

Abstract
Griseofulvin (1) is a spirocyclic fungal natural product used in treatment of fungal dermatophytes. Formation of the spirocycle, or the grisan scaffold, from a benzophenone precursor is critical for the activity of 1. In this study, we have systematically characterized each of the biosynthetic enzymes related to the biogenesis of 1, including the characterization of a new polyketide synthase GsfA that synthesize the benzophenone precursor and a cytochrome P450 GsfF that performs oxidative coupling between the orcinol and the phloroglucinol rings to yield the grisan structure. Notably, the finding of GsfF is in sharp contrast to the copper-dependent dihydrogeodin oxidase that performs a similar reaction in the geodin biosynthetic pathway. The biosynthetic knowledge enabled the in vitro total biosynthesis of 1 from malonyl-CoA using all purified enzyme components. This work therefore completely maps out the previously unresolved enzymology of the biosynthesis of a therapeutically relevant natural product
Keywords: Antifungal, polyketides, cytochrome P450
Griseofulvin (1), a polyketide produced by ascomycetes such as Penicillium aethiopicum,1 is the first oral antifungal drug2-4 and has been in use for many years in medical and veterinary applications.5 While 1 is largely superseded by newer antifungal drugs such as azoles and echinocandins, 1 has a niche in treatment of dermatophytes such as tinea capitis (ringworm of the scalp) and tinea pedis (athlete's foot). 1 recently gained medical attention due to its ability to disrupt mitotic spindle6 and act as potential inhibitor of centrosomal clustering7 in tumor cells. From a biosynthetic perspective, 1 occupies a place in the pantheon of natural products due to its significance in establishing the field of study of polyketide biosynthesis.8 The acetate origin of the carbon skeleton of 1 was demonstrated through the feeding of [1-14C]-acetate to the griseofulvin-producing strain Penicillium griseofulvum Dierckx and the labeling pattern in 1 was determined using degradation experiments.9, 10 The polyketide hypothesis11 was further confirmed through feeding of singly and doubly [13C]-acetate followed by NMR analysis (Figure 1).12 In addition, the formation of the spirocyclic grisan ring of 1 was an examples used to illustrate the prevalence of phenol oxidative coupling reactions in Nature.13 Subsequent studies using feeding of the radiolabeled benzophenone intermediates griseopheone B (10) and C (11) implicated their role in the biosynthesis of 1 and supported further suggests the requirement of oxidative coupling to form the grisan ring.14-16
Figure 1.

Griseofulvin (1) and other secondary metabolites produced by Penicillium aethiopicum. A.) The structure of 1 and dechlorogriseofulvin (2) shown together with the acetate origin of carbon and oxygen atoms. B.) Viridicatumtoxin (3) and tryptoquialanine (4) are the other chemotaxonomic marker in P. aethiopicum; and C.) Arrangement of genes in the biosynthetic gene cluster of 1 in P. aethiopicum.
Despite these pioneering studies, the genetic and biochemical basis of the biosynthetic pathways of 1 have been left unexplored. Considering the importance of 1 to the field of natural products, specifically the polyketide community, the complete understanding of how its carbon backbone is assembled and is subsequently transformed into 1 is therefore compelling. We recently sequenced the genome of P. aethiopicum to map its biosynthetic capacity.17 In addition to 1 and the related metabolite dechlorogriseofulvin (2), P. aethiopicum also produces viridicatumtoxin (3) and tryptoquialanine (4), two other secondary metabolites with spirocyclic moiety (Figure 1B).1, 17, 18 Bioinformatic mining of the P. aethiopicum genome led to the discovery of the putative gsf cluster (Figure 1C), which contains gsfA, a gene for a non-reducing polyketide (NR-PKS) and genes for tailoring enzymes that appear consistent with the modifications required to form 1. Gene deletion of gsfA and flavin-dependent halogenase gene gsfI confirmed the association between gsf cluster and biosynthesis of 1.17 The enzymology of the NR-PKS, which synthesizes a polyketide backbone atypical to the characterized aromatic polyketides, and the enzymes involved in maturation of the griseofulvin scaffold structure, highlighted by the spirocyclic grisan ring, can now be studied using modern genetic and biochemical tools. In this work, we present the comprehensive single-gene knockout and in vitro reconstitution of every enzyme in the gsf pathway. These insights led to the in vitro synthesis of 1 from malonyl-CoA using purified gsf enzymes and cofactors.
RESULTS AND DISCUSSION
GsfA is a norlichexanthone synthase
Our initial genetic studies with P. aethiopicum showed that gsfA, which encodes a thioesterase (TE)-less NR-PKS, is required to produce the heptaketide backbone of 1.17 To biochemically investigate the role of GsfA, gsfA cDNA was cloned into the YEpLac195 yeast-E. coli shuttle vector driven by the ADH2 promoter. The resulting YepLac195-GsfA construct was transformed into S. cerevisiae BJ5464-NpgA strain, which contain a chromosomal copy of npgA, the phosphopantetheine transferase gene from Aspergillus nidulans which post- translationally installs the phosphopantetheine arm to the acyl carrier protein (ACP) domain of PKS.19 Intact holo-GsfA (185 kDa) with a C-terminal His-tag was purified to near homogeneity to a final titer of 2 mg/L of yeast culture.
