

Abstract
Glutathione (GSH) was discovered in yeast cells in 1888. Studies of GSH in mammalian cells before the 1980s focused exclusively on its function for the detoxication of xenobiotics or for drug metabolism in the liver, in which GSH is present at its highest concentration in the body. Increasing evidence has demonstrated other important roles of GSH in the brain, not only for the detoxication of xenobiotics but also for antioxidant defense and the regulation of intracellular redox homeostasis. GSH also regulates cell signaling, protein function, gene expression, and cell differentiation/proliferation in the brain. Clinically, inborn errors in GSH-related enzymes are very rare, but disorders of GSH metabolism are common in major neurodegenerative diseases showing GSH depletion and increased levels of oxidative stress in the brain. GSH depletion would precipitate oxidative damage in the brain, leading to neurodegenerative diseases. This review focuses on the significance of GSH function, the synthesis of GSH and its metabolism, and clinical disorders of GSH metabolism. A potential approach to increase brain GSH levels against neurodegeneration is also discussed.
Keywords: glutathione, cysteine transport, oxidative stress, neurodegeneration, EAAC1, GTRAP3-18
1. Introduction
Glutathione (GSH) was discovered in yeast by two independent scientists over a century ago. In 1888, J. de Rey-Pailhade identified a substance from yeast cells, which he named “philothione” (from the Greek words meaning “love” and “sulfur”) because of its reactivity with sulfur to form hydrogen sulfide [1,2]. Subsequently, F.G. Hopkins reported this substance as a dipeptide containing glutamate and cysteine, and he named it “glutathione” [3], which is actually a tripeptide consisting of glutamate, cysteine, and glycine [4,5]. GSH is useful to weaken the strength of bread dough for baking, by its reaction with gluten in wheat. However, the significance of GSH function in living cells did not receive much attention until the 1970s, when a variety of studies on GSH-related biochemical reactions and its metabolism emerged. The studies of GSH since then have revealed that GSH is involved in pivotal cellular physiological processes, i.e., antioxidant defense, detoxication of xenobiotics, intracellular redox homeostasis, cysteine carrier/storage, cell signaling, protein function, gene expression, and cell differentiation/proliferation. Thus, dysfunction of GSH metabolism can cause lethal cellular events. Early clinical studies of GSH metabolism focused solely on GSH-related enzyme dysfunction or the GSH-related metabolism of drugs and endogenous compounds in the liver. However, accumulating lines of evidence from recent studies indicate that disorder of GSH function is implicated in the etiology of some neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), progressive supranuclear palsy (PSP), Huntington’s disease (HD), and multiple sclerosis (MS). This review focuses on the significance of GSH function, GSH synthesis and its metabolism, clinical disorders of GSH metabolism, and therapeutic strategies for the treatment of GSH-related neurodegenerative diseases.
2. Glutathione Synthesis
GSH is produced intracellularly from three amino acids—glutamate, cysteine and glycine—through two consecutive steps catalyzed by γ-glutamylcysteine ligase (GCL, also known as γ-glutamylcysteine synthetase) and GSH synthetase (GS) (Figure 1). GCL mediates the first step, which is the ATP-requiring reaction with glutamate and cysteine to form a dipeptide, γ-glutamylcysteine (γGluCys). The mammalian GCL is a heterodimer enzyme consisting of an approx. 73-kDa catalytic (heavy) subunit, GCLC, and an approx. 28-kDa modulatory (light) subunit, GCLM. GCLC, but not GCLM, has all the enzymatic activity and is subject to feedback inhibition by GSH [6]. GCLM has no enzymatic activity; however, the association of GCLM with GCLC decreases the Km value for glutamate and increases the Ki value for the feedback inhibition of GSH [7]. GS catalyzes the second step, another ATP-requiring reaction with γGluCys and glycine to form GSH, although much less is known about the precise mechanisms underlying the regulation of GS activity compared to those of GCL.
Figure 1.
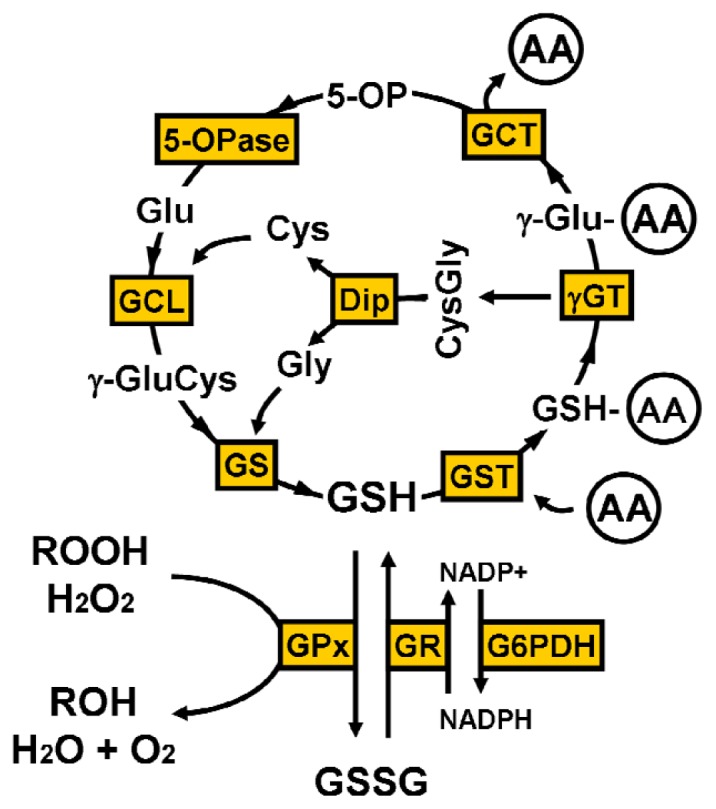
The γ-glutamyl cycle. AA, amino acids; Cys, cysteine; CysGly, cysteinylglycine; Dip, dipeptidase; GCL, γ-glutamylcysteine ligase; GCT, γ-glutamyl cyclotransferase; γGT, γ-glutamyl transpeptidase; γGluCys, γ-glutamylcysteine; Glu, glutamate; Gly, glycine; G6PDH, glucose-6-phosphate dehydrogenase; GPx, glutathione peroxidase; GR, glutathione reductase; GS, glutathione synthetase; GSH, glutathione; GSSG, glutathione disulfide; GST, glutathione-S-transferase; H2O2, hydrogen peroxide; NADPH, nicotinamide adenine dinucleotide phosphate; 5-OP, 5-oxoproline; 5-OPase, 5-oxoprolinase; ROH, alcohol; ROOH, hydroperoxide.
