

Abstract
The localization of phosphorylation sites in peptide sequences is a challenging problem in large-scale phosphoproteomics analysis. The intense neutral loss peaks and the coexistence of multiple serine/threonine and/or tyrosine residues are limiting factors for objectively scoring site patterns across thousands of peptides. Various computational approaches for phosphorylation site localization have been proposed, including Ascore, Mascot Delta score, and ProteinProspector, yet few address direct estimation of the false localization rate (FLR) in each experiment. Here we propose LuciPHOr, a modified target-decoy-based approach that uses mass accuracy and peak intensities for site localization scoring and FLR estimation. Accurate estimation of the FLR is a difficult task at the individual-site level because the degree of uncertainty in localization varies significantly across different peptides. LuciPHOr carries out simultaneous localization on all candidate sites in each peptide and estimates the FLR based on the target-decoy framework, where decoy phosphopeptides generated by placing artificial phosphorylation(s) on non-candidate residues compete with the non-decoy phosphopeptides. LuciPHOr also reports approximate site-level confidence scores for all candidate sites as a means to localize additional sites from multiphosphorylated peptides in which localization can be partially achieved. Unlike the existing tools, LuciPHOr is compatible with any search engine output processed through the Trans-Proteomic Pipeline. We evaluated the performance of LuciPHOr in terms of the sensitivity and accuracy of FLR estimates using two synthetic phosphopeptide libraries and a phosphoproteomic dataset generated from complex mouse brain samples.
Phosphorylation is a common and essential form of post-translational regulation that has been extensively studied via mass spectrometry (1–5). However, tandem mass spectra produced from phosphorylated peptides can be difficult to interpret because of their relatively low abundance within the cell and the presence of intense neutral loss peaks in the MS/MS spectra (6, 7). Correctly determining which residue bears the phosphate group is typically a tedious and error-prone process. Most commonly used database search tools for peptide identification from MS/MS spectra are not optimized for site localization of a post-translational modification, nor do they provide any confidence score for the assigned site. In addition, manual verification of the modification sites is a time-consuming process that requires expertise in mass spectrometry. As a result, the challenges of site localization have been acknowledged by the proteomics community, including within the latest version of the data publication guidelines of this journal (8).
A number of computational approaches that localize phosphorylation sites have been reported in the literature, enabling automated phosphoproteomic analysis (reviewed in Ref. 9). These tools either rescore the MS/MS spectra to assign confidence measures for individual sites based on site-determining ions (10–15) or derive localization scores directly from the search engine output (16, 17). Ascore, a representative tool in the rescoring category, scores each candidate phosphosite based upon the peaks representing the site-determining ions and subsequently reports a confidence score for the phosphopeptide sequence (11). This algorithm uses the binomial distribution to compute the probability of a random (incorrect) localization for each candidate site in each spectrum. PhosphoRS extends the scoring approach of Ascore by adjusting the probability of random peak matching based on the density of peaks in different regions of each spectrum (18). In contrast, the Mascot Delta score (MD-score)1 determines the confidence of phosphosite localization on peptides as the difference in Mascot ion scores between the highest scoring phosphopeptide (the peptide reported by the search engine) and the next best scoring phosphopermutation (same peptide sequence, alternative phosphorylation site (17)). Thus, the MD-score represents the second type of approach, which, instead of rescoring MS/MS spectra for the purpose of improved site localization, derives the scores directly from the database search engine output. A similar idea was implemented in the SLIP score using a modified version of the Batch-Tag search engine of the ProteinProspector suite (16) and in the variable modification localization score of the proprietary software Spectrum Mill (9). These tools, however, apply the logic of delta scoring for individual sites, not for the whole peptide; this is an important consideration in the case of multiply phosphorylated peptides.
Although these tools have significantly improved the quality of published phosphopeptide identification data, several important issues remain. The level of uncertainty in modification site localization varies significantly across different peptides depending on the total number of candidate sites and the number of phosphorylated residues on the peptide. This, in turn, makes it difficult to compare localization scores between different peptides. Secondly, few algorithms provide a direct estimation of the false localization rate (FLR) in filtered data. Thirdly, most existing algorithms are tied to specific search engines and/or require proprietary libraries (e.g. Ascore and MD-score were developed for SEQUEST and Mascot, respectively; PhosphoRS requires proprietary libraries from Thermo Scientific). This makes it difficult to access these tools and to compare their performance.
Here we present LuciPHOr, an alternative approach for site localization and direct FLR estimation. We introduce a novel scoring approach that utilizes both peak intensity and mass accuracy to aid the computation of an objective score for phosphosite determination and dynamically adapts to characteristic peak properties in different types of instrumentation and fragmentation methods. LuciPHOr computes the scores for phosphosite permutations and associated FLR estimates for the best scoring prediction at the peptide level. It also reports site-level scores for multiphosphorylated peptides, with an acknowledgment that it is difficult to rigorously estimate the FLR in such cases. We also highlight the practical utility of LuciPHOr, which is capable of processing the results of any database search tool (including commonly used search engines X! Tandem (19), SEQUEST (20), and Mascot (21)) that is supported by the widely used Trans-Proteomic Pipeline (TPP) (22). We benchmark LuciPHOr using two previously published datasets generated using synthetic phosphopeptide libraries and demonstrate similar or better performance relative to the existing methods. We also demonstrate the high accuracy of the FLR estimated by LuciPHOr obtained using a target-decoy modification site framework. Lastly, the performance of LuciPHOr is further investigated using a complex mouse brain dataset, and we also discuss the issue of site-level scoring in the analysis of multiphosphorylated peptides.
