

Abstract
The timing of vaccine availability is essential for an effective response to pandemic influenza. In 2009, vaccine became available after the disease peak, and this has motivated the development of next generation vaccine technologies for more rapid responses. The SAM® vaccine platform, now in pre-clinical development, is based on a synthetic, self-amplifying mRNA, delivered by a synthetic lipid nanoparticle (LNP). When used to express seasonal influenza hemagglutinin (HA), a SAM vaccine elicited potent immune responses, comparable to those elicited by a licensed influenza subunit vaccine preparation. When the sequences coding for the HA and neuraminidase (NA) genes from the H7N9 influenza outbreak in China were posted on a web-based data sharing system, the combination of rapid and accurate cell-free gene synthesis and SAM vaccine technology allowed the generation of a vaccine candidate in 8 days. Two weeks after the first immunization, mice had measurable hemagglutinin inhibition (HI) and neutralizing antibody titers against the new virus. Two weeks after the second immunization, all mice had HI titers considered protective. If the SAM vaccine platform proves safe, potent, well tolerated and effective in humans, fully synthetic vaccine technologies could provide unparalleled speed of response to stem the initial wave of influenza outbreaks, allowing first availability of a vaccine candidate days after the discovery of a new virus.
Keywords: influenza, RNA vaccine, SAM vaccine, vaccine platform
INTRODUCTION
Human deaths due to avian influenza A (H7N9) virus in China highlight the unpredictability of influenza and the need for counter-measures that can be applied rapidly.1,2 Vaccines for pandemics can reduce morbidity and mortality,3 but are too slow to develop, produce, and distribute to blunt initial pandemic waves. For example, on 11 June 2009, the World Health Organization announced community level outbreaks of a new influenza A (pH1N1) virus in multiple regions of the world, indicating that an influenza pandemic was underway.4 Three months later, several manufacturers had completed vaccine development, received regulatory authorization, scaled up production and begun to supply vaccines.5 Despite the unprecedented speed of the response to this pandemic, substantial quantities of vaccine were only available in November 2009, after the second pandemic wave had peaked.6,7 Furthermore, the amount of vaccine was constrained by the global manufacturing capacity, which is not sufficient to vaccinate the global population. Pandemic vaccine was available in significant quantities only in high-income countries.
The timing of delivery for the 2009 H1N1 pandemic vaccine provided a strong impetus for the development of next-generation vaccine technologies that are more amenable to a rapid response.1,8,9,10 These include synthetic influenza vaccine viruses, recombinant DNA-derived subunits, virus-like particles, nucleic acid vaccines and viral vector-based vaccines. Potency, speed, manufacturing robustness and production capacity are critical parameters that will determine the ability to respond to future influenza outbreaks. Recently, we described the use of an enzymatic, cell-free gene assembly technique to synthesize hemagglutinin (HA) and neuraminidase (NA) genes and their use to generate synthetic vaccine seeds in less than a week from the discovery of a new virus.11 While this technology can greatly advance the speed of production of licensed influenza vaccines, there is still a need for novel approaches that can further transform the vaccine field. Of the new approaches, nucleic acid (plasmid DNA and mRNA) offer the greatest potential for speed. However, DNA-based vaccines have made limited clinical progress during the last 20 years, and they have not been licensed for human use yet. In addition, the dose requirements for plasmid DNA in clinical trials have been high (≥1 mg),12,13 and this will challenge pandemic production capacity for this vaccine platform. Similarly, testing of non-amplifying mRNA in preclinical animal models (mice, ferrets and pigs) has indicated that two doses (80–250 µg) of vaccine are required to elicit protective responses.10 The mRNA vaccine technology is in its infancy, and additional gains in potency can been expected from improved delivery and further optimization of RNA constructs.