For GSH synthesis, the Km value of GCL for cysteine is ~0.15 mM, while that for glutamate is ~1.7 mM, and that of GS for glycine is ~0.8 mM [8]. The levels of intracellular glutamate (1–10 mM) [9] and glycine (2 mM in astrocytes, 10 mM in neurons) [10] are much higher than the Km values, whereas the intracellular cysteine level is around its Km value [8]. Although intracellular amino acid levels vary with tissues or cell types, cysteine concentrations in the brain are maintained at levels that are lower than those of glutamate or glycine [11] because of its neurotoxicity [12,13]. Therefore, the intracellular cysteine level is considered the rate-limiting factor for GSH synthesis in the brain.
3. Glutathione in the Brain
3.1. Distribution of GSH in the Brain
GSH serves approximately 95% of total non-protein thiol groups in living cells [14] and is ubiquitously distributed throughout the human body. However, GSH levels vary according to the organs (0.5–10 mM) [15,16]; the highest levels are found in the liver (5–10 mM), followed by the kidney, spleen, small intestine, brain, pancreas, lung, heart, and muscle [17]. The brain contains GSH at a varying concentration of approximately 2–3 mM [18] in different regions; it is highest in the cortex, followed by the cerebellum, hippocampus, and striatum, and lowest in the substantia nigra (SN) [19]. The GSH level in the cerebrospinal fluid (CSF) is much lower (~5 μM) than those of brain tissues [20,21]. The GSH levels also depend on cell types in the brain. Primarily, the GSH level is lower in neurons than in astrocytes [22]. Microglia contain higher GSH levels than neurons or astrocytes in vitro [23,24]. These findings suggest that the regulatory mechanisms of GSH metabolism/homeostasis are specific to the organs and cell types.
3.2. Thiol Source for the Brain
GSH is a non-toxic storage form of cysteine at levels 10–100 times higher than that of cysteine in mammalian tissues [18]. Approximately one-third to one-half of the total liver GSH serves as a cysteine reservoir that can be released into the blood as necessary [25,26]. However, the released GSH does not reach the brain directly because of the blood-brain barrier (BBB), which consists of a layer of capillary endothelial cells surrounded by astrocytes that functions as a selective barrier protecting the brain from xenobiotics in the blood. GSH can penetrate the BBB only poorly by the mechanism of passive diffusion, because of its hydrophilic property [27]. It is as yet unclear whether a direct GSH transport system exists at the BBB. Intravenously administered GSH is rapidly metabolized in blood [28,29]. Consistently, plasma GSH levels are much lower (2–20 μM) than those in the liver [16].
Cysteine is also unable to penetrate the BBB because of its lack of an acidic omega side chain, which facilitates the transport through the BBB [30]. In contrast, the disulfide form with two cysteines, called cystine, is transported from blood into the endothelial cells at the BBB via a cystine transporter, called system xc−, and is subsequently transported out of the endothelial cells into the CSF via the l-type amino acid transporter LAT1 at the BBB [31,32]. Favorably, cystine presents in the plasma at much higher concentrations (50–100 μM) than cysteine (10–25 μM), GSH, or other thiol derivatives [16,21,33]. These findings suggest that plasma cystine is the main thiol source of the brain [21].
3.3. Thiol Source for Glutathione Synthesis in the Brain
In the CSF, the cystine levels are relatively lower but the cysteine and GSH levels are higher compared to those in the blood [21]. These findings suggest a conversion of plasma cystine into other thiol derivatives in the central nervous system (CNS). Cystine is the primary source of GSH synthesis in astrocytes expressing system xc−, which is a sodium-independent cystine/glutamate antiporter composed of two subunits, xCT and 4F2hc [34], mainly present on glial cells [35,36]. Cystine imported into astrocytes is intracellularly reduced back to cysteine, which is used as a substrate for astroglial GSH synthesis. Astrocytes can also take up intact dipeptides, γGluCys and cysteinylglycine (CysGly), for GSH synthesis [37,38]. Astrocytes reserve high GSH contents at the concentration of approx. 8 mM [39] intracellularly, and they can export 10% of intracellular GSH within 1 h through multidrug resistance proteins (MRPs), which are members of the family of ATP-binding cassette transporters [40]. The released GSH is cleaved into γ-glutamyl moiety and CysGly by the reaction of an ectoenzyme on the glial plasma membrane, γ-glutamyl transpeptidase (γGT). CysGly is hydrolyzed by aminopeptidase N to cysteine and glycine, which are subsequently transported into neurons for GSH synthesis [41]. The released GSH may also react with intracellular cystine, which is transported from plasma, to form cysteine and cysteine-GSH. These metabolic interactions between astrocytes and neurons are essential as the source of cysteine or its precursor for neuronal GSH synthesis in the brain [42].
