48), researchers are able to quantify protein expression on a large scale. More and more high-throughput proteomic data have been generated for the study of a variety of biological processes (49–55). This facilitates studies on gene regulation at the posttranscriptional level. The complexity of miRNA function mechanisms makes it difficult to infer which level miRNA uses to control each individual target. Integrating high-throughput mRNA and protein data provides an opportunity to solve this problem. ProteoMirExpress, taking multiple miRNA function mechanisms into consideration, studies the effect of miRNA on both mRNAs and proteins. ProteoMirExpress reports not only miRNA targets with decreased mRNA levels, but also subgroups of targets whose proteins are suppressed but whose mRNAs are not significantly changed, or whose mRNAs are decreased but whose proteins are not significantly changed, which is possible only with high-throughput proteomic data. Current tools commonly use only mRNA expression data to construct miRNA regulatory networks. With these tools the subgroups of miRNA targets with suppressed proteins but little-changed mRNAs would be completely lost in the network. In the case study of miR-124, TFs SP1 and TFAP4, which are regulated by miR-124, did not display any significant change in their mRNA abundance. Without proteomic data, they might not be detected as targets of miR-124 in the network, even though both of them have several downstream targets that are indirectly regulated by the miRNA. Moreover, when a TF is in a subgroup of miRNA targets with decreased mRNA but little-affected protein, genes with putative binding sites of the TF would be reported as indirect targets of miRNAs by tools using only mRNA expression data. However, because the protein abundance of the TF is not significantly changed, its effects on the downstream targets in the reported network might not be true. The same problem would also occur when miRNA-regulated proteins control downstream pathways, which leads to misunderstanding of the functions of these miRNA-regulated proteins in the network.
In the two case studies analyzed above, out of 51 direct target genes that are significantly suppressed by the overexpression of miR-124 in HeLa cells (either the mRNA level is less than 67% or the protein level is less than 50% of that of the control, as the protein level is measured one day later than the mRNA level, and having at least one miR-124 site), 12 genes (23%) are suppressed in both mRNA and protein level, 9 genes (18%) are suppressed in protein level but not mRNA level, and 30 genes (59%) are suppressed in mRNA level but not protein level. Out of 35 direct target genes that are significantly suppressed by miR-223 (either mRNA in miR-223 knockdown is less than 67% or protein is less than 67% of that in the control neutrophil cells, and having at least one miR-223 site), 6 genes (17%) are suppressed in both mRNA and protein level, 24 genes (69%) are suppressed in protein level but not mRNA level, and 5 genes (14%) are suppressed in mRNA level but not protein level. It seems that the proportion of each subgroup of targets can be different for different miRNAs, or maybe for different cells. The data collection time is also thought to affect the proportion of different subgroups: short time courses after miRNA perturbation but before deadenylation may lead to more observations of the UD group (30, 31, 56, 57), and long time courses after miRNA perturbation, when mRNA deadenylation and decay show strong effects, might lead to more genes being reported from the DD group (58, 59). However, in an experiment with long time scales in which the miRNA effects are steady, fewer direct targets and more indirect targets will be detected. Thus, the selection of a data collection time is an important issue in miRNA target identification studies.
It has been reported that miRNA affects target genes' expression through both translation inhibition and RNA degradation, and the former effect is relatively mild compared with the latter (32). This implies that multiple strategies are used by miRNA to refine their control over their targets in a quantitative manner. In the ProteoMirExpress analysis, we found a subgroup of targets whose mRNAs are suppressed but whose protein levels are not significantly changed. This indicates that the degradation of mRNA might not immediately suppress the protein level. This might be caused by the low protein-degradation rate of these targets. Thus, for this group of targets, miRNA seems to stop the increase in protein level but not immediately decrease it. The three groups of targets detected by ProteoMirExpress demonstrate the limitations of using either mRNA or protein data alone to study the effect of miRNA. Either method may miss a subgroup of targets. Thus, with the integrative approach, ProteoMirExpress provides users with a more complete and detailed regulatory network controlled by miRNAs. Further analyses on the functions, binding site sequences, and expression details of different subgroups of miRNA targets will improve our understanding of the strategy that miRNA uses to precisely control thousands of targets.