We have previously described the SAM vaccine platform.14 This platform, now in preclinical development, is based on a synthetic, self-amplifying mRNA, delivered by a synthetic lipid nanoparticle (LNP). We have shown that a SAM vaccine candidate against human immunodeficiency virus elicited broad, potent and functional immune responses and that a SAM vaccine candidate against respiratory syncytial virus elicited protective immunity. Non-viral delivery systems, such as LNPs, allow RNA delivery to cells without the complication of anti-vector immunity that can thwart repeated immunization with viral vector-based vaccines. Production of RNA by a cell-free enzymatic transcription reaction avoids the use of microorganisms or cultured cells in manufacturing, with the associated quality and safety issues, and enables simple downstream purification and very rapid and cost-effective manufacturing.10,15
In this paper, we demonstrate that a SAM vaccine encoding an influenza H1 HA antigen from H1N1 virus was immunogenic in mice at low doses, and elicited antibody responses that were comparable to a licensed influenza vaccine. In addition, when we used the gene assembly and error correction technique for the rapid and accurate cell-free synthesis of the HA gene, we were able to generate a SAM vaccine encoding an influenza H7 HA antigen from H7N9 virus. This vaccine was ready for immunization 8 days after the availability of the H7 gene sequence and 35 days after immunization elicited mouse antibody titers that are associated with protection of humans from influenza, indicating that the combination of gene synthesis and the SAM vaccine technology could enable unprecedented speed and reliability of future vaccine responses to influenza pandemics.
MATERIALS AND METHODS
RNA synthesis of SAM H1 and H7 vectors
The full-length H1 HA gene was amplified from the reverse-transcribed RNA genome of influenza A/California/07/2009 (H1N1), using forward primer 5′-ATT CCC GTC GAC GCC ACC ATG AAG GCA ATA CTA GTA GTT CT-3′ and reverse primer 5′-ATT TAC GCC TAG GTT ATC AAA TAC ATA TTC TAC ACT GTA GAG AC-3′. The full-length H7 HA gene from A/Shanghai/2/2013 was amplified from a DNA fragment, assembled based on the previously described enzymatic isothermal assembly method with error correction,11 using forward primer 5′-AAT TAA GTC GAC GCC ACC ATG AAC ACT CAA ATC CTG GTA TTC G-3′ and reverse primer 5′-AAT TAA TCT AGA TTA TCA TAT ACA AAT AGT GCA CCG CAT G-3′. Amplicons were cloned into SalI and XbaI sites of a modified replicon construct.16 Plasmid DNA was purified using the QIAfilter plasmid Maxi kit (Qiagen, Valencia, CA, USA) following the manufacturer's instructions and the nucleotide sequence of the inserts was confirmed by Sanger sequencing. DNA was linearized immediately following the 3′ end of the replicon by restriction digest and purified by phenol/chloroform extraction and ethanol precipitation. Linearized DNA template was transcribed into RNA using the MEGAscript T7 high-yield transcription kit (LifeTechnologies, Carlsbad, CA, USA). Transcripts were purified by precipitation in the presence of 2.8 M LiCl, capped using the ScriptCap m7G Capping System (CellScript, Madison, WI, USA) and LiCl precipitated again. H1 HA expression was confirmed by Western blot analysis of transfected baby hamster kidney (BHK) cell lysates.
BHK cell transfection and Western blot analysis of expressed H7 protein
To confirm expression of H7 HA from replicon RNA, 106 BHK cells were transiently transfected with 4 µg of RNA using Lipofectamine 2000 (LifeTechnologies, Carlsbad, CA, USA). At 16 h post transfection cells were lysed in 100 µL radio-immunoprecipitation assay buffer (Boston BioProducts, Ashland, MA, USA) with complete protease inhibitor cocktail (Roche, Madison, WI, USA). Lysates were separated under non-reducing conditions on a 4%–12% Bis-Tris polyacrylamide gel in MOPS electrophoresis buffer (LifeTechnologies, Carlsbad, CA, USA) and blotted to nitrocellulose membrane (LifeTechnologies, Carlsbad, CA, USA). H7 HA was visualized using an H7-specific polyclonal antibody (GeneTex, Irvine, CA, USA).
LNP/RNA formulation
Lipid nanoparticles were prepared as described previously with minor modifications.14 1,2-dilinoleyloxy-3-dimethylaminopropane was synthesized as previously described.17 1,2-diastearoyl-sn-glycero-3-phosphocholine was purchased from Genzyme (Cambridge, MA, USA). Cholesterol was obtained from Sigma-Aldrich (St. Louis, MO, USA). 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-𝓃-[methoxy(polyethylene glycol)-2000] (ammonium salt) (PEG-DMG 2000) was obtained from Avanti Polar Lipids (Alabaster, AL, USA). Briefly, 1,2-dilinoleyloxy-3-dimethylaminopropane, 1,2-diastearoyl-sn-glycero-3-phosphocholine, cholesterol and PEG-DMG 2000 were dissolved in ethanol at a 10:48:2:40 molar ratio. RNA in 100 mM citrate buffer pH 6.0 was combined with the ethanolic lipid solution through a T-mixer, and an additional buffer volume of 100 mM citrate buffer pH 6.0 was added simultaneously to the RNA/lipid suspension resulting in a 33% ethanol solution. LNPs were placed into a 10 000 molecular weight cellulose dialysis cassette and dialyzed overnight at 4 °C against phosphate buffered saline (PBS) pH 7.4. Formulations were characterized for particle size, RNA concentration, encapsulation efficiency and RNA integrity (using gel electrophoresis) as previously described.14 Prior to administration, LNPs were diluted in 1× PBS to the appropriate concentration.