4. Glutathione Function
4.1. γ-Glutamyl Cycle
Reactive oxygen species (ROS) are produced during redox metabolism in cells. A large part (~90%) of ROS production is attributed to mitochondria. Initial observations indicated that approx. 2% of the total oxygen consumption is diverted to generate ROS [43], while more recent studies indicated that the basal value of ROS was reduced to ~0.2% under physiological conditions [44,45]. Superoxide, one of the typical ROS, is generated by the respiratory chain in mitochondria, leading to ATP production [46]. Superoxide is converted to hydrogen peroxide (H2O2) by two types of intracellular superoxide dismutase (SOD) under physiological conditions: Cu/Zn-SOD in the cytosol and Mn-SOD in the mitochondrial matrix. Subsequently, the produced H2O2 is catalyzed into water and molecular oxygen by catalase or GSH peroxidase (GPx). Catalase can react with H2O2 but not other hydroperoxides, whereas GPx can react with both H2O2 and other hydroperoxides. For H2O2 detoxication, the Km value of catalase is high, whereas that of GPx is low [47]. Therefore GPx, but not catalase, mainly compensates for the peroxide detoxication under physiological conditions in neurons [18].
In mammalian tissues, there are four types of selenium-containing GPx, one of which, GPx-1, is widely expressed in most brain areas and various cell types [48]. GPx-1 is the most abundant cytosolic GPx, and it functions as an important antioxidative enzyme to interact with fatty acid hydroperoxides as well as H2O2 in the brain [49,50]. Additional GPx isozymes have been also identified as gastrointestinal GPx (GPx-2), plasma GPx (GPx-3), and phospholipid hydroperoxide GPx (GPx-4) [49]. In contrast to other GPx isozymes, GPx-4 can interact with a wide range of phospholipid hydroperoxides as well as H2O2 in the brain [49,51]. In the processes of H2O2 or organic hydroperoxide detoxication by GPx, GSH is oxidized to GSH disulfide (GSSG) [52,53], which is then regenerated as GSH by the reaction with GSH reductase (GR) [54]. This reaction of GR with GSSG is regulated by nicotinamide adenine dinucleotide phosphate (NADPH), which is provided as a substrate for supplying electrons to GSSG by NADPH regenerating enzymes, such as glucose-6-phosphate dehydrogenase (G6PDH), 6-phosphogluconate dehydrogenase, NADP+-dependent isocitrate dehydrogenase, malic enzyme, and mitochondrial nicotinamide nucleotide transhydrogenase [24]. Although the reaction rate of GR is limited by the supply of NADPH, neuronal GR is sufficiently active to rapidly regenerate GSH from GSSG [55]. GSH-S-transferase (GST) is a family of enzymes that detoxify a variety of electrophilic xenobiotics with GSH-S-conjugation. GST metabolizes anticancer drugs, insecticides, herbicides, carcinogens, and by-products of oxidative stress. In mammalian species, there are seven classes of cytosolic GST isoforms; α, μ, π, σ, θ, ω, and ζ [56]. GSH, GSSG and GSH-S-conjugates are released to the extracellular space via MRPs [57,58]. After the cleavage of GSH and its conjugates into the γ-glutamyl moiety and CysGly by γGT, the γ-glutamyl moiety is degraded to the corresponding amino acid and 5-oxoproline by the reaction with γ-glutamyl cyclotransferase. 5-Oxoproline, also known as pyroglutamate, is then converted to glutamate by the reaction with 5-oxoprolinase (ATP-hydrolysing). GSH is metabolized to the related amino acids, which are reused for GSH synthesis to form the γ-glutamyl cycle (Figure 1).
4.2. Oxidative Stress
Despite these GSH-related antioxidant systems, the brain is especially vulnerable to oxidative stress because of the relatively lower antioxidant enzymatic activities (SOD, GPx, GR, and catalase) compared to those in other tissues [59]; the brain requires a large amount of O2, leading to a high production of ROS and lipid peroxidation. Nitric oxide (NO) is an important molecule that regulates physiological cell function and signaling and also a free radical leading to the generation of reactive nitrogen species (RNS) under pathological conditions. NO is produced by NO synthase (NOS) activation, and its brain concentrations under pathological conditions are approximately 100-fold higher than those under normal conditions [60,61]. Neither superoxide nor NO is toxic unless they react non-enzymatically with each other to form peroxynitrite, which is a potent RNS in the brain [62]. Peroxynitrite is produced at an estimated rate of 50–100 μM per min with a half-life of ~10−2 s and can diffuse within one to two cell diameters (~5–20 μm) [63] to cause lipid peroxidation, antioxidant depletion, antioxidant enzyme inhibition, and DNA damage [62,63]. The rate of peroxynitrite formation is accelerated in a synergistic manner if both superoxide and NO productions are elevated under pathological conditions [62].
