

Abstract
The demonstration of induced pluripotency and direct lineage conversion has led to remarkable insights regarding the roles of transcription factors and chromatin regulators in mediating cell state transitions. Beyond its considerable implications for regenerative medicine, this body of work is highly relevant to multiple stages of oncogenesis, from the initial cellular transformation to the hierarchical organization of established malignancies. Here we review conceptual parallels between the respective biological phenomena, highlighting important inter-relationships among transcription factors, chromatin regulators and pre-existing epigenetic states. The shared mechanisms provide insights into oncogenic transformation, tumor heterogeneity and cancer stem cell models.
Transcription factors and chromatin regulators: partners in specification of cellular fate and identity
Specification of cellular fate during development is a dynamic process by which diverse phenotypes are established in precise temporal and positional patterns. Beginning from a single totipotent cell, successive waves of self-renewal, differentiation and commitment ultimately yield the intricate array of cell types, tissues and organs of a fully formed organism. DNA sequence-specific transcription factors (TFs) play a prominent role in fate specification, as demonstrated by seminal studies of the muscle fate master regulator MyoD(1) and the core TFs that mediate pluripotency(2, 3). The classical dogma by which TFs act within proximal promoters to initiate transcription has been expanded by the identification of staggering numbers of distal ‘enhancer-like’ elements in the human genome, which are activated by TFs in combinatorial and highly cell type-specific patterns(4, 5).
In order to exert their proximal and distal regulatory activities, TFs must contend with the underlying organization of chromatin, a higher-order structure of DNA, RNA, histones and regulatory proteins(6, 7). TFs recruit chromatin regulators (CRs) that modulate the accessibility of target DNA and impart specific ‘chromatin states’ characterized by signature histone modifications and common functional roles(4). However, since TF binding is dependent on chromatin accessibility, CRs and chromatin states also function as gatekeepers that modulate TF regulatory activities. Differentiation events frequently rely on promoters and enhancers that are ‘poised’ by pioneer TFs and characteristic chromatin configurations(4, 8). Thus, a hierarchy of TFs, cooperating CRs and coordinated chromatin states guide successive differentiation and commitment events during developmental specification (Figure 1).
Figure 1. Developmental specification is associated with global alterations in chromatin structure.

A) In pluripotent cells, chromatin is hyper-dynamic and globally accessible. B) Upon differentiation, inactive genomic regions may be sequestered by repressive chromatin enriched for characteristic histone modifications and refractory to regulatory activity. These global structures are regulated by DNA methylation, histone modifications and numerous CRs whose expression levels are dynamically regulated through development. In addition, transcriptional changes are accompanied by focal alterations in chromatin structure at specific gene loci.
Lessons from induced pluripotency
In 2006, Shinya Yamanaka demonstrated induced pluripotency, whereby a differentiated cell can be directly reprogrammed into an ‘induced’ pluripotent stem (iPS) cell by a defined set of TFs(2, 3). The Nobel prizewinning discovery represented a seminal advance for the fields of stem cell and regenerative biology. Yet the finding and a flurry of follow-up studies may have equally profound implications for cancer biology. The body of work demonstrates the dramatic consequence of deploying gene regulatory mechanisms in inappropriate developmental contexts. It provides key insights into the mechanisms of action of TFs, CRs and chromatin states that direct, facilitate or hinder cell fate transitions. A striking number of the implicated factors and mechanisms are now recognized to play critical roles in malignant transformation. This review draws upon these shared themes in an examination of genetic and epigenetic mechanisms that contribute to cellular reprogramming and cancer.
Induced pluripotency was initially demonstrated by reprogramming fibroblasts with four TFs, Oct-4, Sox2, Klf4 and c-Myc. Only the ‘core’ factors Oct-4 and Sox2 are strictly required, whereas the other components may primarily enhance reprogramming efficiency and can be substituted by other genes such as Nanog and Lin28(2, 3, 9). Demonstrations of direct conversion between cell lineages reinforce that master TFs determine cellular identity(10, 11). The right combination of TFs can drive state transitions, binding synergistically to promoters and enhancers to activate gene networks. Reprogramming also involves focal and global changes to chromatin structure as required to reset the epigenetic landscape(12). In iPS reprogramming, de novo chromatin activation, mediated by TF recruitment of CRs and associated transcriptional changes, occurs early(13). In contrast, the formation of bivalent domains and the global chromatin decondensation characteristic of pluripotent cells appear to represent later event(3, 12). These changes involve chromatin modification and remodeling, rendering reprogramming dependent on CRs that catalyze these activities. Moreover, pre-existing chromatin states and DNA methylation can present roadblocks that impede TF binding and gene induction, thus hindering cell state transitions(14–16).