Preparation of the monovalent H1N1 subunit and MF59 adjuvant
The monovalent egg-produced A/California/07/2009 (H1N1) subunit vaccine preparation was produced from the X179A reassortant vaccine virus18 and was standardized for HA content by single radial immunodiffusion. The MF59 emulsion was manufactured as previously described.18,19 The adjuvanted subunit vaccine was prepared by mixing MF59 1:1 with monovalent egg-derived H1N1 subunit antigen to a final concentration of 1 µg/mL of HA in PBS.
In vivo models
Mice were housed in the Novartis Vaccines and Diagnostics Animal Facilities. All animal experiments were performed in accordance with Institutional Animal Care and Use Committee protocols. Female BALB/c mice, aged 6–10 weeks, were obtained from Charles River Laboratories (Wilmington, MA, USA). For the SAM (H1) vaccine study, groups of 12 mice were immunized intramuscularly (50 µL per quadriceps) on days 0 and 21 (3-week interval) or on days 0 and 56 (8-week interval). Serum samples were collected 3 weeks after the first and 2 weeks after the second immunization. For the SAM (H7) vaccine study, groups of 6–8 mice were immunized on days 0 and 21 (3-week interval) or on days 0 and 56 (8-week interval). Serum samples were collected 2, 3, 5 and 8 weeks after the first and 2 weeks after the second immunization.
Enzyme-linked immunosorbent assay (ELISA) for antigen-specific H1N1 serum antibody
A two-step fully automated rapid ELISA (Hamilton Starlet System, Switzerland) was performed with individual sera to titrate total HA-specific immunoglobulin G (IgG). Maxisorp plates (Nunc, Roskilde, Denmark) were coated overnight at 2 °C–8 °C with 0.26 µg/well of monovalent egg-derived A/California/7/2009 (H1N1) antigen in PBS and blocked for 1 h at 37 °C with 200 µL of ‘Smartblock' solution (Candor Bioscience, Germany). Plates were then washed and incubated with 200 µL of a sealer/stabilizer solution ‘Liquid Plate Sealer' (Candor Bioscience) for 1 h at 37 °C and then aspirated. Serum samples and serum standard and control were initially diluted 1:5000–1:20 000 in PBS, 1% bovine serum albumin, 0.05% Tween-20, transferred into coated-blocked plates and serially 2-fold diluted. Antigen-specific total IgG was detected with alkaline phosphatase-conjugated goat anti-mouse IgG (Sigma Chemical Co., St. Louis, MO, USA). Any OD405 below 0.150 was not considered in the calculation. OD405 from each dilution of standard serum was plotted, and a linear regression was made using the least-square methodology. Titers of control and unknown samples were calculated by interpolating from this curve each OD405 that was comprised in the standard curve OD405 range. The value obtained was then multiplied for the dilution factor. Titers were calculated as the average of the points that are comprised in the range described above.
Hemagglutination inhibition (HI) assay
HI for seasonal influenza strains was performed according to standard procedures using a 0.5% suspension of adult turkey erythrocytes. To inactivate nonspecific inhibitors, all serum samples were pre-treated with receptor-destroying enzyme (DENKA, Japan, Tokyo) according to manufacturer's instructions, achieving a final serum dilution of 1:10. Duplicates of individual sera were then serially 2-fold diluted in V-bottom microtiter plates, in a final volume of 25 µL. Inactivated X179A virus or RG-ID-1603, a virus with the HA and NA genes of A/Shanghai/2/2013 (H7N9) on a PR8x backbone,11 was adjusted to 4 HA units per 25 µL, as verified by back titration, and 25 µL of this virus suspension was added to each of the 96 wells. After incubation at room temperature for 60 min, 50 µL of a 0.5% turkey red blood cell suspension was added, and the mixture was incubated for another 60 min at room temperature. The assay was read by visual inspection. The HI titers were calculated as the reciprocal of the last serum dilution that contained non-agglutinated red blood cells. Samples that scored negative at the lowest dilution tested (1:20) were assigned a HI titer of 10.