Although SOD can remove superoxide to prevent peroxynitrite formation, the rate of reaction between superoxide and NO is much faster than that between superoxide and SOD [64,65]. Moreover, peroxynitrite can inactivate Mn-SOD by the nitration of tyrosine residue [66]. GSH reacts non-enzymatically with ROS such as superoxide, NO, hydroxyl radical, and peroxynitrite [67]. H2O2 is generated by the SOD-catalyzing reaction to superoxide in a cell. H2O2 itself is not particularly toxic in the physiological range; however, H2O2 should be removed by catalase or GPx to prevent the subsequent formation of hydroxyl radicals, which is considered a potent oxidant targeting sugars, amino acids, phospholipids, DNA bases, and organic acids [68]. Hydroxyl radicals are a highly toxic ROS produced by peroxynitrite decomposition or the Fenton reaction, leading to the ferrous iron-dependent decomposition of H2O2[62,63]. No known enzymatic defense has been reported against hydroxyl radicals. However, the toxicity of hydroxyl radicals is limited in the cell because of its short half-life (10−9 s) [69]. Hydroxyl radicals can diffuse only within a small distance, which is 10,000 times smaller than that of peroxynitrite, and moreover the rate of Fenton reaction is a million times slower than that of peroxynitrite formation [62,64].
4.3. S-Glutathionylation
All of the amino acid residues can be subject to posttranslational modifications such as oxidation or disulfide formation. Cysteine, which comprises up to 3% of the amino acids in human proteins [70], works as the most reactive nucleophilic residue in proteins. Oxidative/nitrosative damage can alter the redox state of the cell by reacting with thiol residues of redox-sensitive proteins. ROS/RNS induce irreversible protein modifications, such as carbonylation or nitration, leading to permanent loss of the functions of proteins as enzymes, receptors, and transporters [71]. Moreover, these irreversible modifications lead the proteins to their misfolding or aggregation as the targets for degradation by the ubiquitin-proteasome system (UPS) [72].
GSH is the major thiol redox buffer to maintain intracellular redox homeostasis. Under oxidative stress conditions, GSH can lead to the reversible formation of mixed disulfides between protein thiol groups (S-glutathionylation) to prevent irreversible protein oxidation [73]. S-glutathionylation is a reversible modification for restoring these protein functions when the intracellular redox state returns to normal after the insults. Disulfide bonds are also reduced back to thiol residues by the reaction with disulfide reductases, such as thioredoxin or glutaredoxin [74], which facilitate normal protein folding [75]. Protein S-glutathionylation is an important adaptive cellular response to protect crucial protein functions in the cell.
4.4. Thiol Redox State
Although the intracellular GSH/GSSG ratio may vary, ranging from 10 to 300, the ratio is more than 100 under the steady state, while it can transiently shift to ~10 or less under oxidative stress conditions [76,77]. The cellular thiol redox state is usually expressed by the GSH/GSSG ratio, which regulates gene transcription [78]. DNA binding activity of the transcription factors, such as c-Jun, NF-κB, and Fos, are modulated by changes in the GSH/GSSG ratio [79–81]; the decreased ratio induces S-glutathionylation of the cysteine residue in the DNA binding domains, leading to the inhibition of DNA binding. The inhibition of GSH synthesis arrests the cell cycle in the S and G2 phases [82], whereas proliferating cells in the S and G2 phases of the cell cycle showed increased GSH levels in the nucleus [83]. These results indicate that GSH is required at the appropriate period for cell proliferation.
The cellular thiol redox state also regulates programmed cell death [84]. A decreased intracellular GSH/GSSG ratio induces anti-apoptotic protein Bcl-2 loss, cytochrome c release from mitochondria, and caspase activations by the induction of the p38 mitogen-activated protein kinase pathway, whereas an increased intracellular GSH/GSSG ratio prevents the programmed cell death [85].
5. Cysteine Uptake for Neuronal Glutathione Synthesis
5.1. The Excitatory Amino Acid Transporters
Mature neurons use extracellular cysteine, but not cystine, for their GSH synthesis [40,86,87]. Approximately 90% of total cysteine uptake in neurons is mediated by sodium-dependent systems, mainly the excitatory amino acid transporter (EAAT), also known as system XAG- [88,89]. EAATs are high-affinity sodium-dependent glutamate transporters that remove extracellular glutamate in the CNS [88,90]. EAATs form trimers, which co-transport anionic amino acid with three Na+ and one H+ while counter-transporting one K+[91,92]. This transport system can concentrate intracellular glutamate 5 × 106-fold across the plasma membrane under equilibrium conditions [93]. To date, five EAATs have been cloned: glutamate aspartate transporter (GLAST, also termed EAAT1), glutamate transporter-1 (GLT-1, also termed EAAT2), excitatory amino acid carrier 1 (EAAC1, also termed EAAT3), EAAT4 and EAAT5 [88].
GLAST and GLT-1 are expressed mainly in astrocytes [88], and EAAC1, EAAT4 and EAAT5 are expressed in neurons [88]. EAAT4 and EAAT5 are exclusively localized to cerebellar Purkinje cells and the retina, respectively, whereas EAAC1 is widely expressed throughout the brain. Mature neurons in vivo do not express either GLAST or GLT-1, but do express EAAC1. GLAST and GLT-1 are expressed on the plasma membrane of glial processes surrounding the glutamatergic synapses, and EAAC1 is expressed mainly in the neuronal soma and dendrites, but not in the axons or synaptic terminals [88,94–97]. Indeed, the extracellular glutamate in the synaptic clefts is removed by glial EAATs, especially by GLT-1, but not by EAAC1 [97–99]. EAATs can transport not only extracellular glutamate but also cysteine into the cells [100]. In particular, EAAC1 can preferentially transport cysteine, rather than glutamate, into neurons. The relative efficacy of cysteine transport by EAAC1 is 10- to 20-fold higher than that of GLAST or GLT-1 [100]. These findings indicate that the main function of EAAC1 is not related to glutamatergic neurotransmission, but to cysteine metabolism in neurons.