Supplementary Material
Footnotes
* This work was supported by a University Postgraduate Fellowship of the University of Hong Kong, the Food and Health Bureau of Hong Kong (10091262), NIH of USA (GH001696), NSFC of China (91229105, 91019016) and the Research Grants Council of Hong Kong (781511M, N_HKU752/10).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CLIP-Seq
- cross-linking and immunoprecipitation sequencing
- Degradome-Seq
- degradome sequencing
- miRNA
- micro-ribonucleic acid
- mRNA
- messenger ribonucleic acid
- TF
- transcription factor.
REFERENCES
- 1. Carthew R. W., Sontheimer E. J. (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136, 642–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calin G. A., Croce C. M. (2006) MicroRNA signatures in human cancers. Nature Rev. Cancer 6, 857–866 [DOI] [PubMed] [Google Scholar]
- 3. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. (2004) Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. (2007) The role of site accessibility in microRNA target recognition. Nat. Genet. 39, 1278–1284 [DOI] [PubMed] [Google Scholar]
- 5. Betel D., Wilson M., Gabow A., Marks D. S., Sander C. (2008) The microRNA.org resource: targets and expression. Nucleic Acids Res. 36, D149–D153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lewis B. P., Shih I. H., Jones-Rhoades M. W., Bartel D. P., Burge C. B. (2003) Prediction of mammalian microRNA targets. Cell 115, 787–798 [DOI] [PubMed] [Google Scholar]
- 7. Krek A., Grun D., Poy M. N., Wolf R., Rosenberg L., Epstein E. J., MacMenamin P., da Piedade I., Gunsalus K. C., Stoffel M., Rajewsky N. (2005) Combinatorial microRNA target predictions. Nat. Genet. 37, 495–500 [DOI] [PubMed] [Google Scholar]
- 8. Yang J. H., Li J. H., Shao P., Zhou H., Chen Y. Q., Qu L. H. (2011) starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 39, D202–D209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. (2009) miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 37, D105–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Papadopoulos G. L., Reczko M., Simossis V. A., Sethupathy P., Hatzigeorgiou A. G. (2009) The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 37, D155–D158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nam S., Li M., Choi K., Balch C., Kim S., Nephew K. P. (2009) MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 37, W356–W362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hausser J., Berninger P., Rodak C., Jantscher Y., Wirth S., Zavolan M. (2009) MirZ: an integrated microRNA expression atlas and target prediction resource. Nucleic Acids Res. 37, W266–W272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsu S. D., Chu C. H., Tsou A. P., Chen S. J., Chen H. C., Hsu P. W., Wong Y. H., Chen Y. H., Chen G. H., Huang H. D. (2008) miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res. 36, D165–D169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sales G., Coppe A., Bisognin A., Biasiolo M., Bortoluzzi S., Romualdi C. (2010) MAGIA, a web-based tool for miRNA and Genes Integrated Analysis. Nucleic Acids Res. 38, W352–W359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang J. C., Babak T., Corson T. W., Chua G., Khan S., Gallie B. L., Hughes T. R., Blencowe B. J., Frey B. J., Morris Q. D. (2007) Using expression profiling data to identify human microRNA targets. Nat. Methods 4, 1045–1049 [DOI] [PubMed] [Google Scholar]
- 16. Liang Z., Zhou H., He Z., Zheng H., Wu J. (2011) mirAct: a web tool for evaluating microRNA activity based on gene expression data. Nucleic Acids Res. 39, W139–W144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sood P., Krek A., Zavolan M., Macino G., Rajewsky N. (2006) Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. U.S.A. 103, 2746–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng C., Li L. M. (2008) Inferring microRNA activities by combining gene expression with microRNA target prediction. PloS One 3, e1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Dongen S., Abreu-Goodger C., Enright A. J. (2008) Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods 5, 1023–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexiou P., Maragkakis M., Papadopoulos G. L., Simmosis V. A., Zhang L., Hatzigeorgiou A. G. (2010) The