Influenza virus neutralization (VN)
For sera from animals immunized with H1N1-based vaccines, VN was tested on pooled sera that had been heat-inactivated at 56 °C for 30 min. Serial 2-fold dilutions of serum samples were mixed in equal volume with 100-fold the median tissue culture infective dose (TCID50) of H1N1 (X-181) virus, in 96-well tissue culture plates, and incubated 1 h at 37 °C. Subsequently Madin–Darby canine kidney cells (ATCC CCl-34; 5×104 cells per well) were added to the plates, followed by 18–22 h incubation at 37 °C, 5% CO2. Cell monolayers were then washed with PBS and fixed in 2% formaldehyde. Viral antigen was detected by ELISA with a monoclonal antibody against influenza A nucleoprotein (3In5 MAb InA224; Hytest Ltd). Serum titers were expressed as the reciprocal of the serum dilution that inhibited 50% of infection compared to control wells with virus alone. A titer of 10 was assigned to sera that gave a negative result at the first dilution tested (1:20).
For sera from animals immunized with H7N9-based vaccines, VN was performed with some modifications from the H1N1 protocol. VN was tested on individual sera that had been pre-treated with receptor-destroying enzyme and heat-inactivated at 56 °C for 30 min. Serial 4-fold dilutions of serum samples were mixed in 96-well half-area tissue culture plates in equal volume with 300 focus forming units of a H7N9 vaccine virus made using the method described by Dormitzer et al.11 The plates were incubated for 2 h at 37 °C. Subsequently, Madin–Darby canine kidney 33016PF cells (9×103 cells per well) were added to the plates, followed by incubation for 18–22 h at 37 °C and 5% CO2. Cell monolayers were fixed in 50:50 acetone/methanol (v/v), and viral antigen was detected by fluorescence focus assay with a monoclonal antibody against influenza A nucleoprotein (MAB8251; Millipore). Serum titers were expressed as the reciprocal of the serum dilution that inhibited 50% of infection compared to control wells with virus alone. A titer of 20 was assigned to sera that gave a negative result at the first dilution tested (1:40).
Statistical analyses
We used the one-way analysis of variance, Kruskal–Wallis (non-parametric) with Dunn's post-test with a 95% confidence interval and the Mann–Whitney test on selected groups. All statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA) (ns: not significant).
RESULTS
Immunogenicity of the SAM (H1/LNP) vaccine
HA is the primary protective antigen in licensed influenza vaccines and the target of potent neutralizing antibodies.20 As a proof-of-concept for an influenza SAM vaccine, we generated an RNA vector that expresses A/California/07/2009 (H1N1) HA and encapsulated the RNA in an LNP. The immunogenicity of the SAM (H1/LNP) vaccine candidate was compared to that of A/California/07/2009-like (X-179A reassortant) influenza subunit vaccine preparations formulated with or without the MF59 oil-in-water emulsion adjuvant in mice (Figure 1). All mice (24/24) immunized with a single intramuscular injection of 1 µg of the SAM (H1/LNP) vaccine candidate seroconverted, with a HI geometric mean titer (GMT) of 73, and the responses boosted after the second immunization. Increasing the interval between SAM (H1/LNP) injections of either dosage from 3 to 8 weeks significantly (P<0.001) increased H1-specific total serum IgG and HI titers. For the 0.1 µg dose, the HI GMT increased 5.8-fold (214 vs. 1244) and for the 1.0 µg dose, 4.4-fold (415 vs. 1810). Homotypic HA-binding (ELISA), HI and influenza virus neutralizing antibody titers elicited by 0.1 µg or 1.0 µg of the SAM (H1/LNP) vaccine candidate were as high as those elicited by the un-adjuvanted subunit antigen preparation, but they did not reach the levels of the adjuvanted subunit vaccine.
Figure 1.

Comparative mouse immunogenicity study of a SAM (H1/LNP) vaccine. Groups of 12 mice were vaccinated i.m. on days 0 and 21 (3-week interval, 3wk) or 0 and 56 (8-week interval, 8wk), with the SAM (H1/LNP) vaccine (0.1 µg and 1.0 µg), subunit (0.1 µg, +/− MF59 adjuvant) or PBS (0). H1-specific antibodies were quantified in sera 3 weeks and 2 weeks after the first and second vaccination, respectively. (A) Total IgG titers; (B) HI titers; (C) VN titers (pooled sera). *P<0.05, **P<0.01 vs. H1N1, one-way ANOVA non-parametric Dunn's multicomparison test. Data are from individual mice (depicted as dots) and the GMT are solid lines. For determination of GMT, titers below the limit of detection were assigned a value of 10. ANOVA, analysis of variance; i.m., intramuscularly; ns, not significant; PBS, phosphate-buffered saline.