5.2. Regulation of EAAC1 on Neuronal Glutathione Synthesis
Inhibition of EAAC1 expression by an antisense oligonucleotide significantly reduced the cysteine uptake, intracellular GSH levels, and cell viability against oxidative stress in cultured neurons [101]. Consistently, EAAC1-deficient mice show decreased GSH levels in the brain and vulnerability to oxidative stress. Interestingly, EAAC1-deficient mice also show brain atrophy, spatial learning and memory dysfunction, loss of dopaminergic neurons in the SN, and movement disorder at advanced ages but not when younger [102,103]. These findings suggest an involvement of EAAC1 dysfunction in brain GSH depletion leading to neurodegeneration (Figure 2).
Figure 2.
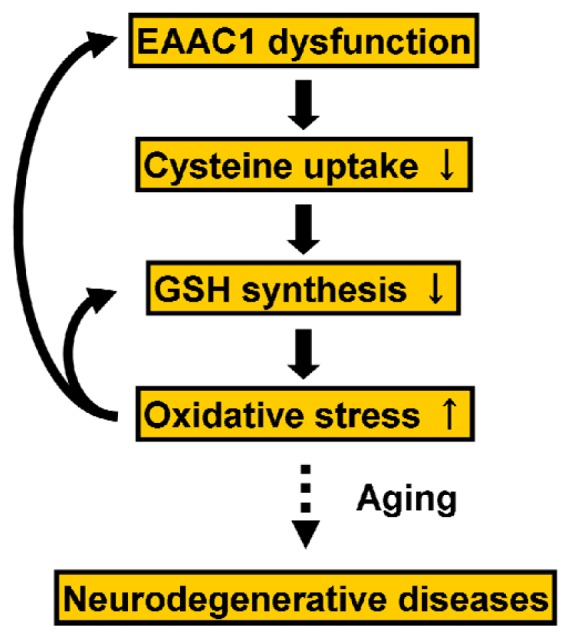
Possible mechanism of neurodegenerative diseases caused by GSH depletion via EAAC1 dysfunction.
The effect of EAAC1 on the glutamate/cysteine uptake into neurons is regulated by its expression on the plasma membrane. At steady state, only ~20% of total EAAC1 is expressed on the plasma membrane, whereas the cell surface expression was increased by twofold when stimulated by a protein kinase C activator, phorbol 12-myristate 13-acetate [104]. The cell surface expression of EAAC1 was also increased by stimulation with a phosphoinositide 3-kinase activator, platelet-derived growth factor [104]. EAAC1 translocation to the plasma membrane is negatively regulated by glutamate transport associated protein 3–18 (GTRAP3–18), which anchors EAAC1 in the endoplasmic reticulum [105]. GTRAP3–18 decreases the EAAC1-mediated cysteine uptake and GSH synthesis in neurons [11,106,107]. The precise regulatory mechanisms of EAAC1 and GTRAP3–18 are discussed in our previous reviews [108,109].
6. Inborn Errors in the GSH-Related Enzymes
6.1. γ-Glutamylcysteine Ligase
Disruption of the mouse GCLC gene causes embryonic lethality [110]. GCLM-deficient mice are viable and fertile with no obvious phenotype, but the GSH levels in their organs and plasma are low [111]. Loss of the GCLM gene induced premature senescence, increased intracellular ROS and DNA damage in primary fibroblasts [112]. Clinically, GCL deficiency is a very rare autosomal recessive disease that has been reported in only nine patients in seven families in the world. The patients have mutations in the gene encoding GCLC and show hemolytic anemia in all cases, and neurological symptoms such as spinocerebellar degeneration, mental retardation, peripheral neuropathy, myopathy and aminoaciduria in some cases [113]. The laboratory data show low GCL activity/levels and low GSH levels in erythrocytes and/or cultured skin fibroblasts. No promising treatment has been established.
6.2. Glutathione Synthetase
Disruption of the mouse GS gene also causes embryonic lethality [114]. Clinically, GS deficiency is the most common inborn error of GSH metabolism, reported in 77 patients in 65 families; it is characterized by autosomal recessive inheritance [114]. Some different mutations or epigenetic modifications of the human GS gene have been reported. The patients present hemolytic anemia, metabolic acidosis, and 5-oxoprolinuria. Severely affected patients also show progressive neurologic symptoms such as psychomotor/mental retardation, seizure, spasticity, ataxia, and intention tremor [113]. Laboratory data show increased γGluCys levels and cysteine in cultured fibroblasts and low GSH levels in erythrocytes and cultured fibroblasts. About 25% of the patients with GS deficiency die in the first year of life. The early administration of vitamin C (100 mg/kg/day) and/or vitamin E (10 mg/kg/day) improves the long-term clinical outcome [113].
6.3. γ-Glutamyl Transpeptidase
Human γGT deficiency is a very rare autosomal-recessive disease which has been reported in seven patients in five families worldwide [113]. The patients display increased GSH levels in plasma and urine, and in some cases, CNS involvement. Leukocytes or cultured fibroblasts from the patients exhibit low γGT activity. No mutation has been found in the patients. Clinically, no specific treatment has been established.
6.4. 5-Oxoprolinase
Human 5-oxoprolinase deficiency is a very rare autosomal-recessive disease reported in eight patients worldwide [113]. The patients exhibit low activity of 5-oxoprolinase in leukocytes or cultured skin fibroblasts, and 5-oxoprolinuria. Clinical manifestations of the patients include mental retardation, microcephaly, microcytic anemia, hypoglycemia, enterocolitis and renal stones. Clinically, no specific treatment has been reported.