MATERIALS AND METHODS
RAW File Conversions
Thermo Fisher RAW files for all datasets were deisotoped and converted to Mascot generic format and mzXML using msconvert (version 3.04140) (23).
Synthetic Phosphopeptide Libraries
RAW files for the two synthetic peptide libraries were generously provided by Dr. Bernhard Kuster and colleagues (17, 24). The first dataset included spectra that were produced using either collision-induced dissociation (CID) or high-energy collision-induced dissociation (HCD). The Mascot generic format files were searched directly using Mascot (version 2.4) with the same parameters as described in the original publications.
The first library consisted of 180 synthetic peptides (17). The data were searched using the SwissProt human protein sequence database (release 2011_10, 86,975 entries) as the search space. The database was inspected to verify that it contained all of the peptides from the synthetic phosphopeptide library. The sequences of any missing peptides together with the common proteomic contaminants and reverse decoy sequences were appended to the database. Searches were performed with carbamidomethyl cysteine as a fixed modification, and oxidized methionine, protein N-terminal acetylation, and phosphorylation of serine, threonine, and tyrosine were specified as variable modifications. Trypsin was specified as the enzyme, and up to three missed cleavages were permitted. The precursor ion tolerance was set at 10 ppm, and the fragment ion tolerance was set at 0.5 Da for the CID data and 0.02 Da for the HCD data.
The second synthetic phosphopeptide MS/MS dataset was generated from a collection of 96 separate libraries containing a total of 57,830 phosphopeptides (24). Mascot searches were performed against the sequence database provided by the authors. This database included the common contaminants and reverse decoy sequences. Carbamidomethyl cysteine was specified as a fixed modification, with oxidized methionine and phosphorylation of serine, threonine, and tyrosine specified as variable modifications. The search was restricted to fully tryptic peptides, allowing up to two missed cleavages. The precursor ion mass tolerance and fragment ion mass tolerance were set at 5 ppm and 0.02 Da, respectively.
For both datasets, Mascot search results were subsequently processed using the TPP to produce the resultant PeptideProphet pepXML files (25). PeptideProphet was run using high mass accuracy binning, and all peptide spectrum matches (PSMs) were reported in the output pepXML file regardless of their computed probability. For the subsequent site localization analysis, PSMs were filtered based upon the PeptideProphet probabilities. In the first and second synthetic libraries, PSMs with PeptideProphet probabilities below 0.1 and 0.99, respectively, were discarded. These thresholds were empirically chosen for each library to remove low-confidence PSMs (the unusually low probability threshold for the first library was chosen because of the very small size of the dataset).
Mouse Brain Data
The mouse brain dataset was originally described in Ref. 26. Mascot searches on these data were performed using the Swiss-Prot mouse protein sequence database (release 2013_04) appended with reverse decoy sequences and common contaminants (101,826 sequences). The search parameters were selected as described in the original work. Carbamidomethyl cysteine was specified as a fixed modification, and oxidized methionine and phosphorylation of serine, threonine, and tyrosine were set as variable modifications. Trypsin was specified as the enzyme, allowing fully tryptic peptides with up to two missed cleavages. The precursor ion mass tolerance was set to 10 ppm, and the fragment ion mass tolerance was set to 0.02 Da.
To estimate the FLR in this dataset using the approach described in Ref. 16 (SLIP score), Mascot searches were performed with and without decoy phosphorylation of proline (P) and glutamic acid (E). Prior to running LuciPHOr on this dataset, TPP results were filtered to remove PSMs with a PeptideProphet probability of less than 0.99.
LuciPHOr Score
The LuciPHOr score calculation is based on the computed scores for all matched peaks in the experimental MS/MS spectrum. The underlying modeling is performed under different assumptions for the distributions of high mass accuracy (HCD) and low mass accuracy (CID) fragment ion data. Within each category (high or low mass accuracy data), the learning step is performed specifically for each dataset (“dynamic training”). This ensures that the model parameters are unique to a given dataset, thus allowing for the variable properties of mass accuracy and signal intensity distributions that are produced.
MS/MS spectrum i is represented by ni peaks, where peak j has intensity Iij and m/z distance Dij to the closest theoretical ion. All experimental peaks are labeled as either matched or random. The matched peak is taken as the most intense experimental peak that can be associated with a theoretical fragment ion within a specified mass tolerance window (by default, ±0.25 Da for CID and ±0.025 Da for HCD data) (see supplemental Fig. S1 for an illustration). After all possible permutations for a phosphopeptide have been iterated over, with the best candidate peaks recorded for each permutation's fragment ion ladder, all matched peaks in the spectrum are identified. All remaining peaks are deemed random peaks (i.e. unmatchable for the given peptide in any phosphosite permutation). This includes peaks that fall within the mass tolerance window of a theoretical fragment ion but are not the most intense candidate peaks in that window.