Production and immunogenicity of the SAM (H7/LNP) vaccine
The outbreak of H7N9 influenza in Shanghai provided the opportunity to demonstrate the rapid response capabilities of the SAM vaccine platform when combined with rapid and accurate cell-free gene synthesis. On 31 March 2013, the China Center for Disease Control and Prevention announced a human influenza outbreak with an H7N9 avian influenza strain and promptly posted HA and NA gene coding sequences on the Global Initiative for Sharing All Influenza Data system.21 On 1 April, we initiated gene synthesis for the A/Shanghai/2/2013 HA gene (Figure 2). The HA gene synthesis was based on the previously described enzymatic isothermal assembly method with error correction.11 On 2 April, gene synthesis was completed, and the synthetic genes were shipped from the gene synthesis laboratory in California to the SAM vaccine laboratory in Massachusetts. On 3 April, the synthetic HA coding sequence was cloned into a DNA template that contained the RNA-dependent RNA polymerase genes, SAM vaccine genetic control elements and a T7 polymerase promoter. On 5 April, the sequence of the cloned construct was verified. On 6 April, in vitro transcription of the SAM RNA, capping of the RNA and transfection of BHK cells with the RNA were carried out. On 7 April, 7 days after the announcement of the outbreak and the posting of the gene sequences, the integrity of the transcribed RNA was demonstrated by agarose gel electrophoresis (Figure 3A), and expression of influenza H7 HA in the transfected BHK cells was demonstrated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis followed by Western blot analysis with an H7-specific antibody (Figure 3B).
Figure 2.
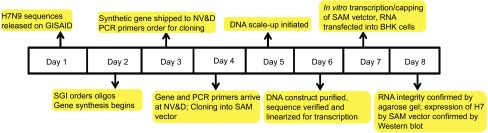
Timeline from electronic gene sequence posting to production of RNA prior to formulation with the LNP delivery system. GISAID, Global Initiative for Sharing All Influenza Data; PCR, polymerase chain reaction.
Figure 3.

RNA quality and confirmation of antigen expression. (A) The SAM vector encoding the H7 antigen was analyzed on a denaturing agarose gel. (B) A SAM vector encoding the H7 antigen was transiently transfected into BHK cells. Cell lysates were separated by SDS–PAGE, and blotted to nitrocellulose membranes. H7 HA was visualized by Western blot using a H7-specific polyclonal antibody. A SAM vector encoding eGFP served as an NC. eGFP, enhanced green fluorescent protein; NC, negative control; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
The RNA was then encapsulated in a LNP delivery system, and the SAM (H7/LNP) vaccine was used to immunize mice (Figure 4). No H7 HA subunit antigen preparation was available to use as a positive control. Therefore, the immunogenicity of the SAM (H7/LNP) vaccine candidate was compared to the same dose of unformulated SAM (H7) RNA (1 µg) and a SAM (H1/LNP) vaccine candidate. Two weeks after the first injection, six of seven H7/LNP-immunized mice seroconverted with H7-specific HI titers ranging from 1:20 to 1:80 (Figure 4A). The HI titers were significantly higher than those elicited by the same dose of unformulated SAM (H7) RNA (1 µg), and the sera were capable of viral neutralization (Figure 4C). The SAM (H1/LNP) vaccine elicited H1-specific seroconversion (Figure 4B), with no crossreactivity with H7 by HI (Figure 4A). The HI titers elicited after two doses of the SAM (H7/LNP) vaccine (1:80–1:640), administered at a 3-week interval, reached the levels associated with human protection from seasonal influenza. HI titers of 1:40 typically provide 50% protection from disease in adults,22,23 although in some populations, such as children, titers as high as 1:110 are required for 50% protection.24 We investigated the kinetics of the antibody response to a single immunization with the SAM (H7/LNP) vaccine (Figure 4D). H7-specific serum HI titers increased over time, with HI GMTs going from 22 at day 13, to 40 at day 21, to 62 at day 35 and to 80 at day 55. At day 56 (8-week interval), the mice were vaccinated a second time and the HI GMT was boosted to 807. This is 3.4-fold higher than mice boosted with a second vaccination on day 21 (3-week interval) (Figure 4A) which reached a HI GMT of 238 by day 35.