6.5. Membrane-Bound Dipeptidase
Membrane-bound dipeptidase hydrolyzes dipeptides such as CysGly, which is produced in the subsequent process of GSH degradation after the reaction with γGT. The dipeptidase also catalyzes the conversion of leukotriene D4 to E4. Human dipeptidase deficiency is an extremely rare autosomal-recessive disease; it has been reported in only one patient worldwide [113]. The patient showed mental retardation, mild motor impairment, and deafness. The biochemical findings consisted of increased urinary excretion of both CysGly and leukotriene D4, and low dipeptidase activity in cultured fibroblasts and/or erythrocytes. No treatment has been reported yet.
7. Disorders of GSH Metabolism in Neurodegenerative Disease
Clinically, inborn errors in the GSH-related enzymes are very rare whereas disorders in GSH metabolism are common in some neurodegenerative diseases showing GSH depletion and increased levels of oxidative stress in the CNS (Figure 2). A recent in vivo method using nuclear magnetic resonance (NMR) spectroscopy makes it possible to measure GSH levels in the living brain [115]. Clinical studies using this method have demonstrated GSH depletion in patients with neurological disorders as described below. It is considered plausible that GSH depletion precedes neurodegeneration [116,117]. Many in vivo studies have shown both a GSH decline and increased ROS/RNS levels with aging in the brain [62,118]. The concept that older cells have less ability to prevent and remove oxidative damage is called “the free radical theory of aging” [119]. Aging also influences GSH homeostasis [118]. GSH depletion enhances oxidative stress, leading to neuronal degeneration [120–122]. Oxidative stress is involved in both normal aging and age-related neurodegenerative diseases [123,124]. Indeed, some neurodegenerative diseases have shown disorders of GSH metabolism in the brain, as discussed below.
7.1. Alzheimer’s Disease (AD)
AD is the most common age-related neurodegenerative disease. It is characterized by progressive dementia occurring in middle-aged or older populations. A recent clinical study using NMR spectroscopy showed reduced GSH levels in the brains of AD patients compared to healthy subjects [125]. No difference has been found in the concentrations of vitamins such as ascorbate or α-tocopherol in the CNS between AD and controls [126,127], suggesting a selective disorder of GSH metabolism in AD pathogenesis. Decreases in GPx and GST activities were observed in AD [128,129]. Genetic polymorphisms in the GPx-1 and GST genes were identified as positive risk factors for AD [130,131]. The Apo E gene has a genetic polymorphism coding three different protein isoforms; ɛ2, ɛ3, and ɛ4. Apo E ɛ4 has been known as a risk factor for AD [132]. Brain tissues from AD patients with the ɛ4 allele of ApoE show decreased GSH levels and GPx and catalase activities compared to those of AD patients homozygous for the ɛ3 allele [133]. Notably, the GSH levels in erythrocytes were reduced not only in AD, but also in mild cognitive impairment (MCI), which is considered the preclinical stage of AD [134]. MCI patients also showed decreased GSH/GSSG ratios and SOD and GST activities in the hippocampus compared to age-matched controls [135]. These results suggest that disorders of GSH metabolism occur before the onset of AD.
AD is pathologically characterized by depositions of amyloid β (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain. In the form of soluble oligomers, Aβ is most toxic, causing oxidative stress that leads to NFT formation and neuronal death [136,137]. Soluble Aβ oligomers inhibited the EAAC1-mediated cysteine uptake, resulting in a decrease in GSH levels in cultured human neuronal cells [138]. Consistently, postmortem brain tissues from AD patients show aberrant EAAC1 accumulation in pyramidal neurons of the hippocampus [139] and decreased GSH/GSSG ratios with the progression of AD [140]. These findings support the notion of EAAC1 dysfunction in AD pathogenesis.
7.2. Parkinson’s Disease (PD)
PD is the second most common age-related neurodegenerative disease. PD is a progressive, late-onset disease characterized clinically by presenting “TRAP” signs (tremor, rigidity, akinesia, and postural instability) [141]. The pathological hallmarks of this disease are the dopaminergic neurodegeneration in the SN and eosinophilic neuronal inclusions, called Lewy bodies, composed mainly of α-synuclein [142]. Most PD cases are sporadic, and less than 10% of the patients have a positive family history [143]. Both genetic and environmental factors are considered important in the etiology of PD [144,145]. PD is also characterized by a selective loss of GSH in the SN, but not in other parts of the brain [146]. As with AD patients, no change has been found in the concentrations of ascorbate or α-tocopherol in the CNS between PD patients and controls [147,148]. These findings also suggest a disorder of GSH metabolism as an underlying cause of PD. Postmortem brain tissues from normal individuals with incidental Lewy bodies and neuronal cell loss in the SN—who would be considered presymptomatic PD subjects—showed decreasing GSH levels in the SN compared to those of age-matched controls without Lewy bodies [149]. GSH depletion is considered an early event in the progression of PD [116]. Oxidative stress accelerates α-synuclein aggregation, which would also be facilitated by GSSG [150]. A decreased GSH/GSSG ratio in the brain may accelerate oxidative stress and Lewy body formation in the brain of an individual with PD.
Approximately 10% of PD patients show inherited forms [151]. Mutations in the α-synuclein gene were found in autosomal-dominant PD. The expression of familial PD-linked mutant human α-synuclein (A53T) in transgenic mice leads to late-onset neurodegeneration with an abnormal aggregation of α-synuclein in neurons [152]. The expression of the mutant A53T form of α-synuclein in dopaminergic neuronal culture caused GSH depletion with mitochondrial dysfunction [153].
Loss of function mutations in parkin were found in autosomal-recessive juvenile PD patients [154]. Parkin protein works as an E3 ubiquitin ligase in the process of ubiquitination to conjugate ubiquitin with specific substrates, including α-synuclein, leading to degradation by the UPS. The active sites of parkin are regions that are cysteine-rich and thereby sensitive to oxidative modification, which alters protein solubility and E3 ligase activity leading to dysfunction of UPS [155–157]. The UPS activity in the midbrain of aged parkin-deficient mice was damaged by GSH depletion [158].