The principle of scoring in LuciPHOr is that the observed distributions of peak intensities and m/z distances (later referred to as mass accuracies) are mixtures of signal (matched peaks) and noise (random peaks) components, and for each peak one can calculate the odds ratio that the peak originates from either distribution. To reflect the fact that the score distributions differ by the type of matched fragment ions, separate distributions are specified for the b and y ions. Peaks representing neutral loss of the phosphate group are also considered in scoring, whereas peaks representing neutral loss of water or ammonia are not. Before any processing begins, the intensity values are normalized by the median intensity of all peaks in each spectrum. Let fbm, fym, and fu denote the intensity distributions for the peaks matched to b-ions, y-ions, and random peaks, respectively. Likewise, define gbm, gym, and gu as the mass accuracy distributions for the corresponding peaks. For CID data, each of the six distributions is defined as a parametric (Gaussian) distribution. This assumption is well supported by empirical observations and allows quick computation. In the case of HCD data, the mass accuracy distribution gu for random peaks is specified as a uniform distribution, followed by nonparametric modeling. This model specification is based on the observation that the m/z distances of randomly matched peaks are uniformly distributed within the mass tolerance window of ±0.025 Da specified for high mass accuracy data.
Following the dynamic training step of establishing the above distributions, the score for each peak j in spectrum i is calculated as follows. For low fragment ion mass accuracy data (CID), if peak j is matched to a b-ion, the log odds score is computed as Sij = wij log(gbm(Dij)/gu(Dij)), where the weighting factor reflects the peak intensity and is obtained using the Bayes rule with equal prior probability of 0.5 (i.e. wij = 1/(1 + exp(−fbm(Iij)/fu(Iij))). This weighting strategy is better suited for the scoring of low mass accuracy fragment ions because the intensity directly and significantly complements the low specificity of matched peaks identified based on m/z distance only. For high fragment ion mass accuracy data (HCD), the log odds score is computed as Sij = log(fbm(Iij)/fu(Iij)) + log(gbm(Dij)/gu(Dij)). This additive scoring strategy is more suitable for HCD data, where it allows the peak intensity to contribute to the score that is otherwise driven by the mass accuracy component—by far the most discriminative scoring component in these data. Peaks matched to y-ions are scored in the same way, using their corresponding distributions.
The total score for the match between spectrum i and peptide phosphopermutation k is then computed as the sum of scores of all matched ions (i.e. as a cumulative log odds for spectrum i), Si(k) = ∑jSij. The delta score is then computed by subtracting the second best scoring permutation k2 from the best scoring permutation k1 (i.e. ΔSi = Si(k1) − Si(k2)). This score is assigned to the best scoring permutation, transforming the score into the odds ratio, which is more comparable across different peptides than the odds score per se.
FLR Estimation
Using the notation x = ΔS for brevity, let hd(x) and hf(x) denote the density functions for decoy and non-decoy PSMs, respectively. The cumulative distribution functions (up to the score threshold δ) for decoys and non-decoys, Hd and Hf, are defined as
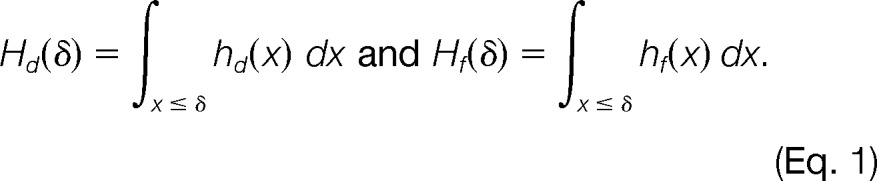 |
The tail probabilities for decoys and non-decoys are defined as
The FLR is then computed at that score threshold as
In addition to the global FLR estimated above, we also estimate the local FLR as
which is equivalent to the local false discovery rate (27). In the derivations above, the factor Nd/Nf is used to approximate the proportion of incorrect localizations in all non-decoy assignments. Here, Nd and Nf are the numbers of PSMs with LuciPHOr delta scores greater than zero having the modification localized to decoy and non-decoys sites, respectively.
Comparison with Representative Existing Algorithms
To obtain the MD-score, Mascot search results (.dat files) were parsed using the MascotDATfile parser (v3.4.9) (28). For singly phosphorylated peptides, the MD-score was computed as defined in Ref. 17 (i.e. as the difference between the Mascot ion scores for the top two alternative phosphosite permutations on the same peptide). For multiply phosphorylated peptides, we extended this definition to permit the site-level MD-score, or the difference between the best Mascot ion score for the phosphopermutation in which a particular site is modified and the best Mascot ion score among all phosphopermutations of the same peptide sequence in which that site is not modified. A small number of MS/MS spectra had a single PSM reported in the Mascot output file. For these, the MD-score was taken as the Mascot ion score of that single PSM. With this modified MD-score definition, we calculated the MD-scores for as many sites as the number of phosphorylations reported on each peptide (i.e. M top scoring sites with positive scores on peptides with M modifications). In the analysis of the mouse brain dataset, the FLR estimation method proposed in Ref. 16 was also implemented, allowing prolines or glutamic acids to be phosphorylated, followed by the scoring steps as described above.
