

Abstract
The presence of lipopolysaccharide (LPS) in the outer leaflet of the outer membrane (OM) of Gram-negative bacteria creates a permeability barrier that prevents the entry of most currently available antibiotics. The seven lipopolysaccharide transport (Lpt) proteins involved in transporting and assembling this glycolipid are essential for growth and division in Escherichia coli; therefore, inhibiting their functions leads to cell death. LptB, the ATPase that provides energy for LPS transport and assembly, forms a complex with three other inner membrane (IM) components, LptC, F, and G. We demonstrate that inhibitors of pure LptB can also inhibit the full IM complex, LptBFGC, purified in detergent. We also compare inhibition of LptB and the LptBFGC complex with the antibiotic activity of these compounds. Our long-term goal is to develop tools to study inhibitors of LPS biogenesis that could serve as potentiators by disrupting the OM permeability barrier, facilitating entry of clinically used antibiotics not normally used to treat Gram-negative infections, or that can serve as antibiotics themselves.
Keywords: Gram-negative, Lipopolysaccharide, Antibiotics, ABC transporter
1. Introduction
There is a need for new antibiotics. Gram-negative bacteria pose a particular problem to treat because the presence of an outer membrane (OM) creates a stringent filter that prevents many of the known classes of antibiotics from reaching their cellular targets. The OM is an asymmetric lipid bilayer with lipopolysaccharide (LPS), a complex glycolipid, occupying the outer leaflet, and phospholipids in the inner leaflet.1,2 The tightly packed, amphipathic LPS molecules in the outer leaflet prevent the entry of both large polar and small hydrophobic molecules into the cell.3 Furthermore, hydrophobic drugs that do penetrate the OM can be efficiently expelled by a variety of drug efflux pumps.4
The inevitable development of bacterial resistance to current antibiotics provides considerable interest in the discovery of new targets to kill Gram-negative organisms. It is known that the OM is essential for cell viability in Gram-negative bacteria, and proper assembly of the OM requires the correct placement of LPS in the outer leaflet.3,5,6 LPS transport and assembly from its site of biosynthesis in the inner membrane (IM), through the aqueous periplasmic space, to its final destination in the OM has been most thoroughly studied in Escherichia coli. There are seven essential proteins, the lipopolysaccharide transport (Lpt) proteins, that span these compartments and are involved in transporting this complex glycolipid in E. coli (Fig. 1a).7–13 These proteins form a transenvelope complex that helps LPS transit the periplasmic compartment and cross the OM.14–16 Compromising the pathway by disrupting the levels of functional Lpt proteins results in a permeabilized OM, and completely abolishing the function of these essential proteins results in cell death.11,17,18 In fact, inhibiting both the biosynthesis19,20 and biogenesis21,22 of LPS using small molecules has been demonstrated to be effective at curing Gram-negative infections in animals. Disrupting OM biogenesis is one of the most exciting strategies for the development of new drugs because these compounds could be antibiotics themselves or could be used to weaken the OM permeability barrier enough to allow entry of existing antibiotics that currently cannot penetrate the OM.
Figure 1.

LPS transport is powered by ATP hydrolysis at the IM. (a) In E. coli, LPS is transported from the outer leaflet of the IM to the outer leaflet of the OM by seven essential lipopolysaccharide transport (Lpt) proteins. Three of these proteins, LptB, F, and G, comprise an ATP-binding cassette (ABC) transporter at the IM. PL: phospholipids; (b) compound 1a was identified in a biochemical screen for inhibitors of the ATPase activity of pure LptB.