DJ-1 is also one of the causative genes for autosomal-recessive juvenile PD. DJ-1 exhibits protein interaction as a redox-dependent molecular chaperone [159] and up-regulates GSH synthesis during oxidative stress [160]. Oxidation of a conserved cysteine residue (Cys106) in DJ-1 regulates its chaperone activity against α-synuclein [159]. However, further oxidation of DJ-1 leads to loss of the ability and thus causes α-synuclein aggregation [159]. Indeed, DJ-1 is oxidatively damaged in the brains of idiopathic PD patients [161].
Dopaminergic neurons express high levels of EAAC1 in human brain [103,162]. EAATs are vulnerable to oxidative stress, resulting in reduced uptake function [163]. DA neurons are more susceptible to EAAC1 dysfunction than non-DA neurons [164]. Our previous study showed that dopaminergic neurotoxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium, damage EAAC1 to reduce the neuronal cysteine uptake, leading to GSH depletion [165]. It is plausible that oxidative stress induces EAAC1 dysfunction, causing GSH depletion in PD.
7.3. Amyotrophic Lateral Sclerosis (ALS)
ALS is a progressive paralytic disorder characterized by the selective loss of motor neurons in the spinal cord and motor cortex. GSH reduction in the spinal cord has been reported in a mouse model of ALS [166]. Clinically, GSH levels and the activities of GR and G6PDH in erythrocytes are reduced in ALS patients [167]. These changes correlate with the disease progression. The spinal cord tissues obtained postmortem from patients with ALS showed increased protein glutathionylation in the gray matter [168] and reduced GST mRNA expression [169] compared to age-matched controls. Loss of GLT-1 has been reported in the spinal cord and motor cortex of ALS patients [170,171]. The expression of EAAC1 was reported to be slightly decreased in ALS motor cortex, although not significantly so [170]. Numerous studies have reported the involvement of oxidative stress in the pathogenesis of ALS [172]. Disorders of GSH metabolism might be a key risk factor in ALS. Riluzole, a neuroprotective drug for ALS patients, inhibits neuronal glutamate release and enhances astroglial glutamate uptake in the CNS [173,174]. A recent study showed an additional effect of riluzole on glial GSH synthesis under oxidative stress [175].
7.4. Progressive Supranuclear Palsy (PSP)
PSP is another age-related neurodegenerative disease; it is characterized by early postural instability, parkinsonism and a vertical supranuclear gaze palsy. PSP patients were recently shown to have decreased GSH levels in the SN [176]. A lipid peroxidation product, 4-hydroxy-2-nonenal (HNE), leads to the formation of cross-linked GSH-related enzymes to impair the enzymatic activities [177,178]. In the CNS of PSP patients, GPx conjugates with HNE, leading to impairment of its enzymatic activity [179]. Although recent studies have provided evidence that oxidative stress is involved in PSP pathogenesis [180], the precise mechanisms of declining brain GSH are not fully understood.
7.5. Huntington’s Disease (HD)
HD is a progressive neurodegenerative disease characterized by choreic movements caused by basal ganglia disorders. In the plasma of HD patients, the lipid peroxidation levels are higher and the GSH levels are lower than those of age- and gender-matched controls [181]. HD is associated with the expansion of a CAG trinucleotide repeat (in excess of 38 repeats) on the gene coding “huntingtin” with autosomal-dominant inheritance. In an in vitro study, decreased levels of GSH with elevated ROS levels were found in primary neurons from a knock-in mouse model of HD (HD140Q/140Q) in which a human huntingtin gene with 140 CAG repeats was inserted [182]. That study also showed EAAC1 dysfunction in the mouse model, which impaired cysteine uptake leading to GSH depletion in the neurons.
7.6. Multiple Sclerosis (MS)
MS is a neurological disorder characterized by inflammatory-mediated demyelination in the CNS. Although it is still arguable whether MS is a neurodegenerative disease or not, increasing lines of evidence support neurodegeneration as the major cause of irreversible neurological disability in MS patients [183]. Neuronal degeneration is a prominent feature in the brain of MS patients accompanied by neuritic transaction, neuronal apoptosis, and reduced neuronal and synaptic density [184]. Clinical studies by NMR spectroscopy have revealed lower brain GSH levels in MS patients than in controls [185,186]. Oxidative stress plays a major role in the pathogenesis of MS [187], however, the precise mechanism of GSH depletion is still unclear.
8. A Potential Approach to Increase GSH Levels in the Brain
A therapeutic strategy to increase neuronal GSH levels in the brain is a potential treatment for GSH-related neurodegenerative diseases, as mentioned above. However, no therapeutic drugs are available for increasing brain GSH levels at present. Orally dosed GSH is rapidly degraded in the gut, and intravenously administered GSH is rapidly oxidized to GSSG in the blood with a half-life of 2–3 min [28,29,188,189]. The administration of crude GSH does not seem promising for the treatment of neurodegenerative diseases.
N-acetylcysteine (NAC) is useful for the treatment of acetaminophen-induced hepatotoxicity by increasing hepatic GSH production or by its direct effect as an antioxidant. The systemic administration of NAC can also increase neuronal GSH levels in the brain by penetrating the BBB and the plasma membrane, even though neurons lack cysteine transporters [102]. Based on the favorable results of NAC treatments in some neurodegenerative models, some clinical trials using NAC have been in process for the treatment of AD or PD in the U.S. (ClinicalTrials.gov identifier: NCT01320527, NCT01370954, NCT01427517, and NCT01470027).