To assay the activity of recombinant GsfA, we incubated the enzyme with either malonyl-CoA directly or with an in situ malonyl-CoA generation system (Figure 2). Furthermore, since GsfA lacks a TE domain that may release the mature polyketide product and no standalone TE is found in the gene cluster, we incubated GsfA with a promiscuous TE domain from the hypothemycin NR-PKS Hpm3.20 Surprisingly, GsfA alone was sufficient for the production and release of a predominant product 5 with m/z=259, [M+H]+ (Figure 2). Addition of the Hpm3 TE domain did not enhance the turnover of the product 5. To purify sufficient amount of 5 for structural characterization, we cultured the BJ5464-NpgA strain expressing GsfA, which was able to produce 5 at a final titer of 10 mg/L. Compound purification followed by NMR characterization identified 5 as norlichexanthone (Figure 2), which bears the expected heptaketide backbone.
Figure 2.

GsfA catalyzes formation of 5 from malonyl-CoA as confirmed by HPLC analysis. i.) No GsfA control ii.) GsfA and the excised TE domain of Hpm3 incubated with in situ generated malonyl-CoA. iii.) GsfA incubated with in situ generated malonyl-CoA using MatB, malonate, CoA and ATP. iv.) GsfA incubated with malonyl-CoA only.
A typical NR-PKS contains starter-unit-ACP transacylase (SAT),21 ketosynthase (KS), malonyl-CoA-ACP transacylase (AT), product template (PT),22, 23 ACP and TE domains.24-28 The cyclization regioselectivity of the nascent polyketide chain is determined by the functions of the PT and the TE domains.22-24 The PT domain, in particular, mediates the regioselective aldol-type cyclization through steric interactions with the newly-formed polyketide.22, 23 Using phylogenetic analysis and domain shuffling experiment, we previously classified ~100 PT domains into distinct clades based on ring size and first aldol condensation regioselectivity.29 Interestingly, the GsfA PT does not classify into any of the clades, in correspondence with its unique cyclization selectivity. Indeed, based on previous labeling studies and the biochemical studies using purified GsfA, we can conclude that GsfA PT must catalyze the unusual C8-C13 aldol condensation as the first cyclization step in the formation of 5.
The enzymatic basis of the Claisen-like condensation (C1-C6) to afford the phloroglucinol ring, however, is unclear. GsfA belongs to a TE-less NR-PKS, of which no terminal TE domain is found in the NR-PKS. For TE-less NR-PKSs, a standalone β-lactamase-like enzyme is typically present in the gene cluster to release the product either through hydrolysis or Claisen-like condensation.30 No gene for such releasing enzyme is present in the gsf cluster, and our reconstitution studies show that GsfA alone is able to turnover products. Therefore the activities to catalyze the C1-C6 cyclization must be within the GsfA NR-PKS. It is reasonable to propose that because of the smaller size of the polyketide, the PT domain, when distorting the polyketide backbone for the aldol cyclization, may also promote the C1-C6 cyclization in its active site. Hence, formation of 5 represents an intriguing example in which a NR-PKS performs a Claisen-like cyclization without using a dedicated, in-line or dissociated TE. While the two cyclization events in the biosynthesis of 5 (and 5a) are unusual, it is far from unique. To wit, compounds bearing the norlichexanthone scaffold have been isolated from lichens.31 Thus one can reasonably speculate that a GsfA homolog may be found in the fungal symbionts in the lichens that produce compounds related to 5.
Methylation of GsfA product by GsfB and GsfC hinders xanthone formation
Although the xanthone 5 is detected as the only polyketide product from the in vitro assay, it is most likely that the true heptaketide product of GsfA is the benzophenone 5a (Table 1), which can undergo a spontaneous dehydration to form 5. Formation of the grisan structure in 1 must proceed through the intermediate 5a, thereby making 5 a likely off-pathway shunt product. Indeed, feeding of 5 to the ΔgsfA mutant did not restore the production of 1 (Figure. S1), thereby ruling out the possibility of 5 undergoing rehydration to open the xanthone ring and form 5a during biosynthesis of 1. We hypothesize that the methylation of the phenols in 5a, especially at 9-OH may hinder dehydration and suppress xanthone formation. To investigate this hypothesis, genetic inactivation and in vitro reconstitution of the three O-methyltransferases (MTs) GsfB, GsfC and GsfD were performed. Deletion of gsfB led to the production of 5 as a minor product, 7 (obs. m/z= 291, [M+H]+), 8 (obs m/z= 325, [M+H]+), and 9 (obs. m/z= 337, [M+H]+) (Figure 3, ii). Elucidation of the structures of the compounds using 1D 1H and 13C and 2D HSQC and HMBC NMR revealed the identity of 7 as griseophenone E, 8 as griseophenone F and 9 as desmethyl-dehydrogriseofulvin B. 7-9 share the characteristic lack of methylation at 3-OH, suggesting that GsfB is the 3-OH MT. The isolation of 9 also suggests that downstream steps, including the formation of the grisan structure, are not dependent on the 3-OH methylation step. On the other hand, the ΔgsfC mutant showed 5 as a more prominent product in comparison to the metabolic profile of the ΔgsfB, along with 6 (obs. m/z = 291, [M+H]+), 16 (obs. m/z = 273, [M+H]+) and 17 (obs. m/z= 325, [M+H]+), which were identified as griseophenone D, griseoxanthone C and griseophenone H respectively (Figure 3, iv). In contrast to the metabolites found in ΔgsfB mutant, 6, 16 and 17 are methylated at 3-OH, but lack methylation at 9-OH, thereby confirming that GsfC is the 9-OH MT. Isolation of these benzophenone intermediates in the ΔgsfB and ΔgsfC mutants confirms our hypothesis that methylations at 3-OH and more so at 9-OH, can suppress the formation of the off-pathway shunt product 5.