Three of these Lpt proteins, LptB, F, and G, comprise an ATP-binding cassette (ABC) transporter located in the IM.10,11,23 ATP hydrolysis in the cytoplasm by the dimeric nucleotide-binding domain LptB is thought to provide energy to extract LPS from the outer leaflet of the IM.6,11,24 However, because there is no ATP present in the periplasmic space, ATP hydrolysis is believed to power the entire LPS transport and assembly process. LptB belongs to one of the largest protein superfamilies,25,26 and the primary sequences of LptB in various pathogenic Gram-negative organisms are highly conserved (Fig. S1). Furthermore, LptB is the only Lpt component with a known enzymatic activity (ATP hydrolysis). Over the years, an enormous number of compound libraries have been assembled based on core structures that are designed to inhibit nucleotide-binding proteins. While there might be some concern that it could be difficult to reach a protein inside the cell, it is also true that LpxC, the cytoplasmic enzyme that catalyzes the first committed step of LPS biosynthesis, has been established to be an effective target in Gram-negative bacteria.19,20 Of course, the quinolones have cytoplasmic targets as well.27,28 Therefore, we believe that targeting LptB is a reasonable starting point for finding inhibitors of LPS biogenesis.
Previously, we developed an assay to monitor the ATPase activity of LptB.29 This assay can be formatted to screen large libraries of compounds. Here, we establish that this high-throughput screen can provide lead compounds that inhibit LptB in the IM complex present in cells. We hope to use these tools as part of a larger effort to find and evaluate inhibitors that target the ABC transporter responsible for releasing LPS from the IM.
2. Materials and methods
2.1. Inhibitor synthesis
All inhibitors were synthesized and characterized as previously described.30,31
2.2. Bacterial strains and plasmids
E. coli strain BL21(λDE3) [F− dcm ompT hsdS ( ) gal (λDE3)] (Novagen) was used for overexpression of LptB and LptBFGC. NovaBlue cells [endA1 hsdR17 ( ) supE44 thi-1 recA1 gyrA96 relA1 lac F′ [proA+B+ lacIqZΔM15::Tn10] (TetR)] (Novagen) were used for DNA manipulation. Strains MC4100 [F− araD139 Δ(argF-lac) U169 rpsL150 relA1 flbB5301 ptsF25 deoC1 ptsF25 thi] and NR69832 were used for minimal inhibitory concentration measurements (these are defined as ‘wild type’ and ‘leaky’ E. coli, respectively, in the text).
Plasmid pET22/42-lptBhis construction has already been reported. 29 To construct pCDFDuet-lptCABhisFG, the DNA fragment encoding LptFG was amplified by PCR from MC4100 genomic DNA using primers N-NdeI-LptF (5′-GGAATTCCATATGATAATCATAAGATATCTGGTGCGG-3′) and LptG-KpnI-C (5′-CGGGGTACCTTACGATTTTCTCATTAACAGCCACA-3′). The amplified fragment encoding LptFG was digested with restriction enzymes Nde I and Kpn I (New England Biolabs) and ligated into pCDFDuet-1 (Novagen) between the respective restriction sites to make pCDFDuet-lptFG. The DNA fragment encoding LptCAB-His6 was amplified by PCR from MC4100 genomic DNA using primers N-NcoI-LptC (5′-CATGCCATGGGTAAAGCCAGACGTTGGGTT-3′) and LptB-His6-EcoRI-C (5′-GGAATTCTTAATGATGATGATGATGATGCTCGAGTCTGAAGTCTTCCCCAAGG-3′). The amplified fragment was then digested with restriction enzymes Nco I and EcoR I and ligated into pCDFDuet-lptFG to make pCDFDuet-lptCABhisFG. In order to amplify the DNA fragment for LptCAB-His6 with the correct restriction site, the amino acid at the second position of LptC was mutated from serine to glycine.