Figure 4.
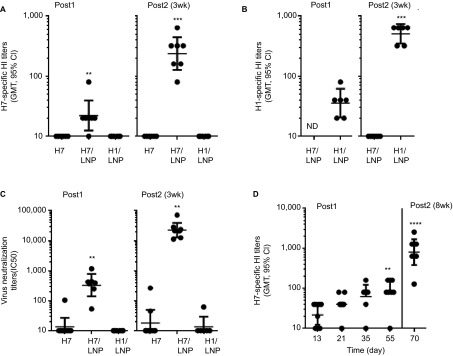
Comparative mouse immunogenicity study of a SAM (H7/LNP) vaccine. Groups of 6–8 mice were vaccinated i.m. on days 0 and 21, with unformulated SAM (H7) RNA (1.0 µg), a SAM (H7/LNP) vaccine (1.0 µg) or SAM (H1/LNP) vaccine (1.0 µg). HA-specific antibodies were quantified in sera 14 days after the first and second vaccinations. (A) H7-specific HI titers; (B) H1-specific HI titers; (C) H7N9 VN titers. **P<0.01, ***P<0.001 vs. H7 RNA, one-way ANOVA non-parametric Dunn's multicomparison test. (D) Kinetics of H7 HI titers after vaccination. Mice were vaccinated on day 0 and 56 with a SAM (H7/LNP) vaccine (1.0 µg), and H7-specific HI titers were measured in sera 13, 21, 35 and 55 days after the first immunization, and 14 days post second vaccination; **P<0.01, ****P<0.0001 vs. day 13. Data are from individual mice (depicted as dots) and the GMTs are solid lines. For determination of GMT, titers below the limit of detection were assigned a value of 10. ANOVA, analysis of variance; i.m., intramuscularly; ND, not determined.
DISCUSSION
The SAM vaccine candidate encoding H1 HA was immunogenic in mice at a low dose (0.1–1.0 µg) and elicited HI and neutralization titers equivalent to those elicited by an un-adjuvanted subunit vaccine preparation. In contrast, in previous reports for non-amplifying mRNA, two 80 µg doses were required for comparable immunogenicity.10 Although unformulated self-amplifying RNA can also protect mice from influenza challenge, two 10 µg doses were required and the HA-specific antibody titers were highly variable between mice.25 In response to the H7N9 influenza outbreak in China, we combined cell-free gene synthesis, enzymatic error correction and enzymatic SAM RNA production to rapidly generate a vaccine candidate that was immunogenic in mice. With further streamlining, it should be possible to produce similar vaccine candidates within 5 days of the gene sequence of the virus being available. We recently demonstrated similarly rapid generation of vaccine viruses for conventional inactivated influenza vaccines.11 However, the generation of a vaccine virus is an early step in the production of conventional influenza vaccines. A synthetic SAM RNA formulated with LNP is the final vaccine product.
One might consider the ideal influenza pandemic or outbreak response platform to have the following attributes: (i) completely synthetic processes that do not require cell culture and are amenable to automation; (ii) robust, generic processes to manufacture vaccines against any influenza strain reproducibly; (iii) a small manufacturing footprint with standard, disposable equipment; and (iv) raw materials that can be stockpiled with production equipment that can be colocated in a single facility, enabling rapid progression from gene sequence to vaccine product. Rapid cell-free antigen gene synthesis followed by enzymatic production of self-replicating RNA and non-viral lipid-mediated delivery meets these specifications. For this initial proof-of-concept experiment, the RNA was produced using a commercial in vitro transcription kit, which is capable of producing milligram quantities of RNA. Ongoing work is focused on establishing a GMP-compliant manufacturing process that will enable the production of much larger quantities of RNA that meets regulatory requirements for human use. The DNA templates for in vitro transcription in these experiments were produced using standard microbiology techniques and amplification in Escherichia coli. There is now substantial effort focused on non-fermentation-based approaches for high-yield DNA production.26 Modern nucleic acid amplification technologies such as polymerase chain reaction. or isothermal techniques such as rolling circle amplification could make the SAM vaccine production process completely cell-free.27
The feasibility of using RNA as the basis for a nucleic acid vaccine was initially regarded as questionable, due to the inherent instability of mRNA in the presence of tissue fluids, the uncertainty of developing reasonable manufacturing processes yielding a stable formulation and the anticipated high cost of the product.28 However, these drawbacks are being addressed, increasing the likelihood that RNA-based vaccines will be commercially viable (for a review, see, references 15 and 28–30). RNA is generally considered to be an unstable molecule because of the presence of the hydroxyl group at the 2′ position on the ribose sugar, allowing the phosphodiester bond to be cleaved by intramolecular transesterification. Therefore, it is possible that stable RNA formulations may require frozen, freeze-dried or lyophilized formualtions.29 Recently, Jones et al.31 reported that a self-amplifying RNA freeze dried in trehalose was stable for 10 months when stored at 4 °C, and Petsch et al.10 reported that a lyophilized mRNA influenza vaccine was stable at 37 °C for 3 weeks. Both preliminary studies indicate the feasibility of using lyophilization to stabilize RNA during storage, although more research and development will be required to develop a commercially viable format.