Table 1.
Putative functions of the genes in the 1 biosynthetic pathway and identified metabolites from single-gene knockout strains.*
| Gene | Function | Identified metabolites from KO strains |
|---|---|---|
| gsfA | NR-PKS (SAT-KS-AT-PT-ACP) | None* |
| gsfB | (3-OH) O-methyltransferase | 5, 7, 8, 9 |
| gsfC | (9-OH), O-methyltransferase | 6, 16, 17 |
| gsfD | (5-OH), O-methyltransferase | 13, 14, 15 |
| gsfE | short-chain dehydrogenase/reductase | 18, 19 |
| gsfF | cytochrome P450 | 10, 11, 12 |
| gsfI | halogenase | 2 * |
| gsfK | ketoreductase | 1, 2 |
Knockout of gsfA and gsfI was reported in a previous study by our group (ref 17).
Figure 3.

The O-methyltransferases GsfB and GsfC tailor the nascent GsfA product. i.) Secondary metabolic profile of Penicillium aethiopicum showing the production of 1-4. ii.) Deletion of gsfB led to the production of 5, 7, 8, and 9. iii.) Coupled in vitro assay of GsfA and GsfC led to the production of 7 thereby confirming 9-OH regioselectivity of GsfC iv.) Deletion of gsfC led to the production of 5, 6, 16 and 17. v.) Coupled in vitro assay of GsfA and GsfB led to the production of 5, 6 and 16 thereby confirming the 3-OH regioselectivity of GsfB.
In order to verify the roles of the MTs, in vitro assay of GsfA together with each of the recombinant MT was performed. The coupled reaction with GsfA generates the labile benzophenone 5a in situ that serve as substrates for the MTs. Assay containing GsfA and GsfB produced predominantly xanthones 5 and 16, as well as 6, demonstrating partial suppression of dehydration after methylation of 3-OH (Figure 3, v). Presumably, 6 undergoes dehydration to form 16. Alternatively, the nascent polyketide product of GsfA undergoes dehydration to 5 followed by methylation by GsfB to form 16. The coupled assay of GsfA with GsfC, on the other hand, produced predominantly 7 as expected, with a trace amount of 5, further confirming the 9-OH methylation step as a means to keep the GsfA product 5a on pathway during biosynthesis of 1 (Figure 3, iii). Taken together, these studies with MTs confirm that the GsfA product undergoes methylation by both GsfB and GsfC before forming the grisan ring, which is in agreement of the previous feeding studies.14, 15
On the other hand, knockout of gsfD yielded the production of grisan-containing compounds desmethyl-dechlorogriseofulvin (13) (obs. m/z = 305, [M+H]+), desmethyl-dehydrogriseofulvin A (14) (obs. m/z = 337, [M+H]+), and desmethyl-griseofulvin (15) (obs. m/z = 339, [M+H]+) (Figure 4, iii). These metabolites represent late-stage shunt products (13 and 15) and intermediate (14) during the formation of 1. The structures of 13-15 show the lack of methylation at the 5-OH position, implying that GsfD regioselectively targets 5-OH. Based on these results, we propose the role of GsfD is to methylate of 14 to produce dehydrogriseofulvin (18).
Figure 4.

Tailoring reactions in biosynthesis of 1. i.) Wild-type P.aethiopicum showing the production of 1-4. ii.) Knockout of cytochrome P450 gene gsfF led to the production of the dimethylated phenones 10, 11 & 12 containing different degrees of chlorination. iii.) Knockout of the O-methyltransferase gsfD led to the production of 13, 14 and 15. iv.) Knockout of the short-chain dehydrogenase /reductase gene gsfE led to the production of 18, and 19.
Biochemical confirmation of the role of GsfI
Previously, loss of 1 and sole production of 2 were observed when the gene gsfI encoding the flavin-dependent halogenase was deleted in P. aethiopicum.17 To biochemically reconstitute the halogenation reaction, we cloned, expressed and purified GsfI and performed an in vitro assay using 10, the proposed intermediate from the previous labeling studies,15 as substrate. In order to regenerate the reduced flavin cofactor of GsfI, the NADPH-dependent flavin reductase SsuE was added to the reaction. As expected, assay of the non-chlorinated 10 with GsfI led to the production of chlorinated phenone 11 at ~50% conversion(Figure S4), reflecting the ratio of the chlorinated final compound 1 and its dechlorinated counterpart 2 in the original host.
GsfF performs phenolic coupling to afford the grisan scaffold
Oxidation of the phenone ring to form the grisan ring dramatically transforms the phenone scaffold of griseophenone B (11)into the spirocyclic grisan compound 14, thereby dearomatizing the orcinol ring of 11 and creating the cyclohexadienone in 14. An analogous reaction during biosynthesis of geodin in A. terreus was proposed to be catalyzed by copper-centered enzyme dehydrogeodin oxidase.32 However, such a copper laccase enzyme is not encoded in the gsf cluster, thus we proposed that the cytochrome P450 (CYP) GsfF may instead catalyze the phenolic coupling reaction of 11 to afford 14.17 To probe the role of GsfF, we deleted gsfF in P. aethiopicum and characterized the resulting metabolites via LC-MS and NMR. The ΔgsfF strain produced griseophenone C (10) (obs. m/z = 305, [M+H]+), monochlorinated 11 (obs. m/z = 339, [M+H]+) and dichlorinated griseophenone G (12) (obs. m/z = 373, [M+H]+) (Figure 4, ii). Accumulation of 10 and 11 as major intermediates supports the coupling role of GsfF in the pathway. The degrees of chlorination of the metabolites were further ascertained based on the presence of the expected 3:1 isotopic distribution of the monochlorinated 11 and the 1.5:1 isotopic distribution of the dichlorinated 12 (Supporting Information). The C2-regioselectivity of the first chlorination reaction was determined by the loss of the symmetry along the C3-C6 axis of 11 in comparison to 10, as evident in comparative NMR analysis. The symmetry along C3-C6 axis as shown in the NMR spectra was restored upon further chlorination of 11 to afford 12, thus confirming the regioselectivity of the second chlorination at C4.