Reprogramming and cancer epigenetics
Oncogenic transformation frequently involves de novo acquisition of developmental programs, analogous to cellular reprogramming, and yields cells with unlimited self-renewal potential, a feature shared with iPS and other directly reprogrammed stem cells. This parallelism is supported at a mechanistic level by facilitators and barriers shared between the processes(12). Several reprogramming TFs represent bona fide oncogenes, while many genes that act as barriers to reprogramming correspond to known tumor suppressors, including p53 and Ink4A/Arf, whose effects on proliferation and apoptosis impede both processes(12). Similarly, CRs play essential roles as effectors and modulators of reprogramming, and have established functions in oncogenesis(7, 12). These general principles suggest that epigenetic rewiring necessary for cellular reprogramming may be recapitulated in part during cellular transformation.
Transcription factors
Each of the iPS reprogramming factors has established roles in oncogenesis (Figure 2A). Oct-4 plays a driving role in initiating germ cell tumors, and represents an important diagnostic marker(17). Sox2 is amplified in squamous cell carcinoma of the lung and esophagus(18), and small cell lung carcinomas(19), acting as a lineage-survival oncogene in each of these tumors. Sox2 is also an essential driver of cancer stem cell sub-populations in glioblastoma, breast cancer and Ewing sarcoma, consistent with its vital role in pluripotent and tissue stem cells (Figure 2A)(20, 21).
Figure 2. Genes involved in both iPS nuclear reprogramming and cancer.

List of TFs (A) and CRs (B) implicated in iPS reprogramming together with the malignancies in which they have established roles. These include bona fide oncogenes and tumor suppressors directly affected by genetic alterations as well as other genes with mechanistic roles in cancer.
c-Myc functions in a wide range of human malignancies. Its expression may also explain the tendency of mice generated from iPS clones to spontaneously develop tumors(2). c-Myc may drive proliferation through induction of common gene expression programs in ES cells and various malignancies. Alternatively, c-Myc may function as a global amplifier of gene expression through its potent effect on the RNA polymerase II elongation factor P-TEFb(22, 23). This alternative view may explain how a single oncogene can reinforce the pluripotency network, while also driving a wide range of malignant programs(24).
Klf4 plays dual roles in cancer, promoting the development of breast and skin malignancies but suppressing gastric, colorectal, and bladder cancer(25). The mechanism by which Klf4 promotes tumor initiation may relate to its functions in nuclear reprogramming. These include functional links to c-Myc and simultaneous repression of p53, which both enhance cellular proliferation. Klf4 also induces telomerase activity in ES cells and carcinoma cells by promoting β-catenin binding to the TERT gene promoter(26). Conversely, suppression of cancer initiation by Klf4 in specific contexts may be mediated through induction of the pro-apoptotic gene CDKN1A(25).
Nanog is expressed in multiple malignancies and appears to have critical functions in cancer stem cell subpopulations. In hepatocellular carcinoma, it is required for CD24-positive stem-like cells, where its expression is maintained by STAT3 signaling(27). In glioblastoma, induction of Gli1 by Nanog increases the clonogenic and tumorigenic potential of the CD133-positive stem cell fraction(28). Nanog is also expressed in stem-like populations in colon and prostate carcinomas, and Ewing sarcoma (Figure 2A)(20, 29, 30).
Lin28 plays central roles in stem cell biology, accelerating iPS reprogramming and promoting ES cell proliferation(31). This RNA-binding protein is notable for expression across a significant proportion of human cancers, particularly undifferentiated and advanced malignancies. Lin28 potently inhibits maturation of the Let-7 microRNA family leading to derepression of multiple oncogenes, including c-Myc, K-Ras, Sall4 and Hmga2. Recent evidence supports additional mechanisms of action, including a direct effect on Oct-4 translation. These roles may explain the ability of Lin28 to supplement c-Myc in reprogramming, and also underlie its oncogenic properties.
In addition to these pluripotency factors, Olig2, Foxg1 and other TFs that mediate direct lineage conversion are implicated in human malignancies, thus generalizing the link between reprogramming and oncogenic transformation(10). It is important to note, however, that the parallels are not complete and that the roles of an individual factor are not necessarily identical in the two processes, and may even diverge within different malignant settings. Moreover, the strict analogy to reprogramming fails to appreciate the oncogenic roles of many other TFs, such as transforming fusion proteins identified in hematopoietic malignancies and, more recently, in solid tumors.