2.3. Overexpression and purification of LptB and LptBFGC
LptB-His was over-expressed as previously described.29 Cells were pelleted and resuspended in Tris–buffered saline (TBS; 20 mM Tris, pH 8.0, 150 mM NaCl), 20% glycerol, 0.5 mM Tris(hydroxypropyl)phosphine (THP, EMD Millipore) (LptB buffer) supplemented with 1 mM phenylmethylsulfonylfluoride (PMSF; Sigma), 100 μg/mL lysozyme (Sigma), and 50 μg/mL DNase I (Sigma). The cells were lysed by three passages through a French pressure cell (Thermo Electron) at 16,000 psi. After separating unbroken cells, the lysate was centrifuged at 100,000×g for 30 min to remove membranes. The supernatant was applied to Ni-NTA Superflow resin (Qiagen) that had been equilibrated with LptB buffer supplemented with 10 mM imidazole. The resin was washed with 20 column volumes of LptB buffer containing 20 mM imidazole. LptB-His was then eluted in one batch with 2 column volumes of LptB buffer with 200 mM imidazole. The remainder of the procedure was performed as previously reported, 29 except that the LptB buffer described here was used for size exclusion chromatography.
LptBFGC was over-expressed and purified using essentially the reported method.23 The plasmid described in Section 2.2 was transformed into BL21(λDE3) cells, which were grown in Luria Bertani (LB) broth supplemented with 50 μg/mL spectinomycin (Sigma) at 37 °C until they reached OD600 ~1. At this time, 50 mu;M isopropyl β-D-1-thiogalactopyranoside (IPTG; Gold Biotechnology) was added to the media to induce expression of LptBFGC. Cells were grown for an additional 2 h at 37 °C, at which point they were harvested by centrifugation at 5200×g for 10 min. Cells were resuspended in 50 mM Tris–HCl, pH 7.4, supplemented with 1 mM PMSF, 100 μg/mL lysozyme, and 50 μg/mL DNase I. Harvested cells were lysed by three passages through a French pressure cell at 16,000 psi. After removal of unbroken cells, membranes were recovered by centrifugation at 100,000×g for 1 h. Membranes were resuspended in 20 mM Tris–HCl, pH 7.4, 10% glycerol. At this point, membranes were flash frozen in liquid nitrogen and stored at −80 °C. Thawed membranes were solubilized with 20 mM Tris–HCl, pH 7.4, 5 mM MgCl2, 300 mM NaCl, 1% n-dodecyl-β-D-maltopyranoside (DDM; Anatrace), 10% glycerol, and 2 mM ATP at 4 °C for 1 h, followed by centrifugation at 100,000×g for 30 min. The remainder of the purification is the same as the reported protocol.23
2.4. Minimal inhibitory concentration (MIC) determination
MIC values were determined using a previously reported colorimetric method.29,33
2.5. ATPase activity assay
ATPase activity was measured using a modified molybdate method for detecting inorganic phosphate release.23,34 The LptB reaction mixture contained 7 μM LptB in 50 mM Tris–HCl, pH 8.0, 500 mM NaCl, and 10% glycerol. The LptBFGC reaction mixture contained 0.2 μM LptBFGC in 50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 10% glycerol, and 0.05% DDM. For inhibition assays, all compounds were dissolved and diluted in DMSO. The reaction mixtures were pre-incubated with inhibitors (or DMSO) for 5 min at 25 °C. Reactions were then started at 25 °C with the addition of the indicated amount of ATP. All reactions contained a final concentration of 2% DMSO. The linear time range of activity was determined for both LptB and LptBFGC. Reactions were stopped within the linear range by the addition of an equal volume of 12% SDS. Inorganic phosphate was measured using the reported method.34 Absorbance values were measured using a Spectramax Plus 384 plate reader (Molecular Devices). For all inhibition assays, activity was normalized relative to an uninhibited DMSO control. Data were analyzed and kinetic parameters were determined using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
2.6. Other techniques
SDS–PAGE analysis was done as reported using 14% Tris–HCl polyacrylamide gels.35
3. Results
3.1. Compounds identified in a screen for inhibition of LptB also inhibit LptBFGC in vitro
We have previously developed a biochemical assay to monitor the ATPase activity of LptB. Using this assay, we identified two LptB inhibitors from a library of 244 small molecule kinase inhibitors.29 The best compound, 1a (Fig. 1b), had previously been characterized as a potent inhibitor of Wee1, a eukaryotic kinase that regulates entry into mitosis.36 Because this primary screening assay reports on the ATPase activity of pure LptB, we wondered whether this compound class would inhibit LptB in a more physiologically relevant context. In the active LPS exporter, LptB is a part of an ABC transporter with transmembrane domains LptF and LptG. Together with LptC, this complex releases LPS from the outer leaflet of the IM.24 Therefore, screening for LptB inhibitors can reveal initial hit compounds, but testing inhibitors with the pure LptBFGC complex would provide a better indication of how the inhibitors might behave in vivo. We overexpressed and purified LptBFGC in detergent (Fig. 2a). Although the overexpression plasmid for the IM complex encodes for LptCAB, LptA did not co-purify with the the complex in detergent. Measured the ATPase activity of the IM complex using a molybdate assay, which detects inorganic phosphate release. LptBFGC demonstrates comparable rates of ATP hydrolysis to those previously reported (Fig. 2b).23 We determined that LptBFGC is 10– 20 times more active than LptB alone by comparing the maximum reaction velocities. This suggests that interactions between the individual components of the transporter stabilize the proteins and promote formation of an active conformation of dimeric LptB.