To perform a comparison with Ascore (compatible with the SEQUEST search engine only), MS data for the first synthetic dataset were searched with SEQUEST (version 28, rev. 13) using the same protein database and search parameters as described above for Mascot searches. The Ascore program was downloaded from the Harvard Medical School website (download date: February 22, 2013). The SEQUEST OUT and DTA files were then converted to an XML file for input into Ascore using the Out2XML program provided in the download. The output from Ascore was a comma-delimited file that contained scores for up to six residues in a phosphopeptide. This file was parsed, and only the PSMs of peptide sequences from the synthetic library were retained for the downstream analysis.
RESULTS
Overview of the LuciPHOr Algorithm
LuciPHOr takes as input the database search results processed using PeptideProphet (pepXML files) and the corresponding MS data files in mzXML or mzML format. It records all phosphopeptide identifications and extracts the associated MS/MS spectra. It then proceeds to the training step to learn the distributions of the peak intensities and mass accuracies of all matched and random peaks (Fig. 1). This training step is carried out for each and every dataset (dynamic training), allowing the model parameters to be adjusted to better reflect the properties of the data (see “Materials and Methods”).
Fig. 1.

Scoring framework. The algorithm first goes through a training step in which it learns the peak feature distributions for matched and random peaks. With this information, each phosphosite permutation is scored using all the peaks in the given spectrum, and the FLR is computed using the target-decoy framework for various score thresholds. On the right, two permutations derived from the same peptide are shown with their detailed scores using the information from peak feature distributions, along with the final delta score.
Second, for each high-scoring PSM in the dataset, all possible phosphosite permutations for peptide sequences of these PSMs are generated. The total number of permutations for a phosphopeptide is computed from the binomial coefficient, known as “n choose k.” A peptide with n candidate sites, k of which are reported to be phosphorylated, will have C(n,k) permutations to be considered for localization scoring. The theoretical ion fragments for each permutation are generated, and the algorithm then tries to match these ions to the peaks in the spectrum. For any given phosphosite permutation, a matched peak is scored based on its intensity and mass accuracy, quantifying its contribution to support the given permutation. Using the densities of the positive (correctly matched peaks) and negative (random peaks) distributions estimated in the training step, the score for each matched peak is computed as the ratio of its positive versus negative density functions evaluated at the given value. This ratio corresponds to the odds of the peak being a correct match for the corresponding fragment ion given its observed intensity and mass accuracy. The score of each permutation is calculated as a cumulative sum of the log-odds scores over all matched peaks. The phosphosite permutation with the highest score is taken to be the best permutation. The confidence for the reported permutation is measured by the difference between the scores of the top two scoring permutations, the LuciPHOr delta score (see “Materials and Methods”).
Next, LuciPHOr computes the FLR at the peptide level using the delta scores. The FLR estimation procedure is based on a nonparametric empirical Bayes method that models the score distribution with a mixture of two distributions: one for the permutations with correct localization, and the other for those with incorrect localization (27). Decoy permutations are generated by artificially placing phosphorylation on non-S/T/Y residues in the peptide sequences. For multiphosphorylated peptides, the decoys are generated with the same number of non-S/T/Y residues modified. All of the possible decoy permutations of a peptide are then scored (Fig. 2A). From the resulting scores of both decoy and non-decoy permutations, the density functions of the two score distributions are estimated separately (Fig. 2B), using a standard technique for nonparametric density estimation with Gaussian kernel and normal distribution approximation for bandwidth selection (29, 30). The density estimation step performs smoothing of FLRs, ensuring that these estimates are not rugged between similar score thresholds. Then the mixing proportion is computed as the number of decoy permutations with positive delta scores divided by the number of such forward permutations. Finally, FLR estimates are computed for every delta score threshold as the ratio of the right tail areas under the two distributions (Fig. 2B). See “Materials and Methods” for the details of the FLR estimation procedure.
Fig. 2.

Decoy generation and FLR estimation. A, decoy generation and scoring, illustrated using the singly phosphorylated peptide LQTVHSIPLTINK. The peptide harbors three potential sites (highlighted in red), and all other sites (in green) were used to generate decoy permutations with one modified site. In the first scenario, the non-decoy permutation with modification on the first threonine (T) scores the highest, followed by another non-decoy permutation with modification on the serine (S). In the second scenario, the decoy permutation with valine (V) scores the highest, followed by a non-decoy permutation with the first threonine (T) modified. B, the FLR is computed using the statistical method for estimating local false discovery rates based on the empirical Bayes method. The mixture model serves as a smoothing step to overcome the granularity of simple counts of decoys. The proportion of decoys is estimated as the number of effective (positive scoring) decoy permutations divided by the number of all scored non-decoy permutations.
For peptides with multiple (M) phosphorylations, LuciPHOr can also compute the site-level scores and report the delta scores for M highest scoring sites in addition to the single peptide-level score for the best permutation. These scores are defined as the ratio of the odds score between the best scoring (non-decoy) permutation with phosphorylation on a given site and the best scoring (non-decoy) permutation with no phosphorylation on that site. Constructed from the odds at the permutation level, the site-level score carries the meaning of the odds ratio of phosphorylation, which is a standardized relative confidence of the modification on a particular site over the best alternative. This way, the site-level relative confidence scores can still be compared across different peptides, just as the peptide-level scores can. Note that the site-level score defined here is always identical to the peptide-level score for singly phosphorylated peptides.
LuciPHOr was designed to run in a two-step fashion: (i) scoring all phosphosite permutations, including decoy permutations, followed by the FLR estimation at various delta-score threshold values, as described above, and (ii) scoring using the same procedure but without decoy permutations, followed by generation of the final report, which includes the associated FLR rates estimated in step (i).