DNA methylation
DNA methylation is a relatively stable epigenetic modification that mediates silencing of repetitive elements and certain gene promoters(32). Methylation patterns must be reset during cellular reprogramming, and can present a potent barrier to the process(12, 14). In cancer, hyper-methylation of CpG islands is a well recognized epigenetic event(32). Genetic inactivation of the DNA methyltransferase Dnmt3a has also been documented in leukemia and lymphoma(32, 33). Although the mechanisms remain obscure, DNA methylation may negatively influence transformation by impeding the induction of genes needed for epigenetic reprogramming or, alternatively, may promote oncogenesis by silencing genes that mediate differentiation or apoptosis. Hence, this epigenetic feature could play markedly different roles in alternate malignant settings. Indeed, seemingly contradictory roles are observed for several CRs in different tumor types, as discussed below.
Chromatin regulators (CRs)
CRs have established roles in cellular reprogramming and oncogenesis (Figure 2B). Their contributions to malignancy involve multiple modalities. Like specific TFs, many CRs represent bona fide oncogenes or tumor suppressors, and are directly affected by gain- and loss-of-function genetic mutations or by translocations(7, 32). Even in the absence of direct genetic alterations, CRs may be co-opted by fusion proteins or other oncogenic factors to modulate gene expression. CRs also regulate other cancer-relevant processes, including epithelial-to-mesenchymal transition (EMT), senescence, genome stability and metastasis. These diverse modalities and the fact that some CRs have opposite effects in different malignant settings present a challenge for predicting how a given CR mutation, altered chromatin state or epigenetic therapy will impact a particular tumor.
Repressive chromatin states
In differentiated cells, inactive portions of the genome are partitioned between different forms of repressive chromatin. Repressive structures with a compact organization refractory to regulatory activity can affect large genomic regions (Figure 1). Canonical repressive states include classical heterochromatin marked by histone H3 lysine 9 trimethylation (H3K9me3), broad Polycomb-repressed regions enriched for H3K27me3, and highly condensed regions associated with the nuclear lamina and H3K9me2(4, 6).
H3K9 methylation and associated CRs play important roles in chromosomal stability, EMT and cellular senescence(32, 34, 35). Large H3K9me3 domains that arise in certain differentiated cells appear to impede the initial binding of reprogramming TFs and may be inefficiently reset during iPS generation(15, 16). Accordingly, suppression of CRs that catalyze H3K9 methylation, including Suv39h1, Setdb1 and G9a, increases reprogramming efficiency(36, 37). H3K27me3 and associated Polycomb repressors play critical roles in tissue-specific gene regulation(38). Ezh2 and Utx, which catalyze the addition and removal of H3K27me3, respectively, are both required for efficient iPS reprogramming(3, 36, 39).
Suv39h1 is required for oncogene-induced senescence, and its loss is associated with decreased viability, genomic instability and increased tumor risk in mice(35). Recruitment of Suv39h1 to aberrant gene targets by fusion proteins promotes acute myeloid leukemia(40). Setdb1 and G9a have established role in a number of malignancies (Figure 2B), and G9a has also been implicated in metastasis and EMT(41, 42). Polycomb regulators are subject to genetic mutation or over-expression in a wide range of malignancies (Figure 2B)(43–49). In particular, Ezh2 and Bmi1 mediate CDKN2A epigenetic silencing and are essential in cancer stem cells (7, 32, 38).
Active chromatin states
Active genes and regulatory elements are associated with characteristic chromatin states. Enhancers and promoters are marked by varying degree of H3K4 methylation and histone acetylation (Figure 1). The methylation mark is catalyzed by complexes that contain Mixed Lineage Leukemia (Mll) homologs (related to Drosophila trithorax) and accessory subunits such as Wdr5(7). These CRs play important roles in reprogramming and cancer. Wdr5 directly interacts with the pluripotency TF Oct4 and is essential for iPS cell generation(12, 50). Mll fusion proteins represent potent leukemic drivers, although they typically lack the catalytic domain and instead appear to function via co-factor recruitment(7). Inactivating mutations of MLL homologs have also been identified in multiple tumors (Figure 2B). The H3K4 demethylase Lsd1, which functions as a coactivator or corepressor in different contexts(7), also has diverse roles in cancer (Figure 2B)(7, 51, 52).