Figure 2.

Pure LptBFGC shows high levels of ATPase activity and a similar inhibition profile to LptB alone. (a) Purified LptB (lane 1) and LptBFGC (lane 2) were analyzed by SDS–PAGE and stained with Coomassie brilliant blue; (b) LptBFGC is 10–20 times more active than LptB. The following kinetic parameters for LptBFGC can be obtained from the graph (all values represent mean ± standard error): Vmax = 203.3 ± 8.0 mol ATP hydrolyzed/min mol LptBFGC, K′= 0.7 ± 0.1 mM, and h = 1.1 ± 0.1; (c) LptB activity was measured with 1 mM ATP and 100 μM of the respective inhibitor. (d) LptBFGC activity was measured with 0.5 mM ATP and 100 μM of the respective inhibitor. Relative activity compares the rates of ATP hydrolysis in the presence and absence of inhibitors. Error bars represent standard deviations from 3 independent experiments done in duplicate.
There have been extensive medicinal chemistry efforts to optimize the potency of 4-phenylpyrrolocarbazoles, like 1a, against their kinase target, Wee1.30,31,37 These analogs allowed us to investigate the inhibition by a set of compounds comprising two scaffolds (1 and 2, see Table 1), both closely related to our initial hit 1a. Therefore, we used analogs of 1a to evaluate if this compound class is a useful starting point for finding inhibitors of the IM complex.
Table 1.
MIC values of two structural classes of small molecules that kill an OM-permeabilized strain of E. coli
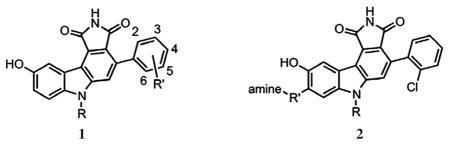
| ||||||
|---|---|---|---|---|---|---|
| Compound | R | R′ | Amine | MIC (μM)
|
References | |
| Wild typea | Leakya | |||||
| 1a | H | 6-Cl | — | >100 | 25 | 29 |
| 1b | H | 2-Cl, 6-Cl | — | >100 | 12.5 | 30 |
| 1c | H | 2-Br | — | >100 | 12.5 | 30 |
| 1d | H | 2-SMe | — | >100 | 25 | 30 |
| 2a | Me | O(CH2)3 | NMe2 | >100 | 25 | 31 |
| 2b | Me | O(CH2)3 | Npyrrol | >100 | 100 | 31 |
| 2c | (CH2)2OH | O(CH2)3 | Npyrrol | >100 | 100 | 31 |
| 2d | Me | SO2(CH2)3 | Npyrrol | >100 | 50 | 31 |
Wild type cells are E. coli strain MC4100; leaky cells are E. coli strain NR698.32
As shown in Figure 2c, the tested compounds inhibit LptB to different extents. The three compounds from the class more closely related to our initial hit (1b–d) all inhibit LptB better than those in group 2. Furthermore, the relative potencies of the two groups display similar inhibition patterns between LptB and LptBFGC (Fig. 2d). The best compound against LptB, 1b, is also the best inhibitor against LptBFGC; the least effective compounds against LptB are also the least effective compounds against LptBFGC (2a– d). Therefore, screening for inhibitors of pure, soluble LptB leads to compounds that also inhibit the four-protein membrane complex containing a stable dimer of LptB.