To functionally confirm the role of GsfF, the CYP was expressed in S. cerevisiae BJ5464-NpgA strain together with the A. terreus CYP oxidoreductase (CPR) using Gal1 and Gal10 bidirectional promoter,33 allowing the temporal coexpression of the CYP and the CPR that is required for turnover of the CYP. Following induction of GsfF and CPR expression, 11 was supplemented to the yeast culture at a final concentration of 1 μM. The organic metabolites were extracted twenty-four hours later with ethyl acetate and analyzed by LCMS. S. cerevisiae cells expressing both CPR and GsfF were able to completely convert 11 to a product that is identical (UV, mass and retention time) to 14 isolated from ΔgsfD mutant (Figure 5, ii). In contrast, yeast cells expressing only CPR did not show any conversion of 11(Figure 5, i). To further prove the conversion of 11 to 14 is catalyzed by GsfF, microsomal fraction from yeast cells coexpressing CPR and GsfF was isolated and in vitro assay was performed in the presence of NADPH. Microsomes containing both GsfF and CPR facilitated 90% conversion of 11 to 14 after overnight incubation (Figure 5, iv). Time course kinetics of GsfF revealed conversion of 11 to 14 at a turnover rate of 0.437 μM min−1·mg−1 of microsomal protein (Supporting Information). In contrast, identically prepared microsomes containing only CPR did not shown any conversion (Figure 5, iii). Therefore, we unequivocally confirmed the role of GsfF in the oxidative formation of the spirocyclic grisan.
Figure 5.

The cytochrome P450 GsfF catalyzes the coupling of orcinol and phloroglucinol rings in griseophenone B (11) to form desmethyl-dehydrogriseofulvin A(14). i.) BJ5464-NpgA expressing CPR only did not convert 11 to 14. ii.) BJ5464-NpgA expressing both CPR and GsfF completely converted 11 to 14 after 24 hours. iii.) Yeast microsomes containing CPR do not convert 11 in vitro. iv.) Yeast microsomes containing both CPR and GsfF converted 11 to 14 in the presence of NADPH. v.) Standard of 14 purified from ΔgsfD deletion strain.
In this coupling reaction, the orcinol ring is contorted leading to the metamorphosis of the di-aromatic 11 into the tricyclic 14. Based on the feeding studies in the biosynthesis of 1 using [1,1-18O]-labeled acetate, all of the oxygen atoms in 1 originated from acetate.34 Based on known reactions catalyzed by cytochrome P450, one can propose two different mechanisms for GsfF reaction (Scheme 2): A.) a di-radical mechanism and B.) a mechanism involving the formation of an arene oxide intermediate (11b). In the first mechanism, the reaction is initiated by the formation of Compound I, the active form of the heme cofactor of GsfF. Compound I then abstracts a hydrogen atom from 11 to form the resonance-stabilized phenoxy radical on the orcinol ring. Thereafter, Compound II form of GsfF abstracts the second hydrogen atom from 11a to form the phenoxy radical on the phloroglucinol ring with water as the by-product of the reaction. The incipient phloroglucinol radical then couples with the orcinol radical in a stereospecific fashion to form 14. This di-radical mechanism, first envisioned by Barton and Cohen,13 has also been proposed for the mechanisms of the coupling between the isoquinoline and phenolic rings in reticuline by salutaridine synthase during morphine biosynthesis,35 the coupling of the two indole rings of chromopyrrolic acid by StaP to form indolocarbazole in the biosynthesis of staurosporine,36 and the dimerization of coniferyl alcohol monomers in the biosynthesis of lignin.37 Incidentally, both salutaridine synthase and StaP are CYP-type oxygenases as well.35, 36 Moreover, this proposed mechanism is in agreement with the [1,1-18O]-acetate labeling study since all oxygen atoms are retained during the transformation of 11 to 14.34
Scheme 2.

Putative mechanisms of the orcinol and phloroglucinol ring coupling by GsfF. A.) Di-radical ring coupling mechanism of GsfF. B.) Alternative mechanism for the ring coupling involving an arene oxide intermediate.