The transition from transcriptional initiation to elongation is a critical regulatory step. Dot1l, an H3K79 methyltransferase that promotes this transition, impedes iPS reprogramming(36) and is required for leukemogenesis(53). Mll fusion proteins mediate their aberrant regulatory programs through recruitment of Dot1l. Accordingly, Mll-rearranged leukemias are sensitive to small molecule Dot1l inhibitors(53). The multiple myeloma fusion protein Mmset and Kdm2b respectively catalyze the addition and removal of H3K36 methylation, a characteristic mark of elongating transcripts(7, 54, 55). Kdm2b promotes iPS formation(56), can immortalize primary cells(57), and contributes to the development and maintenance of leukemic stem cells(58).
Many other CRs associated with active chromatin states or chromatin remodeling have been implicated in neoplastic transformation. The CBP/p300 histone acetyltransferase complex functions as a coactivator for specific TFs at proximal and distal regulatory elements, driving epigenetic programs involved in cellular proliferation, apoptosis, differentiation and DNA stability(59). CBP/p300 also acetylates non-histone proteins, including p53, Rb and c-Myc, increasing their transcriptional activity. Chromosomal rearrangements involving CBP/p300 promote hematopoietic malignancies, whereas loss-of-function mutations have been identified in solid tumors, including colorectal and breast carcinomas(7, 59). Dynamic chromatin regulation also requires nucleosome remodeling. The BAF remodeling complex markedly increases reprogramming efficiency by facilitating Oct4 binding to target loci(60). SWI/SNF homologs, including ARID1A, are subject to recurrent genetic inactivation in a broad range of tumors, though the underlying mechanisms remain unclear (Figure 2B)(7, 61–63).
These and other studies thus document widespread roles for CRs in human cancer, complementing similarly pervasive functions in development and reprogramming. The fact that CRs with similar catalytic activities and developmental phenotypes can lead to apparently contradictory outcomes in different malignant settings may relate to their diverse roles in gene regulation, differentiation, senescence, telomere regulation and other processes. The same CR might function in alternate contexts to facilitate epigenetic reprogramming, to arrest epigenetic state of a progenitor cell by differentiation block, or to counteract apoptosis or senescence. Substantial variability in the epigenetic makeups of pre-malignant target cells or the maintenance requirements of a particular tumor may also help explain varied and sometimes incongruous roles for TFs and CR mutations across the spectrum of human cancers.
From reprogramming to cancer stem cells and beyond
Here we have explored conceptual parallels between reprogramming and cancer, and highlighted shared TFs and CRs unveiled by mechanistic studies and cancer genome sequencing. The analogy to reprogramming suggests that some CR alterations in cancer may represent early events that render a cell-of-origin susceptible to epigenetic rewiring required for transformation and refractory to differentiation, proliferation arrest or apoptosis. Subsequent hits, occurring in variable orders and combinations, may then confer the definitive transformed state and the dynamic cellular hierarchy within a tumor (Figure 3). In certain cancers, “stem-like” cells critical for tumor initiation and growth occupy the apex of this hierarchy(64). Just as the reprogramming field is deciphering the combinatorial TF code for cellular identity, cancer research is increasingly focused on the determinants of heterogeneous epigenetic states in tumors. By activating specific TF drivers and modulating collaborating CRs, cancer cells may dynamically regulate their epigenetic circuits to rewire differentiated cancer cells into “stem-like” cells, thus refueling cancer growth. Although speculative, such dynamic bidirectional transitions could provide a unifying view of cellular organization within tumors, compatible with both the cancer stem cell and the stochastic models. Regardless of which models best explain a given malignancy, principles and mechanisms shared between cellular reprogramming and oncogenic transformation provide fundamental insights into tumor biology with potential to guide biological understanding, diagnosis and therapeutic strategies.
Figure 3. Cellular hierarchies in normal tissues and malignancies.