DIANA-mirExTra web server: from gene expression data to microRNA function. PloS One 5, e9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang G. T., Athanassiou C., Benos P. V. (2011) mirConnX: condition-specific mRNA-microRNA network integrator. Nucleic Acids Res. 39, W416–W423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho S., Jun Y., Lee S., Choi H. S., Jung S., Jang Y., Park C., Kim S., Lee S., Kim W. (2011) miRGator v2.0: an integrated system for functional investigation of microRNAs. Nucleic Acids Res. 39, D158–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maragkakis M., Vergoulis T., Alexiou P., Reczko M., Plomaritou K., Gousis M., Kourtis K., Koziris N., Dalamagas T., Hatzigeorgiou A. G. (2011) DIANA-microT Web server upgrade supports fly and worm miRNA target prediction and bibliographic miRNA to disease association. Nucleic Acids Res. 39, W145–W148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiromatzo A. O., Oliveira T. Y., Pereira G., Costa A. Y., Montesco C. A., Gras D. E., Yosetake F., Vilar J. B., Cervato M., Prado P. R., Cardenas R. G., Cerri R., Borges R. L., Lemos R. N., Alvarenga S. M., Perallis V. R., Pinheiro D. G., Silva I. T., Brandao R. M., Cunha M. A., Giuliatti S., Silva W. A., Jr. (2007) miRNApath: a database of miRNAs, target genes and metabolic pathways. Genet. Mol. Res. 6, 859–865 [PubMed] [Google Scholar]
- 25. Wightman B., Ha I., Ruvkun G. (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862 [DOI] [PubMed] [Google Scholar]
- 26. Chen X. (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303, 2022–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doench J. G., Sharp P. A. (2004) Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu L., Fan J., Belasco J. G. (2008) Importance of translation and nonnucleolytic ago proteins for on-target RNA interference. Curr. Biol. 18, 1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brodersen P., Sakvarelidze-Achard L., Bruun-Rasmussen M., Dunoyer P., Yamamoto Y. Y., Sieburth L., Voinnet O. (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320, 1185–1190 [DOI] [PubMed] [Google Scholar]
- 30. Bazzini A. A., Lee M. T., Giraldez A. J. (2012) Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Djuranovic S., Nahvi A., Green R. (2012) miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336, 237–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Selbach M., Schwanhausser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 [DOI] [PubMed] [Google Scholar]
- 33. Eulalio A., Rehwinkel J., Stricker M., Huntzinger E., Yang S. F., Doerks T., Dorner S., Bork P., Boutros M., Izaurralde E. (2007) Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev. 21, 2558–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vinther J., Hedegaard M. M., Gardner P. P., Andersen J. S., Arctander P. (2006) Identification of miRNA targets with stable isotope labeling by amino acids in cell culture. Nucleic Acids Res. 34, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baek D., Villen J., Shin C., Camargo F. D., Gygi S. P., Bartel D. P. (2008) The impact of microRNAs on protein output. Nature 455, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li M. J., Sham P. C., Wang J. (2010) FastPval: a fast and memory efficient program to calculate very low P-values from empirical distribution. Bioinformatics 26, 2897–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Griffiths-Jones S., Grocock R. J., van Dongen S., Bateman A., Enright A. J. (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qin J., Li M. J., Wang P., Zhang M. Q., Wang J. (2011) ChIP-Array: combinatory analysis of ChIP-seq/chip and microarray gene expression data to discover direct/indirect targets of a transcription factor. Nucleic Acids Res. 39, W430–W436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao Y., Liu H., Li Y., Wu J., Greenlee A. R., Yang C., Jiang Y. (2011) The role of miR-506 in transformed 16HBE cells induced by anti-benzo[a]pyrene-trans-7,8-dihydrodiol-9,10-epoxide. Toxicol. Lett. 205, 320–326 [DOI] [PubMed] [Google Scholar]