First Synthetic Phosphopeptide Library Dataset
We used two datasets generated using synthetic phosphopeptide libraries to examine the accuracy of LuciPHOr and to compare LuciPHOr to MD-score and Ascore as representative existing methods for the two major categories of site localization tools. The advantage of using data generated using synthetic libraries is that the true phosphorylation sites are known for all peptides a priori, and as such, the true FLR can be computed. MS/MS database searches for each phosphopeptide library dataset were performed using Mascot and SEQUEST, and the MD-score and Ascore were used to compute site localization scores for each PSM (see “Materials and Methods”). The Mascot results were post-processed with PeptideProphet to generate the pepXML files that are the main input for LuciPHOr.
The first library consisted of 180 synthetic peptides analyzed by both CID and HCD (17) (see “Materials and Methods”). The PSMs assigned to any of the phosphopeptides from the library were collected, resulting in 1194 and 1120 PSMs for the CID and HCD datasets, respectively. Although two different search engines were used (Mascot and SEQUEST), the total numbers of PSMs used for localization in each tool were similar in both CID and HCD datasets. After the database search, spectra with PeptideProphet probabilities less than 0.1 were removed to filter out poor-quality matches.
Fig. 3 reports the proportion of correctly localized phosphosites as a function of the FLR computed based on the known modification location as specified in the library annotation file. One limitation of this dataset was the relatively small size of the phosphopeptide library. Thus, the estimated FLR was sensitive to a small number of incorrectly localized phosphosites in the high-confidence region. As such, false localization on a few peptides could significantly affect the performance evaluation of any of the three tools we examined. Despite this caveat, Fig. 3 shows that LuciPHOr and the other methods could localize an equivalent number of sites at all FLR levels in both datasets (CID and HCD), suggesting that all three methods performed equally for these data. For the CID data (left-hand panel of Fig. 3A), LuciPHOr, the modified MD-score (extended to the site level to accommodate multiphosphorylated peptides), and Ascore correctly predicted 708 (78%), 620 (70%), and 708 (77%) sites at the FLR of 1%, respectively. For the HCD data (right-hand panel of Fig. 3B), LuciPHOr, the modified MD-score, and Ascore correctly predicted 544 (61%), 437 (49%), and 309 (32%) PSMs, respectively. The MD-score eventually identified more sites than LuciPHOr, but in the higher error rate region (FLR above 2%). See supplemental Tables S1 (CID data) and S2 (HCD data) for the top-scoring localizations for each spectrum matched to a phosphopeptide from the library.
Fig. 3.

Analysis of the first synthetic library dataset. A, B, the proportion of correctly localized sites obtained using Ascore (red curve), LuciPHOr (black), and MD-score (blue) plotted as a function of the true FLR in CID (A) and HCD (B) data. Shown in parenthesis are the total numbers of sites considered by each of the methods. The vertical dashed gray line indicates the FLR threshold of 0.01 (1%). C, D, comparison between the FLR estimated by LuciPHOr (dashed line) and the true FLR (solid line) at various delta score thresholds.
Although the small size of this library prevented a more in-depth comparison, the results suggest that LuciPHOr's performance is at least comparable to that of existing methods with respect to the sensitivity (the number of correctly localized sites). An important advantage of LuciPHOr, however, is the fact that it can directly estimate the FLR from each dataset. We evaluated the accuracy of these FLR estimates by comparing them with the true FLR obtained using the known site information (see Figs. 3C and 3D for the CID and HCD data, respectively). In the CID dataset, the FLRs estimated by LuciPHOr were close to the true values across all thresholds. In the HCD data, LuciPHOr somewhat underestimated the FLR in the score range of 5–9, which is in a high-confidence region. A closer examination of the data revealed that the incorrect localizations in this score range all resulted from five scans of the peptide ESKsSPRPTAEK and two scans of the peptide SSsPTQYGLTK, suggesting that the locally inaccurate estimates can be attributed in part to the small size of this library (and possibly to the problems with the synthetic library itself).
Second Synthetic Phosphopeptide Library Dataset
The second dataset was generated using a much larger phosphopeptide library that recently became available (24). This dataset consisted of MS/MS spectra generated using peptides from 96 separate libraries. Each library contained variants of the same peptide motif with one phosphorylation event and all possible variations of the flanking residues. This yielded 57,830 non-redundant singly phosphorylated peptides, which were analyzed using HCD fragmentation. Our Mascot search of this dataset initially reported 60,354 PSMs. When processed by the TPP, 35,459 PSMs were assigned a PeptideProphet probability equal to or greater than 0.99, accounting for 15,300 unique phosphopeptides. These high-confidence PSMs were then analyzed by LuciPHOr and MD-score (see supplemental Table S3 for the top-scoring localizations for each PSM).
Fig. 4 summarizes the result of this analysis. Fig. 4A shows the distribution of LuciPHOr delta scores when analyzed with decoys. The red bars indicate the scores of decoy permutations outscoring their non-decoy alternatives. The mound on the right-hand side of the histogram primarily represents unambiguous cases (i.e. peptide sequences that contain only one possible phosphorylation site). Note that inclusion or exclusion of the unambiguous phosphopeptides in the analysis does not affect the accuracy of the FLR estimates. The main effect of removing the unambiguous cases is a uniform increase in both the true FLR values and the FLRs estimated by LuciPHOr. We included the unambiguous cases in the FLR estimates because we were presenting all phosphopeptide identifications as a single file.