Alternatively, the oxidation of 11 by GsfF can go through an arene oxide intermediate (11b), which will then be subjected to nucleophilic attack by the hydroxyl of the phloroglucinol ring to form 11c. The hemiacetal intermediate then undergoes dehydration to afford 14. During the dehydration of the hemiacetal intermediate, the oxygen atom that originated from molecular oxygen is removed. Thus, this mechanism agrees as well with the labeling studies.34 An analogous mechanism was proposed, along with a di-radical mechanism, in the ring coupling reaction by the CYP OxyB in the biosynthesis of the vancomycin family of antibiotics.38, 39
GsfE performs stereospecific reduction of enol 18 to afford the final product 1
Having functionally characterized the PKS GsfA, the MTs GsfB-D, the chlorinase GsfI and the P450 GsfF, the only uncharacterized biosynthetic step in the pathway is reduction of the cyclohexadienone ring in 18 or dechloro-dehydrogriseofulvin (19) to afford 1 or 2, respectively. Two of the remaining uncharacterized genes in the cluster, gsfE and gsfK; are for proteins that contain a conserved binding site for a nicotinamide cofactor and one of these may be capable of performing the final enoylreduction. Thus, single gene deletion mutants of gsfE and gsfK were constructed followed by metabolite analysis. All of the ΔgsfK mutants isolated retained the ability to produce 1 and 2. On the other hand, knockout of gsfE led to the production of 18 and 19 (Figure 4, iv), both of which were structurally verified by extensive NMR characterization (Supporting Information). Interestingly, while initial bioinformatic analysis of GsfE suggested that it is related to nucleoside-sugar epimerases based on sequence alignment, further analysis of the GsfE protein sequence via structural homology prediction revealed that it shares an overall folding, as well as conserved catalytic lysine and tyrosine as the progesterone-5β-reductase (POR) from the cardenolide biosynthetic pathway in Digitalis lanata.40 POR catalyzes the stereospecific reduction of progesterone to form 5β-pregnane-3,20-dione, a reaction that is analogous to the reduction of the cyclohexadienone ring of 18 to form 1. To verify the genetic and bioinformatic results, we reconstituted the reaction in vitro using His-tag fused-GsfE expressed and purified from E. coli BL21 (DE3), and 18 or 19 as substrates. NADPH was added as the reducing cofactor of GsfE. As shown in Figure 6A and Figure S5, GsfE fully converted 18 or 19 to 1 or 2, respectively, thereby confirming its role as the last enzyme in the pathway. Moreover, this confirms GsfE performs a 1,4- (Michael-type) hydride addition instead a 1,2-addition performed by nucleoside sugar epimerases (Figure 6B).
Figure 6.

Verifying the activity of GsfE. A.) Incubation of 18 with 10 M GsfE and 1 mM NADPH led to the production of 1. B.) Putative mechanism of GsfE showing the stereospecific 1,4-(Michael) addition of the hydride from NADPH to 18, followed by protonation of the resulting cyclohexadienolate to form 1.
Total in vitro biosynthesis of 1 and 2
Upon identifying all enzymatic components of the biosynthetic pathway, we attempted in vitro synthesis of 1 and 2 using purified Gsf enzymes (see Methods). Incubation of GsfA-F with malonyl-CoA, SAM and NADPH in PBS resulted in the expected production of 2 as a major product, along with 5, 10 and 13. Addition of GsfD-F after three-hour preincubation of GsfA-C with malonyl-CoA and SAM led to the production of 2 and 10 only. Finally, addition of the chlorinase GsfI and SsuE together with GsfA-F led to the production of the desired product 1 as a major product along with 2, 10, 11 and griseophenone A (20) (Figure 7, iii). The final yield of of 1 with respect to malonyl-CoA was ~1%. The presence of 10 and 11 in all assays indicate the microsomal GsfF-catalyzed oxidative coupling is the rate limiting step in the multi-enzyme reactions.
Figure 7.

In vitro total biosynthesis of 1 and 2. i.) GsfA-F, SAM and malonyl-CoA added in one step. ii.) GsfA-C, SAM and malonyl-CoA incubated together for 3 hours prior to addition of NADPH, GsfF ,GsfD and GsfE. iii.) GsfA-C, GsfI, SsuE, SAM, malonyl-CoA and NADPH incubated together for 3 hours prior to addition of GsfF, GsfD and GsfE.
Based on the results from the genetic and enzymatic data, we can establish the individual steps in the biosynthetic pathway for 1 (Scheme 3). The biosynthesis is initiated by the formation of the heptaketide by GsfA using one acetyl-CoA starter unit and six malonyl-CoA extender units. Thereafter, the product template (PT) domain of GsfA catalyzes the unprecedented C8-C13 aldol-type cyclization followed by C1-C6 Claisen-type cyclization to form 5a, wherein dehydration of 5a can readily take place in the absence of downstream steps to form the xanthone shunt product 5. Meanwhile, on pathway processing of 5a continues through the methylation at 3-OH and 9-OH by GsfB and GsfC, respectively, to form 10. Although feeding studies by Harris et al. suggest that 3-OH methylation has priority over 9-OH methylation,16 results from the knockout of gsfB and gsfC indicate that either can perform the first methylation reaction on the GsfA product 5a. Following the two methylation steps, 10 undergoes chlorination by the flavin-dependent halogenase GsfI to form 11. Following chlorination, 11 undergoes phenolic ring coupling by the cytochrome P450 GsfF to form the grisan compound 14. Subsequently, 14 is subjected to two additional tailoring steps: methylation at 5-OH and reduction of the cyclohexadienone to afford 1. Based on the presence of 14 in the ΔgsfD mutant, indicating the inefficient reduction by GsfE of the 5-OH-desmethyl substrates, it can be proposed that methylation by GsfD to yield 18 takes place before GsfE-catalyzed enoylreduction, which is in agreement with previous feeding studies on the biosynthesis of 1.14-16, 34 The chlorination step by GsfI is apparently incomplete in P. aethiopicum and in vitro (Figure 7). Enzymes downstream of GsfI, including GsfF, GsfD and GsfE, can all act on the respective dechlorinated substrates to yield the final product 2. In conclusion, we performed comprehensive single-gene deletion of the gsf genes and functional characterization of the corresponding enzymes to elucidate the biosynthetic pathway of 1. Several grisan analogues of 1 were isolated from the different gene deletion mutants (Scheme 1), hinting at the overall flexibility of the pathway and its potential for biosynthetic engineering. Moreover, this study has uncovered the novel and interesting reactions within this pathway, such as the unorthodox C1-C6 and C8-C13 cyclization of 5a by GsfA, and P450-mediated ring coupling reaction of the orcinol and phloroglucinol rings of 11 to form 14. These two reactions warrant further mechanistic investigations that may possibly lead to a better understanding of the biosynthesis of natural products with similar features.