In summary, we have shown that self-amplifying mRNA vaccines encoding HA can be rapidly produced and are immunogenic in mice. If this vaccine platform proves safe, potent, well tolerated and effective in humans, fully synthetic vaccine technologies could provide unparalleled speed of response to stem the initial wave of influenza outbreaks, allowing first availability of a vaccine candidate days after the discovery of a new virus. In addition, the SAM vaccine platform could offer a potential solution for a wide range of additional pathogens.
Acknowledgments
We thank the RNA Vaccine Platform Team at Novartis Vaccines and Diagnostics, in particular, Nisha Chander, Simona Mangiavacchi and Michelle Chan for coordinating the delivery of formulations for the animal studies; Mary Schaefer and Casey Judge for coordinating the in vivo studies in the United States; Marco Tortoli for performing the in vivo experiment in Italy and NIBR LAS staff for performing the in vivo experiment in the United States; Yevgeniya Teplova and Paola Fontani for running the ELISA assay; Ilaria Vicenti and Marua Prevato for running HI and VN assays; Tim Stockwell and Dave Wentworth for DNA sequence design; Giuseppe Del Giudice and Yasushi Uematsu from Siena for helpful discussions; and Peter Bogner, President of Global Initiative for Sharing All Influenza Data, for facilitating collaboration between NV&D and the China Center for Disease Control and Prevention. The influenza SAM vaccine research was supported in part by funding from the Defense Advanced Research Project Agency under agreement HR0011-12-3-001. The development of the rapid and accurate gene synthesis was funded in part by the Biomedical Advanced Research and Development Authority contract HHSO100201000061C and by funds from the Novartis Foundation.
References
- Rappuoli R, Dormitzer PR. Influenza: options to improve pandemic preparation. Science. 2012;336:1531–1533. doi: 10.1126/science.1221466. [DOI] [PubMed] [Google Scholar]
- Gao R, Cao B, Hu Y, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- Dawood FS, Iuliano AD, Reed C, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12:687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- New influenza A (H1N1) virus: global epidemiological situation, June 2009. Wkly Epidemiol Rec. 2009;84:249–257. [PubMed] [Google Scholar]
- Abelin A, Colegate T, Gardner S, Hehme N, Palache A. Lessons from pandemic influenza A(H1N1): the research-based vaccine industry's perspective. Vaccine. 2011;29:1135–1138. doi: 10.1016/j.vaccine.2010.11.042. [DOI] [PubMed] [Google Scholar]
- Neumann G, Kawaoka Y. The first influenza pandemic of the new millennium. Influenza Other Respi Viruses. 2011;5:157–166. doi: 10.1111/j.1750-2659.2011.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tizzoni M, Bajardi P, Poletto C, et al. Real-time numerical forecast of global epidemic spreading: case study of 2009 A/H1N1pdm. BMC Med. 2012;10:165. doi: 10.1186/1741-7015-10-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormitzer PR, Tsai TF, del Giudice G. New technologies for influenza vaccines. Hum Vaccin Immunother. 2012;8:45–58. doi: 10.4161/hv.8.1.18859. [DOI] [PubMed] [Google Scholar]
- Mooney AJ, Tompkins SM. Experimental vaccines against potentially pandemic and highly pathogenic avian influenza viruses. Future Virol. 2013;8:25–41. doi: 10.2217/fvl.12.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petsch B, Schnee M, Vogel AB, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol. 2012;30:1210–1216. doi: 10.1038/nbt.2436. [DOI] [PubMed] [Google Scholar]
- Dormitzer PR, Suphaphiphat P, Gibson DG, et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci Transl Med. 2013;5:185ra168. doi: 10.1126/scitranslmed.3006368. [DOI] [PubMed] [Google Scholar]