Fig. 4.

Analysis of the second synthetic library dataset. A, histogram of LuciPHOr delta scores in non-decoy (white) and decoy (red) permutations used to estimate the FLR. B, estimated and true FLR as a function of delta score threshold values. C, number of spectra with correct localization obtained using LuciPHOr at various true FLR (solid black line) or estimated FLR (dashed line) thresholds. Also shown is the number of correct localizations obtained using MD-score as a function of true FLR (blue line). D, same as C for the number of unique peptides with correct localization.
The FLR accuracy plot obtained using the knowledge about the sites of modifications in the synthetic phosphopeptide library is shown in Fig. 4B. The solid line shows the true FLR plotted against the threshold values, whereas the dashed line shows the estimated FLR as reported by LuciPHOr at the same thresholds. The plot suggests that the estimated FLR is very accurate up to 1.5%, where localization is assigned on the majority of the peptides. As indicated in Ref. 24, the entire list of phosphopeptide identifications as reported by the Mascot search tool was thought to carry an FLR of less than 2%. This was consistent with our analysis, as all critical delta score thresholds corresponding to the FLR range between 0.5% and 2% were constrained within a small window of values (delta scores of 3 to 5).
The impact of the FLR accuracy in the most relevant region of delta scores is illustrated in the (pseudo)receiver operating characteristic plots in Figs. 4C and 4D. The black solid lines indicate the number of spectra and unique peptides, respectively, having correct localization. The dashed lines show what would have been achieved if the estimated FLR were used as the horizontal coordinate in the plots. It is evident that the whole trajectory of these estimated FLR values as a function of the LuciPHOr delta score threshold was very close to that observed for the true FLR. For the same set of PSMs, we also computed the MD-score directly from the Mascot search results. The comparison between the numbers of correct localizations computed at various FLR threshold values shows that LuciPHOr made far fewer errors in the high-confidence regions (<1% FLR) and identified more spectra and unique peptides with correct localizations at fixed FLR rates than the MD-score.
Mouse Brain Dataset
To demonstrate the performance of LuciPHOr on a dataset with a more realistic sample complexity, we analyzed a mouse brain dataset generated using an LTQ-Orbitrap Velos instrument with HCD fragmentation (26). For the purpose of comparison, we analyzed the same data using the MD-score (modified to allow site-level scoring; see “Materials and Methods”) and we also used the FLR estimation method proposed by Baker et al. (16) for the SLIP score of ProteinProspector. In that study, the searches were performed allowing phosphorylation on proline (P) and glutamic acid (E), and the FLR was estimated using the proportion of phosphorylated prolines or glutamic acids in the data (referred to as FLR-E/P below). This strategy was adopted here for the MD-score.
Fig. 5A shows the LuciPHOr delta score plotted against the LuciPHOr-estimated FLR (see supplemental Table S4 for the top-scoring localizations for each PSM). In this dataset, the FLR estimates were slightly higher than those in the synthetic library datasets at the same delta score threshold values. For example, a delta score of 3 corresponded to an FLR of less than 2% in the second synthetic library dataset, whereas the FLR was estimated to be close to 4% here. This likely reflects the increased amount of noise in complex datasets at borderline delta score thresholds. Fig. 5B shows the pseudo-receiver operating characteristic curve, in which the number of correct localizations was estimated by multiplying the total number of localized sites by (1 − FLR). The figure also shows the results of applying the MD-score, in which case the FLR was estimated using the proportion of either proline or glutamic acid residues reported to be phosphorylated. Unlike the finding reported in Ref. 16, the FLR-E/P estimates varied considerably depending on which residue was used to quantify the errors (the FLR-P estimates were more conservative than FLR-E). Furthermore, in this complex dataset, the FLR-E/P of 1% corresponded approximately to the MD-score threshold of around 5 (see Fig. 5B), whereas the same FLR values were observed for MD-score thresholds around 15 in the synthetic library datasets (data not shown). Typically, as the complexity of the dataset increases, the score threshold corresponding to a particular fixed FLR value (such as at 1%) also increases. The opposite trend observed here suggests that the FLR-E/P may be underestimated.
Fig. 5.
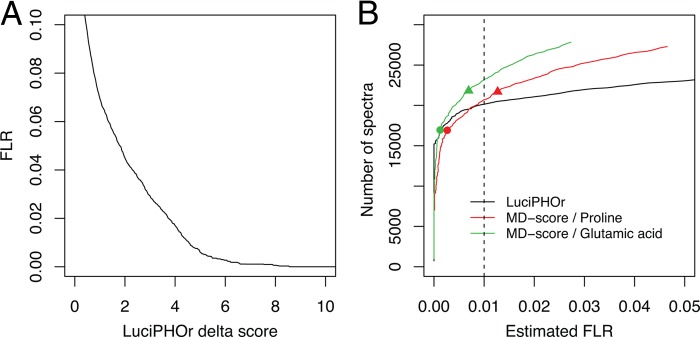
Analysis of the mouse brain dataset. A, the estimated FLR plotted against LuciPHOr delta score thresholds. B, the estimated number of spectra with correct site localization obtained using LuciPHOr plotted against the LuciPHOr-estimated FLR (black curve). Also shown are the results obtained using MD-score plotted against the FLR, with the FLR estimated using the number of decoy localizations on proline (FLR-P approach) and glutamic acid residues (FLR-E approach). The circles indicate the MD-score threshold of 10, and the triangles the MD-score threshold of 5, with the corresponding FRL-P and FLR-E values being lower than the true FDR values observed at these same score thresholds in the synthetic datasets.