The data in Figure 2 represent inhibition at a single inhibitor concentration. A concern when identifying hydrophobic inhibitors is that they could act by nonspecifically denaturing their protein target. By varying the concentration of our best inhibitor, 1b, we could determine whether our compounds display concentration-dependent inhibition. The best compound (1b) shows standard concentration-dependent inhibition of both LptB and the complex (Fig. 3). The IC50 of 1b against LptBFGC is 6 times higher than that of LptB. This is consistent with the fact that LptB forms a stable, active dimer in the protein complex, rendering it more difficult to inhibit in that form. This suggests that our primary screen for inhibitors against LptB alone is a more sensitive method for finding weak inhibitors of the IM complex.
Figure 3.

Compound 1b is 6-times more potent against LptB alone (a) than LptBFGC; (b) relative activity is calculated as described in Figure 2. Error bars represent standard deviations from 3 independent experiments.
3.2. LptB inhibitors kill leaky E. coli
Having established that we can identify compounds that will inhibit the four-protein IM complex using a screen against the pure ATPase component, we wanted to see if these compounds show activity in cells. We chose to work with a strain of E. coli in which the OM has been compromised by mutating a gene involved in OM biogenesis. E. coli containing this mutation has a more permeable (leaky) OM. This strain has been used to characterize the activity of toxic molecules that normally cannot penetrate through the OM.38 Using this strain, we can evaluate how the compounds perform as inhibitors of the IM complex in its physiological membrane without the additional requirement that these compounds be able to penetrate the OM. The MIC values for compounds 1a–d and 2a–d are given in Table 1. The MIC values roughly correlate with their levels of inhibition of LptB and LptBFGC in vitro. These results are consistent with an interpretation that LptB could be a target for the compounds in the cell. The fact that the compounds show activity against the leaky E. coli strain suggests that they can diffuse across the IM to reach their cytoplasmic target(s). None of these compounds are able to kill wild type E. coli, suggesting that they are unable to penetrate an intact OM barrier. These compounds are likely too hydrophobic to diffuse across the OM into the periplasmic compartment, consistent with the belief that the OM is an effective permeability barrier to small, hydrophobic compounds.
4. Discussion
LptBFGC is a promising antibiotic target because blocking the ATPase responsible for releasing LPS from the outer leaflet of the IM will block the entire transport process. These results suggest that it is possible to screen for inhibitors of LptB to find compounds that can also inhibit LptB complexed with LptC, F, and G. The inhibitors that we describe represent the first class of small molecule inhibitors of LptBFGC. Whether it is worthwhile to carry out further medicinal chemistry on this class is a reasonable question. Our best compound was discovered in a screen of a limited number of known kinase inhibitors. It is possible that screening larger collections of kinase inhibitors would produce more potent leads as starting points. It might also be worthwhile to find other structural classes with better physical properties that might better penetrate the OM (more polar or charged compounds). At this point, we could use our assay to screen much larger collections of compounds, without biasing our efforts towards kinase inhibitors. ATP-competitive kinase inhibitors are engineered to bind in a highly conserved kinase active site, which is structurally distinct from a nucleotide-binding domain active site.39–41
ATPases have been established to be important drug targets in both prokaryotes and eukaryotes.42 Inhibitors against ABC transporters in particular have been used to treat cancer, but to the best of our knowledge, only one inhibitor of a bacterial ABC transporter with antimicrobial activity has been reported. 43,44 Given that the assembly of LPS on the surface of Gram-negative organisms by the Lpt proteins is conserved and essential, it seems as though inhibitors of this ABC transporter could be very important compounds as antibiotics. We have validated the use of an ATPase screen to find lead compounds that inhibit the activity of the entire IM complex, containing the ABC transporter responsible for LPS export. The biochemical tools reported here can be used to characterize other inhibitors that we find in our screen. Eventually, we would like to have an in vitro reconstitution using pure IM components to be able to correlate inhibition of ATPase activity with inhibited release of LPS from the IM.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant GM066174 and AI081059(to D.K.) and an NSF Fellowship (to D.J.S.). We acknowledge Dr. Natividad Ruiz for comments on the manuscript.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.04.020.