- Lalor PA, Webby RJ, Morrow J, et al. Plasmid DNA-based vaccines protect mice and ferrets against lethal challenge with A/Vietnam/1203/04 (H5N1) influenza virus. J Infect Dis. 2008;197:1643–1652. doi: 10.1086/588431. [DOI] [PubMed] [Google Scholar]
- Ledgerwood JE, Wei CJ, Hu Z, et al. DNA priming and influenza vaccine immunogenicity: two phase 1 open label randomised clinical trials. Lancet Infect Dis. 2011;11:916–924. doi: 10.1016/S1473-3099(11)70240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geall AJ, Verma A, Otten GR, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci USA. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolo S. Vaccination with messenger RNA (mRNA) Handb Exp Pharmacol. 2008;183:221–235. doi: 10.1007/978-3-540-72167-3_11. [DOI] [PubMed] [Google Scholar]
- Perri S, Greer CE, Thudium K, et al. An alphavirus replicon particle chimera derived from venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J Virol. 2003;77:10394–10403. doi: 10.1128/JVI.77.19.10394-10403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release. 2005;107:276–287. doi: 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Dormitzer PR, Rappuoli R, Casini D, et al. Adjuvant is necessary for a robust immune response to a single dose of H1N1 pandemic flu vaccine in mice. PLoS Curr. 2009;1:RRN1025. doi: 10.1371/currents.RRN1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito LA, Chan M, Baudner B, et al. An alternative renewable source of squalene for use in emulsion adjuvants. Vaccine. 2011;29:6262–6268. doi: 10.1016/j.vaccine.2011.06.067. [DOI] [PubMed] [Google Scholar]
- de Jong JC, Palache AM, Beyer WE, Rimmelzwaan GF, Boon AC, Osterhaus AD. Haemagglutination-inhibiting antibody to influenza virus. Dev Biol (Basel) 2003;115:63–73. [PubMed] [Google Scholar]
- Chen Y, Liang W, Yang S, et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WR, Coleman MT, Mostow SR, Kaye HS, Schoenbaum SC. Inactivated influenza vaccines. 2. Laboratory indices of protection. Postgrad Med J. 1973;49:159–163. doi: 10.1136/pgmj.49.569.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin SA. Vaccines: correlates of vaccine-induced immunity. Clin Infect Dis. 2008;47:401–409. doi: 10.1086/589862. [DOI] [PubMed] [Google Scholar]
- Black S, Nicolay U, Vesikari T, et al. Hemagglutination inhibition antibody titers as a correlate of protection for inactivated influenza vaccines in children. Pediatr Infect Dis J. 2011;30:1081–1085. doi: 10.1097/INF.0b013e3182367662. [DOI] [PubMed] [Google Scholar]
- Fleeton MN, Chen M, Berglund P, et al. Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J Infect Dis. 2001;183:1395–1398. doi: 10.1086/319857. [DOI] [PubMed] [Google Scholar]
- Cai Y, Rodriguez S, Hebel H. DNA vaccine manufacture: scale and quality. Expert Rev Vaccines. 2009;8:1277–1291. doi: 10.1586/erv.09.84. [DOI] [PubMed] [Google Scholar]
- Gill P, Ghaemi A. Nucleic acid isothermal amplification technologies: a review. Nucleosides Nucleotides Nucleic Acids. 2008;27:224–243. doi: 10.1080/15257770701845204. [DOI] [PubMed] [Google Scholar]
- Ulmer JB, Mason PW, Geall A, Mandl CW. RNA-based vaccines. Vaccine. 2012;30:4414–4418. doi: 10.1016/j.vaccine.2012.04.060. [DOI] [PubMed] [Google Scholar]
- Geall AJ, Mandl CW, Ulmer JB.RNA: The new revolution in nucleic acid vaccines Semin Immunol 2013. June 1. 10.1016/j.smim.2013.05.001 [DOI] [PubMed]
- Pascolo S. Messenger RNA-based vaccines. Expert Opin Biol Ther. 2004;4:1285–1294. doi: 10.1517/14712598.4.8.1285. [DOI] [PubMed] [Google Scholar]
- Jones KL, Drane D, Gowans EJ. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques. 2007;43:675–681. doi: 10.2144/000112593. [DOI] [PMC free article] [PubMed] [Google Scholar]