This dataset was also used to further investigate the utility of computing the site-level scores in the case of multiphosphorylated peptides. In total, about 4919 (27%) of all the peptides (18,112) were multiphosphorylated. The delta score threshold of 4.57 corresponded to a 1% FLR, and among these, 23,642 sites were from the peptides surpassing the threshold. In addition, nearly 15% more sites (3391) could be reported as confidently localized (site-level delta scores passing the 4.57 score threshold) even though their peptide-level delta scores were below that threshold (supplemental Fig. S2). This shows that there were a significant number of peptides with multiple phosphorylations, but not all the phosphosites on those peptides could be localized with confidence.
DISCUSSION
In this work we introduced LuciPHOr, a novel approach for phosphosite localization and FLR estimation. LuciPHOr learns the properties of good matched peaks from the highest quality spectra in such a way that the scoring dynamically adjusts to varying peak properties across different types of instruments. The method then computes a cumulative log-odds ratio score for every theoretical phospho-permutation of a peptide and reports the phospho-permutation with the highest score as the localization. It also derives confidence scores at the site level to allow the extraction of additional sites on multiphosphorylated peptides that are partially localizable. When tested on two synthetic phosphopeptide library datasets, including a large dataset that has just become available (24), LuciPHOr was able to estimate the FLR accurately in all datasets.
With continuing improvements in mass spectrometry, the number of identified phosphopeptides, and especially multiphosphorylated species, will continue to increase. As a result, proper scoring and FLR control should be regarded as important components of data analysis. In this context, we demonstrated statistically powerful scoring and accurate FLR estimation using the target-decoy phosphosite localization strategy. A major benefit of the peptide-level localization scores computed by LuciPHOr is that these scores are log odds ratios and thus are standardized across different spectra, enabling straightforward estimation of the FLR with the help of appropriately generated decoys. In addition, confidence scores can be derived for individual sites and used for localizing sites on partially localizable multiphosphorylated peptides.
The key aspect of the decoy phosphosite generation process, important for obtaining accurate FLR estimates, is to ensure that a random match to a decoy site is equally as likely as an incorrect match to a non-decoy site. Only under this scenario is the proportion of localizations on the decoy sites with a score passing a certain threshold representative of the real FLR in the non-decoy data. It is inherently difficult to generate the representative set of decoy modification sites satisfying this condition, because the numbers of candidate sites and actual phosphorylated sites on each peptide are usually different. Nonetheless, because our method attempts to score all possible decoy permutations, the FLR estimates are at least likely to be on the conservative side of the true FLR. In the two synthetic phosphopeptide datasets used in this work, LuciPHOr's FLR estimates were reasonably accurate in the most critical region (and, as expected, more conservative beyond that point).
Although the principle and implementation vary across different methods, currently available localization tools all share the common goal of assigning confidence scores for individual candidate sites. To do so, they contrast the best scoring localization on the peptide of interest against the next best alternative site on the same peptide. One key motivation behind this work was to investigate the probabilistic assumptions behind the site localization process, including computation of site-level scores in the case of multiphosphorylated peptides. A score for each serine, threonine, and tyrosine can be computed as some form of likelihood of that residue being phosphorylated in the peptide. For singly phosphorylated peptides, this delta score represents the relative confidence (based on the information in the MS/MS spectrum) of one site compared with the next best site on the same peptide. However, this is not the case for multiply phosphorylated peptides: a localization score obtained this way for one site is dependent on the concurrent modification status of other sites. This is because some, if not all, site-determining ions for one site will have different mass shifts depending on the modification status of the other sites. Thus, the probability that a particular site is phosphorylated on a multiphosphorylated peptide is the confidence score not of just that site alone, but of the simultaneous configuration of all sites. In addition to the peptide-level delta score as the main score, LuciPHOr provides the site-level scores, which are useful for the identification of partially localizable multiphosphorylated peptides. However, it is important to bear in mind that considering these site-level scores as confidence measures associated with specific sites is a probabilistic approximation.
One of the main motivations behind the development of LuciPHOr was that few existing methods directly estimate the FLR in each and every dataset. From the user's point of view, an interpretable estimate of the statistical error is a useful summary for determining a score threshold for reporting phosphosite localization. To the best of our knowledge, direct estimation of the FLR from the data was published only as a part of the SLIP score method presented in Ref. 16. In that work, the authors allowed proline or glutamic acid to be phosphorylated in the database search and reported the frequency of localization on each residue (P or E) as an estimate of the FLR in a particular mouse dataset (31). In this work, we presented an alternative decoy generation and FLR estimation strategy and showed that it was able to provide accurate estimates in the datasets tested.