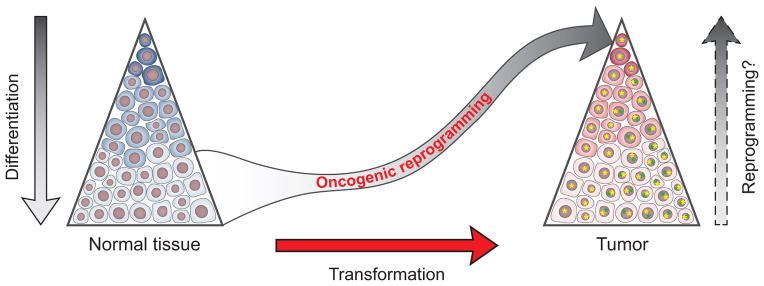
Normal tissues (left) and a growing list of malignancies (right) have established epigenetic hierarchy, with rare populations of stem cells giving rise to more differentiated cellular progeny through intermediate steps (color shades). Reprogramming experiments have shown that differentiation is reversible (left and right arrows). Cellular transformation (red arrow) is a stepwise process involving accumulation of genetic and epigenetic hits. Once initiated, additional and potentially divergent alterations may occur, establishing a tumor with genetic heterogeneity (illustrated by yellow and green *) and, within each genetic subclone, an epigenetic hierarchy (color shades). Altered activity of key regulators, including CRs and TFs, can play dual roles in cancer, contributing to transformation and epigenetic state transitions (“oncogenic reprogramming”). We speculate that the same network of regulators may then act within the established tumor to rewire differentiated cancer cells into stem-like cells, thus establishing a dynamic equilibrium between differentiation and reprogramming.
Acknowledgments
We thank E. Rheinbay, R. Ryan, M. Rivera, I. Bernstein and A. Meissner for critical comments, and L. Gaffney for figure design. Supported by the Howard Hughes Medical Institute, the National Human Genome Research Institute, the NIH Common Fund, the National Heart, Lung and Blood Institute, the Harvard Stem Cell Institute, the Burroughs Wellcome Fund, the Medic Foundation, the Nuovo Soldati Foundation, the Swiss National Science Foundation and Oncosuisse.
References
- 1.Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- 2.Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell stem cell. 2012;10:678. doi: 10.1016/j.stem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–25. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 5.Consortium EP, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Smale ST. Designing an enhancer landscape. Cell. 2012;151:929. doi: 10.1016/j.cell.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 10.Morris SA, Daley GQ. A blueprint for engineering cell fate: current technologies to reprogram cell identity. Cell Res. 2013;23:33. doi: 10.1038/cr.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vierbuchen T, Wernig M. Direct lineage conversions: unnatural but useful? Nat Biotechnol. 2011;29:892–907. doi: 10.1038/nbt.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koche RP, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell stem cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, et al. Genome-wide Chromatin State Transitions Associated with Developmental and Environmental Cues. Cell. 2013;152:642. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gidekel S, Pizov G, Bergman Y, Pikarsky E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell. 2003;4:361. doi: 10.1016/s1535-6108(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 18.Bass AJ, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudin CM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riggi N, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010;24:916. doi: 10.1101/gad.1899710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell stem cell. 2013;12:15. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin CY, et al. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell. 2012;151:56. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nie Z, et al. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell. 2012;151:68. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang CV. MYC on the path to cancer. Cell. 2012;149:22. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nature reviews Cancer. 2006;6:11. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmeyer K, et al. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- 27.Lee TK, et al. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell stem cell. 2011;9:50. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Zbinden M, et al. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ibrahim EE, et al. Embryonic NANOG Activity Defines Colorectal Cancer Stem Cells and Modulates through AP1- and TCF-dependent Mechanisms. Stem Cells. 2012;30:2076. doi: 10.1002/stem.1182. [DOI] [PubMed] [Google Scholar]
- 30.Jeter CR, et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell. 2010;140:445. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews Cancer. 2011;11:726. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nature structural & molecular biology. 2011;18:867. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peters AH, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 36.Onder TT, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet. 2012 doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 38.Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell stem cell. 2010;7:299. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mansour AA, et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488:409. doi: 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- 40.Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene. 2010;29:576. doi: 10.1038/onc.2009.361. [DOI] [PubMed] [Google Scholar]
- 41.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–8. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ceol CJ, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson G, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Haaften G, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41:521. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ntziachristos P, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18:298. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sneeringer CJ, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ang YS, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris WJ, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 53.Bernt KM, Armstrong SA. A role for DOT1L in MLL-rearranged leukemias. Epigenomics. 2011;3:667. doi: 10.2217/epi.11.98. [DOI] [PubMed] [Google Scholar]
- 54.Kuo AJ, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Molecular cell. 2011;44:609. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez-Garcia E, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang G, He J, Zhang Y. Kdm2b promotes induced pluripotent stem cell generation by facilitating gene activation early in reprogramming. Nat Cell Biol. 2012;14:457. doi: 10.1038/ncb2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tzatsos A, et al. Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J Biol Chem. 2011;286:33061. doi: 10.1074/jbc.M111.257667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117:3869. doi: 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 60.Singhal N, et al. Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell. 2010;141:943. doi: 10.1016/j.cell.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 61.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parsons DW, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nature reviews Cancer. 2012;12:133–43. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]