Scheme 3.

Biosynthetic pathway of 1.
Scheme 1.

Shunt products and intermediates that contain the grisan scaffold.
MATERIALS AND METHODS
In vitro assay of GsfA
For the in vitro synthesis of 5, 10 μM GsfA was incubated with 2 mM malonyl-CoA and 15 μM tailoring enzyme (Hpm TE, GsfB, GsfC or GsfD) in 100 mM phosphate buffered saline pH 7.4 in a 100 μL reaction. The reaction was incubated overnight and extracted twice with ethyl acetate. For the reactions using malonyl-CoA generated in situ by MatB, the assay was done essentially the same as above except malonyl-CoA was substituted with 20 μM MatB, 5mM CoA, 100 mM malonate, 5 mM MgCl2, 5mM DTT and 20 mM ATP. The organic phase was dried and dissolved in 20 μL methanol and subjected to LCMS analysis as described in Supplemental Information.
Chemical analysis and compound isolation from P. aethiopicum gene deletion mutants
For small-scale secondary metabolic profile analysis, the P.aethiopicum wild-type and transformants were grown in 10-20 mL YMEG liquid medium for 7 days at 28 °C without shaking. The cultures were extracted with equal volume of ethyl acetate with 1% acetic acid, evaporated to dryness and redissolved in methanol for LCMS analysis in the same manner as the in vitro characterization of GsfA (). For preparative scale compound isolation, gene deletion strain of P.aethiopicum was grown in 3 L YMEG in the same manner as the small-scale cultures. The culture was extracted twice with equal volume of ethyl acetate and was evaporated to dryness and subjected to Sephadex and HPLC purification and NMR characterization in the same manner as 5.
In vitro assay of GsfI
The assay for the chlorinase was done essentially the same as described in Zhou et al.41 Briefly, 50 μM of GsfI was incubated with 200 μM Griseophenone C (10) in 100 mM sodium phosphate buffer (pH 7.4) and 50 mM NaCl. In order to regenerate the reduced flavin in GsfI, the flavin reductase SsuE (15 μM), FAD (5 μM) and NADPH (2 mM) were added to the reaction mix. After overnight incubation, the reaction mix was extracted twice with equal volume ethyl acetate, dried in vacuo and subjected to LCMS analysis as described above for 5.
In vitro assay of GsfE
For in vitro synthesis of 1 from 18, 10 μM of GsfE was incubated with 100 μM and 1 mM NADPH in 100 mM Tris-HCl (pH 7.5) buffer and 100 μL reaction volume. After 5 hour incubation in room temperature, the reaction mix was extracted twice with equal volume of ethyl acetate, dried in vacuo and analyzed by LCMS in the same manner as 5 .
Total in vitro biosynthesis of 1 and 2
For the total in vitro synthesis of 2, 4 μM of GsfA, 2 μM each of the methyltransferases (GsfB, GsfC and GsfD), 2 μL of microsomal protein containing GsfF and AtCPR and 2 μM of GsfE were incubated with 2 mM of malonyl-CoA, 100 μM of S-adenosyl methionine and 2 mM of NADPH in 100 μL reaction mix buffered with 100 mM sodium phosphate (pH 7.4) and 50 mM NaCl. After overnight incubation, the reaction mix was extracted twice with equal volume ethyl acetate, dried and subjected to the same LCMS analysis as 5 (vide supra). The total in vitro biosynthesis of 1 was done in the same manner as 2, except GsfA-C were incubated with 10 μM of GsfI and 2 μM SsuE for 3 hours prior to addition of GsfF, GsfE and GsfD. The total yield of 1 was measured by comparing the area under the chromatogram peak of 1 against a standard curve of known amount of 1 injected and analyzed by LCMS.
Supplementary Material
Table 2.
Griseophenones characterized in this study.
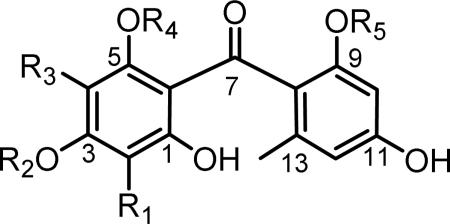
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 |
| 5a | H | H | H | H | H |
| griseophenone D (6) | H | Me | H | H | H |
| griseophenone E (7) | H | H | H | H | Me |
| griseophenone F (8) | Cl | H | H | H | Me |
| griseophenone C (10) | H | Me | H | H | Me |
| griseophenone B (11) | Cl | Me | H | H | Me |
| griseophenone G (12) | Cl | Me | Cl | H | Me |
| griseophenone H (17) | Cl | Me | H | H | H |
| griseophenone A (20) | Cl | Me | H | Me | Me |
ACKNOWLEDGMENT
This work is supported by NIH grants 1R01GM085128 and 1DP1GM106401 to Y.T.; and NRSA GM-0846 and the UCLA Graduate Division to R.A.C. NMR instrumentation was supported by the NSF equipment grant CHE-1048804. We thank D-K Ro for providing the pESC-Leu-AtCPR plasmid for the P450 reconstitution. We express our sincere gratitude to J.C. Vederas for helpful discussion on the biosynthetic routes.