To summarize, we believe that the method described in this work provides an improved foundation for phosphosite scoring and FLR estimation. We demonstrated the performance of our algorithm on two synthetic phosphopeptide library datasets and on a mouse brain dataset representative of complex protein samples. Our algorithm was able to identify more correct phosphosites at equivalent FLRs than several representative existing methods. Last but not least, the software tool LuciPHOr has an important practical advantage: because of its compatibility with the file formats used by the TPP suite of tools, LuciPHOr allows the analysis of peptide identifications obtained using all commonly used database search engines supported by that pipeline.
LuciPHOr was written for Linux in C++ and uses the ProteoWizard library for reading mzXML, mzML, and other open-source formats (32). The program is multithreaded and is available for download online.
Supplementary Material
Acknowledgments
We thank Bernhard Kuster and Harald Marx for sharing their phosphopeptide library datasets and for useful discussions.
Footnotes
* This research has been supported in part by a Singapore Ministry of Education AcRF Tier1 grant (to H.C.) and NIH Grant No. R01-GM94231 (to A.I.N. and A.C.G.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CID
- collision-induced dissociation
- FLR
- false localization rate
- HCD
- high-energy collision-induced dissociation
- MD-score
- Mascot Delta score
- PSM
- peptide spectrum match
- TPP
- Trans-Proteomic Pipeline.
REFERENCES
- 1. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villen J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A., Jørgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., Samson L. D., Woodgett J. R., Russell R. B., Bork P., Yaffe M. B., Pawson T. (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 4. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3(104) [DOI] [PubMed] [Google Scholar]
- 5. Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R., McCartney RR., Schmidt MC., Rachidi N., Lee S. J., Mah A. S., Meng L., Stark M. J., Stern D. F., De Virgilio C., Tyers M., Andrews B., Gerstein M., Schweitzer B., Predki P. F., Snyder M. (2005) Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 [DOI] [PubMed] [Google Scholar]
- 6. Witze E. S., Old W. M., Resing K. A., Ahn N. G. (2007) Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806 [DOI] [PubMed] [Google Scholar]
- 7. White F. M. (2008) Quantitative phosphoproteomic analysis of signaling network dynamics. Curr. Opin. Biotechnol. 19, 404–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bradshaw R. A., Burlingame A. L., Carr S., Aebersold R. (2006) Reporting protein identification data—the next generation of guidelines. Mol. Cell. Proteomics 5, 787–788 [DOI] [PubMed] [Google Scholar]
- 9. Chalkley R. J., Clauser K. R. (2012) Modification site localization scoring: strategies and performance. Mol. Cell. Proteomics 11, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bailey C. M., Sweet S. M. M., Cunningham D. L., Zeller M., Heath J. K., Cooper H. J. (2009) SLoMo: automated site localization of modifications from ETD/ECD mass spectra. J. Proteome Res. 8, 1965–1971 [DOI] [PubMed] [Google Scholar]
- 11. Beausoleil S. A., Villen J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 12. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 13. Phanstiel D. H., Brumbaugh J., Wenger C. D., Tian S., Probasco M. D., Bailey D. J., Swaney D. L., Tervo M. A., Bolin J. M., Ruotti V., Stewart R., Thomson J. A., Coon J. J. (2011) Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 8, 821-U884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruttenberg B. E., Pisitkun T., Knepper M. A., Hoffert J. D. (2008) PhosphoScore: an open-source phosphorylation site assignment tool for MSn data. J. Proteome Res. 7, 3054–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanner S., Payne S. H., Dasari S., Shen Z., Wilmarth P. A., David L. L., Loomis F., Briggs S. P., Bafna V. (2008) Accurate annotation of peptide modifications through unrestrictive database search. J. Proteome Res. 7, 170–181 [DOI] [PubMed] [Google Scholar]
- 16. Baker P. R., Trinidad J. C., Chalkley R. J. (2011) Modification site localization scoring integrated into a search engine. Mol. Cell. Proteomics 10(7), M111.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Savitski M. M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B. (2011) Confident phosphorylation site localization using the Mascot Delta Score. Mol. Cell. Proteomics 10(2), M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taus T., Kocher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 19. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 20. Eng J. K., Mccormack A. L., Yates J. R. (1994) An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 21. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 22. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marx H., Lemeer S., Schliep J. E., Matheron L., Mohammed S., Cox J., Mann M., Heck A. J., Kuster B. (2013) A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 2013. June; 31(6): 557-64 [DOI] [PubMed] [Google Scholar]
- 25. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 26. Jedrychowski M. P., Huttlin E. L., Haas W., Sowa M. E., Rad R., Gygi S. P. (2011) Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol. Cell. Proteomics 10(12), M111.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Efron B., Tibshirani R., Storey J. D., Tusher V. (2001) Empirical Bayes analysis of a microarray experiment. J. Am. Stat. Assoc. 96, 1151–1160 [Google Scholar]
- 28. Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2007) MascotDatfile: an open-source library to fully parse and analyse MASCOT MS/MS search results. Proteomics 7, 364–366 [DOI] [PubMed] [Google Scholar]
- 29. Parzen E. (1962) On estimation of a probability density function and mode. Ann. Math. Stat. 33, 1065–1076 [Google Scholar]
- 30. Silverman B. W. (1998) Density Estimation for Statistics and Data Analysis, Chapman & Hall, London [Google Scholar]
- 31. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 32. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.