Contributor Information
David J. Sherman, Email: dsherman@fas.harvard.edu.
Suguru Okuda, Email: okuda@fas.harvard.edu.
William A. Denny, Email: b.denny@auckland.ac.nz.
Daniel Kahne, Email: kahne@chemistry.harvard.edu.
References and notes
- 1.Muhlradt PF, Golecki JR. Eur J Biochem. 1975;51:343. doi: 10.1111/j.1432-1033.1975.tb03934.x. [DOI] [PubMed] [Google Scholar]
- 2.Kamio Y, Nikaido H. Biochemistry. 1976;15:2561. doi: 10.1021/bi00657a012. [DOI] [PubMed] [Google Scholar]
- 3.Nikaido H. Microbiol Mol Biol Rev. 2003;67:593. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikaido H. Annu Rev Biochem. 2009;78:119. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bos MP, Robert V, Tommassen J. Annu Rev Microbiol. 2007;61:191. doi: 10.1146/annurev.micro.61.080706.093245. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz N, Kahne D, Silhavy TJ. Nat Rev Microbiol. 2009;7:677. doi: 10.1038/nrmicro2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sampson BA, Misra R, Benson SA. Genetics. 1989;122:491. doi: 10.1093/genetics/122.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bos MP, Tefsen B, Geurtsen J, Tommassen J. Proc Natl Acad Sci USA. 2004;101:9417. doi: 10.1073/pnas.0402340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun M, Silhavy TJ. Mol Microbiol. 2002;45:1289. doi: 10.1046/j.1365-2958.2002.03091.x. [DOI] [PubMed] [Google Scholar]
- 10.Sperandeo P, Cescutti R, Villa R, Di Benedetto C, Candia D, Deho G, Polissi A. J Bacteriol. 2007;189:244. doi: 10.1128/JB.01126-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz N, Gronenberg LS, Kahne D, Silhavy TJ. Proc Natl Acad Sci USA. 2008;105:5537. doi: 10.1073/pnas.0801196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu T, McCandlish AC, Gronenberg LS, Chng SS, Silhavy TJ, Kahne D. Proc Natl Acad Sci USA. 2006;103:11754. doi: 10.1073/pnas.0604744103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sperandeo P, Pozzi C, Deho G, Polissi A. Res Microbiol. 2006;157:547. doi: 10.1016/j.resmic.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Tefsen B, Geurtsen J, Beckers F, Tommassen J, de Cock H. J Biol Chem. 2005;280:4504. doi: 10.1074/jbc.M409259200. [DOI] [PubMed] [Google Scholar]
- 15.Chng SS, Gronenberg LS, Kahne D. Biochemistry. 2010;49:4565. doi: 10.1021/bi100493e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freinkman E, Okuda S, Ruiz N, Kahne D. Biochemistry. 2012;51:4800. doi: 10.1021/bi300592c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperandeo P, Deho G, Polissi A. Biochim Biophys Acta. 2009;1791:594. doi: 10.1016/j.bbalip.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Sperandeo P, Lau FK, Carpentieri A, De Castro C, Molinaro A, Deho G, Silhavy TJ, Polissi A. J Bacteriol. 2008;190:4460. doi: 10.1128/JB.00270-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CR. Science. 1996;274:980. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 20.Barb AW, Zhou P. Curr Pharm Biotechnol. 2008;9:9. doi: 10.2174/138920108783497668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Kach A, Eberl L, Riedel K, DeMarco SJ, Robinson JA. Science. 2010;327:1010. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 22.Werneburg M, Zerbe K, Juhas M, Bigler L, Stalder U, Kaech A, Ziegler U, Obrecht D, Eberl L, Robinson JA. ChemBioChem. 2012;13:1767. doi: 10.1002/cbic.201200276. [DOI] [PubMed] [Google Scholar]