ABBREVIATIONS
- PKS
polyketide synthase
- TE
thioesterase domain
- CYP
cytochrome P450
- SAM
S-adenosyl methionine
- SAH
S-adenosyl homocysteine
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
Footnotes
ASSOCIATED CONTENT
Supporting Information. Supplemental material and methods. NMR spectrum tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Frisvad JC, Samson RA. Polyphasic taxonomy of Penicillium subgenus Penicillium. A guide to identification of food and air-borne terverticillate Penicillia and their mycotoxins. Studies in Mycology. 2004;49:1–174. [Google Scholar]
- 2.Gentles JC. Experimental ringworm in guinea pigs: oral treatment with griseofulvin. Nature. 1958;182:476–477. doi: 10.1038/182476a0. [DOI] [PubMed] [Google Scholar]
- 3.Pillsbury DM. Griseofulvin theraphy in dermatophytic infections. Trans Am Clin Climatol Assoc. 1960;71:52–57. [PMC free article] [PubMed] [Google Scholar]
- 4.Gull K, Trinci APJ. Griseofulvin inhibits fungal mitosis. Nature. 1973;244:292–294. doi: 10.1038/244292a0. [DOI] [PubMed] [Google Scholar]
- 5.Finkelstein E, Amichai B, Grunwald MH. Griseofulvin and its uses. Int J Antimicrob Agents. 1996;6:189–194. doi: 10.1016/0924-8579(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 6.Panda D, Rathinasamy K, Santra MK, Wilson L. Kinetic suppression of microtubule dynamic instability by griseofulvin: Implications for its possible use in the treatment of cancer. Proc Natl Acad Sci USA. 2005;102:9878–9883. doi: 10.1073/pnas.0501821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebacz B, Larsen TO, Clausen MH, Rønnest MH, Löffler H, Ho AD, Krämer A. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res. 2007;67:6342–6350. doi: 10.1158/0008-5472.CAN-07-0663. [DOI] [PubMed] [Google Scholar]
- 8.Hutchinson CR. The use of isotopic hydrogen and nuclear magnetic resonance spectroscopic techniques for the analysis of biosynthetic pathways. J Nat Prod. 1982;45:27–37. [Google Scholar]
- 9.Birch AJ, Massy-Westropp RA, Rickards RW, Smith H. 66. Studies in relation to biosynthesis. Part XIII. Griseofulvin. J Chem Soc. 1958;0:360–365. [Google Scholar]
- 10.Birch A, Donovan F. Studies in relation to biosynthesis. I. Some possible routes to derivatives of orcinol and phloroglucinol. Aus J Chem. 1953;6:360–368. [Google Scholar]
- 11.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 12.Simpson TJ, Holker JSE. 13C-NMR studies on griseofulvin biosynthesis and acetate metabolism in Penicillium patulum. Phytochemistry. 1977;16:229–233. [Google Scholar]
- 13.Barton DHR, Cohen T. In Festschrift Prof. Dr. Arthur Stoll. Birkhauser; Basel: 1957. [Google Scholar]
- 14.Rhodes A, Boothroyd B, Mc GP, Somerfield GA. Biosynthesis of griseofulvin: the methylated benzophenone intermediates. Biochem J. 1961;81:28–37. doi: 10.1042/bj0810028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhodes A, Somerfield GA, McGonagle MP. Biosynthesis of griseofulvin. Observations on the incorporation of [14C]Griseophenone C and [36Cl]Griseophenones B and A. Biochem J. 1963;88:349–357. doi: 10.1042/bj0880349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris CM, Roberson JS, Harris TM. Biosynthesis of griseofulvin. J Am Chem Soc. 1976;98:5380–5386. doi: 10.1021/ja00433a053. [DOI] [PubMed] [Google Scholar]
- 17.Chooi Y-H, Cacho R, Tang Y. Identification of the viridicatumtoxin and griseofulvin gene clusters from Penicillium aethiopicum. Chem Biol. 2010;17:483–494. doi: 10.1016/j.chembiol.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao X, Chooi Y-H, Ames BD, Wang P, Walsh CT, Tang Y. Fungal indole alkaloid biosynthesis: genetic and biochemical investigation of the tryptoquialanine pathway in Penicillium aethiopicum. J Am Chem Soc. 2011;133:2729–2741. doi: 10.1021/ja1101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma SM, Li JW-H, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Complete reconstitution of a highly reducing iterative polyketide synthase. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Qiao K, Gao Z, Meehan MJ, Li JWH, Zhao X, Dorrestein PC, Vederas JC, Tang Y. Enzymatic synthesis of resorcylic acid lactones by cooperation of fungal iterative polyketide synthases involved in hypothemycin biosynthesis. J Am Chem Soc. 2010;132:4530–4531. doi: 10.1021/ja100060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crawford JM, Dancy BCR, Hill EA, Udwary DW, Townsend CA. Identification of a starter unit acyl-carrier protein transacylase domain in an iterative type I polyketide synthase. Proc Natl Acad Sci USA. 2006;103:16728–16733. doi: 10.1073/pnas.0604112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crawford JM, Korman TP, Labonte JW, Vagstad AL, Hill EA, Kamari-Bidkorpeh O, Tsai S-C, Townsend CA. Structural basis for biosynthetic programming of fungal aromatic polyketide cyclization. Nature. 