- 41. Izzotti A., Calin G. A., Steele V. E., Croce C. M., De Flora S. (2009) Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 23, 3243–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y., Klijn J. G., Zhang Y., Sieuwerts A. M., Look M. P., Yang F., Talantov D., Timmermans M., Meijer-van Gelder M. E., Yu J., Jatkoe T., Berns E. M., Atkins D., Foekens J. A. (2005) Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365, 671–679 [DOI] [PubMed] [Google Scholar]
- 43. Marti E., Pantano L., Banez-Coronel M., Llorens F., Minones-Moyano E., Porta S., Sumoy L., Ferrer I., Estivill X. (2010) A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 38, 7219–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lagana A., Forte S., Giudice A., Arena M. R., Puglisi P. L., Giugno R., Pulvirenti A., Shasha D., Ferro A. (2009) miRo: a miRNA knowledge base. Database (Oxford) 2009, bap008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 46. Gruhler S., Kratchmarova I. (2008) Stable isotope labeling by amino acids in cell culture (SILAC). Methods Mol. Biol. 424, 101–111 [DOI] [PubMed] [Google Scholar]
- 47. Kislinger T., Rahman K., Radulovic D., Cox B., Rossant J., Emili A. (2003) PRISM, a generic large scale proteomic investigation strategy for mammals. Mol. Cell. Proteomics 2, 96–106 [DOI] [PubMed] [Google Scholar]
- 48. Zhu W., Smith J. W., Huang C. M. (2010) Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010, 840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Everley P. A., Krijgsveld J., Zetter B. R., Gygi S. P. (2004) Quantitative cancer proteomics: stable isotope labeling with amino acids in cell culture (SILAC) as a tool for prostate cancer research. Mol. Cell. Proteomics 3, 729–735 [DOI] [PubMed] [Google Scholar]
- 50. Graumann J., Hubner N. C., Kim J. B., Ko K., Moser M., Kumar C., Cox J., Scholer H., Mann M. (2008) Stable isotope labeling by amino acids in cell culture (SILAC) and proteome quantitation of mouse embryonic stem cells to a depth of 5,111 proteins. Mol. Cell. Proteomics 7, 672–683 [DOI] [PubMed] [Google Scholar]
- 51. Gronborg M., Kristiansen T. Z., Iwahori A., Chang R., Reddy R., Sato N., Molina H., Jensen O. N., Hruban R. H., Goggins M. G., Maitra A., Pandey A. (2006) Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol. Cell. Proteomics 5, 157–171 [DOI] [PubMed] [Google Scholar]
- 52. Romijn E. P., Christis C., Wieffer M., Gouw J. W., Fullaondo A., van der Sluijs P., Braakman I., Heck A. J. (2005) Expression clustering reveals detailed co-expression patterns of functionally related proteins during B cell differentiation: a proteomic study using a combination of one-dimensional gel electrophoresis, LC-MS/MS, and stable isotope labeling by amino acids in cell culture (SILAC). Mol. Cell. Proteomics 4, 1297–1310 [DOI] [PubMed] [Google Scholar]
- 53. Tian R., Wang S., Elisma F., Li L., Zhou H., Wang L., Figeys D. (2011) Rare cell proteomic reactor applied to stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative proteomics study of human embryonic stem cell differentiation. Mol. Cell. Proteomics 10, M110.000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lundberg E., Fagerberg L., Klevebring D., Matic I., Geiger T., Cox J., Algenas C., Lundeberg J., Mann M., Uhlen M. (2010) Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 6, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cui Z., Chen X., Lu B., Park S. K., Xu T., Xie Z., Xue P., Hou J., Hang H., Yates J. R., 3rd, Yang F. (2009) Preliminary quantitative profile of differential protein expression between rat L6 myoblasts and myotubes by stable isotope labeling with amino acids in cell culture. Proteomics 9, 1274–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mathonnet G., Fabian M. R., Svitkin Y. V., Parsyan A., Huck L., Murata T., Biffo S., Merrick W. C., Darzynkiewicz E., Pillai R. S., Filipowicz W., Duchaine T. F., Sonenberg N. (2007) MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science 317, 1764–1767 [DOI] [PubMed] [Google Scholar]
- 57. Thermann R., Hentze M. W. (2007) Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature 447, 875–878 [DOI] [PubMed] [Google Scholar]
- 58. Guo H., Ingolia N. T., Weissman J. S., Bartel D. P. (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hendrickson D. G., Hogan D. J., McCullough H. L., Myers J. W., Herschlag D., Ferrell J. E., Brown P. O. (2009) Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 7, e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.