- 23.Narita S, Tokuda H. FEBS Lett. 2009;583:2160. doi: 10.1016/j.febslet.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 24.Okuda S, Freinkman E, Kahne D. Science. 2012;338:1214. doi: 10.1126/science.1228984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson AL, Dassa E, Orelle C, Chen J. Microbiol Mol Biol Rev. 2008;72:317. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dassa E, Bouige P. Res Microbiol. 2001;152:211. doi: 10.1016/s0923-2508(01)01194-9. [DOI] [PubMed] [Google Scholar]
- 27.Lesher GY, Froelich EJ, Gruett MD, Bailey JH, Brundage RP. J Med Pharm Chem. 1962;91:1063. doi: 10.1021/jm01240a021. [DOI] [PubMed] [Google Scholar]
- 28.Oliphant CM, Green GM. Am Fam Physician. 2002;65:455. [PubMed] [Google Scholar]
- 29.Gronenberg LS, Kahne D. J Am Chem Soc. 2010;132:2518. doi: 10.1021/ja910361r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer BD, Thompson AM, Booth RJ, Dobrusin EM, Kraker AJ, Lee HH, Lunney EA, Mitchell LH, Ortwine DF, Smaill JB, Swan LM, Denny WA. J Med Chem. 2006;49:4896. doi: 10.1021/jm0512591. [DOI] [PubMed] [Google Scholar]
- 31.Smaill JB, Lee HH, Palmer BD, Thompson AM, Squire CJ, Baker EN, Booth RJ, Kraker A, Hook K, Denny WA. Bioorg Med Chem Lett. 2008;18:929. doi: 10.1016/j.bmcl.2007.12.046. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz N, Falcone B, Kahne D, Silhavy TJ. Cell. 2005;121:307. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Fridman M, Balibar CJ, Lupoli T, Kahne D, Walsh CT, Garneau-Tsodikova S. Biochemistry. 2007;46:8462. doi: 10.1021/bi700433v. [DOI] [PubMed] [Google Scholar]
- 34.Chifflet S, Torriglia A, Chiesa R, Tolosa S. Anal Biochem. 1988;168:1. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli UK. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Parker LL, Piwnica-Worms H. Science. 1955;1992:257. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 37.Smaill JB, Baker EN, Booth RJ, Bridges AJ, Dickson JM, Dobrusin EM, Ivanovic I, Kraker AJ, Lee HH, Lunney EA, Ortwine DF, Palmer BD, Quin J, III, Squire CJ, Thompson AM, Denny WA. Eur J Med Chem. 2008;43:1276. doi: 10.1016/j.ejmech.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 38.Eggert US, Ruiz N, Falcone BV, Branstrom AA, Goldman RC, Silhavy TJ, Kahne D. Science. 2001;294:361. doi: 10.1126/science.1063611. [DOI] [PubMed] [Google Scholar]
- 39.Noble ME, Endicott JA, Johnson LN. Science. 2004;303:1800. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Yang PL, Gray NS. Nat Rev Cancer. 2009;9:28. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oswald C, Holland IB, Schmitt L. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:385. doi: 10.1007/s00210-005-0031-4. [DOI] [PubMed] [Google Scholar]
- 42.Chene P. Nat Rev Drug Disc. 2002;1:665. doi: 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- 43.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Nat Rev Drug Disc. 2006;5:219. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 44.Swoboda JG, Meredith TC, Campbell J, Brown S, Suzuki T, Bollenbach T, Malhowski AJ, Kishony R, Gilmore MS, Walker S. ACS Chem Biol. 2009;4:875. doi: 10.1021/cb900151k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.