2009;461:1139–1143. doi: 10.1038/nature08475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawford JM, Thomas PM, Scheerer JR, Vagstad AL, Kelleher NL, Townsend CA. Deconstruction of iterative multidomain polyketide synthase function. Science. 2008;320:243–246. doi: 10.1126/science.1154711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korman TP, Crawford JM, Labonte JW, Newman AG, Wong J, Townsend CA, Tsai S-C. Structure and function of an iterative polyketide synthase thioesterase domain catalyzing Claisen cyclization in aflatoxin biosynthesis. Proc Natl Acad Sci USA. 2010;107:6246–6251. doi: 10.1073/pnas.0913531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujii I, Watanabe A, Sankawa U, Ebizuka Y. Identification of Claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. Chem Biol. 2001;8:189–197. doi: 10.1016/s1074-5521(00)90068-1. [DOI] [PubMed] [Google Scholar]
- 26.Cox RJ. Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 27.Chooi Y-H, Tang Y. Navigating the fungal polyketide chemical space: from genes to molecules. J Org Chem. 2012;77:9933–9953. doi: 10.1021/jo301592k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townsend CA, Crawford JM. New insights into the formation of fungal aromatic polyketides. Nat Rev Micro. 2012;8:879–889. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Xu W, Tang Y. Classification, prediction, and verification of the regioselectivity of fungal polyketide synthase product template domains. J Biol Chem. 2010;285:22764–22773. doi: 10.1074/jbc.M110.128504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Chooi Y-H, Sheng Y, Valentine JS, Tang Y. Comparative characterization of fungal anthracenone and naphthacenedione biosynthetic pathways reveals an α-hydroxylation-dependent Claisen-like cyclization catalyzed by a dimanganese thioesterase. J Am Chem Soc. 2011;133:15773–15785. doi: 10.1021/ja206906d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peres V, Nagem TJ. Trioxygenated naturally occurring xanthones. Phytochemistry. 1997;44:191–214. doi: 10.1016/s0031-9422(00)00303-4. [DOI] [PubMed] [Google Scholar]
- 32.Huang K.-x., Fujii I, Ebizuka Y, Gomi K, Sankawa U. Molecular cloning and heterologous expression of the gene encoding dihydrogeodin oxidase, a multicopper blue enzyme from Aspergillus terreus. J Biol Chem. 1995;270:21495–21502. doi: 10.1074/jbc.270.37.21495. [DOI] [PubMed] [Google Scholar]
- 33.Partow S, Siewers V, Bjørn S, Nielsen J, Maury J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast. 2010;27:955–964. doi: 10.1002/yea.1806. [DOI] [PubMed] [Google Scholar]
- 34.Lane MP, Nakashima TT, Vederas JC. Biosynthetic source of oxygens in griseofulvin. Spin-echo resolution of oxygen-18 isotope shifts in carbon-13 NMR spectroscopy. J Am Chem Soc. 1982;104:913–915. [Google Scholar]
- 35.Gesell A, Rolf M, Ziegler J, Díaz Chávez ML, Huang F-C, Kutchan TM. CYP719B1 Is salutaridine synthase, the C-C phenol-coupling enzyme of morphine biosynthesis in opium poppy. J Biol Chem. 2009;284:24432–24442. doi: 10.1074/jbc.M109.033373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makino M, Sugimoto H, Shiro Y, Asamizu S, Onaka H, Nagano S. Crystal structures and catalytic mechanism of cytochrome P450 StaP that produces the indolocarbazole skeleton. Proc Natl Acad Sci USA. 2007;104:11591–11596. doi: 10.1073/pnas.0702946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W. Lignin biosynthesis and structure. Plant Physiol. 2010;153:895–905. doi: 10.1104/pp.110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zerbe K, Woithe K, Li DB, Vitali F, Bigler L, Robinson JA. An oxidative phenol coupling reaction catalyzed by OxyB, a cytochrome P450 from the vancomycin-producing microorganism. Angew Chem Int Ed. 2004;43:6709–6713. doi: 10.1002/anie.200461278. [DOI] [PubMed] [Google Scholar]
- 39.Holding AN, Spencer JB. Investigation into the mechanism of phenolic couplings during the biosynthesis of glycopeptide antibiotics. ChemBioChem. 2008;9:2209–2214. doi: 10.1002/cbic.200800303. [DOI] [PubMed] [Google Scholar]
- 40.Thorn A, Egerer-Sieber C, Jäger CM, Herl V, Müller-Uri F, Kreis W, Muller YA. The crystal structure of progesterone 5β-reductase from Digitalis lanata defines a novel class of short chain dehydrogenases/reductases. J Biol Chem. 2008;283:17260–17269. doi: 10.1074/jbc.M706185200. [DOI] [PubMed] [Google Scholar]
- 41.Zhou H, Qiao K, Gao Z, Vederas J, Tang Y. Insights into radicicol biosynthesis via heterologous synthesis of intermediates and analogs. J Biol Chem. 2010;285:41412–41421. doi: 10.1074/jbc.M110.183574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.