Considering EAAC1-mediated cysteine uptake as the rate-limiting step for neuronal GSH synthesis, a compound facilitating EAAC1 function might be a potential strategy for the treatment of GSH-related neurodegenerative diseases. To date, there is no promising drug for clinical use to modulate EAAC1 function exogenously. Alternatively, the endogenous modulation of protein-protein interactions might be crucial for enhancing EAAC1 function. GTRAP3–18 would be a potential target leading to an increase in neuronal GSH levels in the brain, but the physiological and pathological roles of human GTRAP3–18 function should be elucidated before it is used clinically.
9. Conclusions
GSH has a variety of pivotal functions in cells. Genetic disorders of GSH-related enzymes are rare, but GSH depletion is present in the pathogenesis of most major neurodegenerative diseases. Considering recent basic and clinical studies indicating that GSH depletion precedes neurodegeneration, neuronal GSH depletion would be a primary cause of neurodegenerative diseases (Figure 2). The development of drugs that target neuronal GSH synthesis would be a promising approach as a therapeutic strategy for neurodegenerative diseases.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.De Rey-Pailhade M.J. Sur un corps d’origine organique hydrogénant le soufre á froid. C. R. Acad. Sci. 1888;106:1683–1684. [Google Scholar]
- 2.Meister A. On the discovery of glutathione. Trends Biochem. Sci. 1988;13:185–188. doi: 10.1016/0968-0004(88)90148-x. [DOI] [PubMed] [Google Scholar]
- 3.Hopkins F.G. On an autoxidisable constituent of the cell. Biochem. J. 1921;15:286–305. doi: 10.1042/bj0150286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopkins F.G. On glutathione: A reinvestigation. J. Biol. Chem. 1929;84:269–320. [Google Scholar]
- 5.Kendall E.C., Mason H.L., McKenzie B.F. A study of glutathione. J. Biol. Chem. 1930;88:409–423. [Google Scholar]
- 6.Richman P.G., Meister A. Regulation of γ-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J. Biol. Chem. 1975;250:1422–1426. [PubMed] [Google Scholar]
- 7.Chen Y., Shertzer H.G., Schneider S.N., Nebert D.W., Dalton T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005;280:33766–33774. doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- 8.Griffith O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 9.Erecinska M., Silver I.A. Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 1990;35:245–296. doi: 10.1016/0301-0082(90)90013-7. [DOI] [PubMed] [Google Scholar]
- 10.Roux M.J., Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron. 2000;25:373–383. doi: 10.1016/s0896-6273(00)80901-0. [DOI] [PubMed] [Google Scholar]
- 11.Aoyama K., Wang F., Matsumura N., Kiyonari H., Shioi G., Tanaka K., Kinoshita C., Kikuchi-Utsumi K., Watabe M., Nakaki T. Increased neuronal glutathione and neuroprotection in GTRAP3–18-deficient mice. Neurobiol. Dis. 2012;45:973–982. doi: 10.1016/j.nbd.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Puka-Sundvall M., Eriksson P., Nilsson M., Sandberg M., Lehmann A. Neurotoxicity of cysteine: Interaction with glutamate. Brain Res. 1995;705:65–70. doi: 10.1016/0006-8993(95)01139-0. [DOI] [PubMed] [Google Scholar]
- 13.Janaky R., Varga V., Hermann A., Saransaari P., Oja S.S. Mechanisms of l-cysteine neurotoxicity. Neurochem. Res. 2000;25:1397–1405. doi: 10.1023/a:1007616817499. [DOI] [PubMed] [Google Scholar]
- 14.Sedlak J., Lindsay R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 15.Pastore A., Federici G., Bertini E., Piemonte F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta. 2003;333:19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- 16.Wu G., Fang Y.Z., Yang S., Lupton J.R., Turner N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 17.Commandeur J.N., Stijntjes G.J., Vermeulen N.P. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev. 1995;47:271–330. [PubMed] [Google Scholar]
- 18.Cooper A.J., Kristal B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem. 1997;378:793–802. [PubMed] [Google Scholar]
- 19.Kang Y., Viswanath V., Jha N., Qiao X., Mo J.Q., Andersen J.K. Brain γ-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J. Neurosci. Res. 1999;58:436–441. [PubMed] [Google Scholar]
- 20.Anderson M.E., Underwood M., Bridges R.J., Meister A. Glutathione metabolism at the blood-cerebrospinal fluid barrier. FASEB J. 1989;3:2527–2531. doi: 10.1096/fasebj.3.13.2572501. [DOI] [PubMed] [Google Scholar]
- 21.Wang X.F., Cynader M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- 22.Makar T.K., Nedergaard M., Preuss A., Gelbard A.S., Perumal A.S., Cooper A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994;62:45–53. doi: 10.1046/j.1471-4159.1994.62010045.x. [DOI] [PubMed] [Google Scholar]
- 23.Hirrlinger J., Gutterer J.M., Kussmaul L., Hamprecht B., Dringen R. Microglial cells in culture express a prominent glutathione system for the defense against reactive oxygen species. Dev. Neurosci. 2000;22:384–392. doi: 10.1159/000017464. [DOI] [PubMed] [Google Scholar]
- 24.Dringen R., Pawlowski P.G., Hirrlinger J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 25.Tateishi N., Higashi T., Naruse A., Nakashima K., Shiozaki H. Rat liver glutathione: Possible role as a reservoir of cysteine. J. Nutr. 1977;107:51–60. doi: 10.1093/jn/107.1.51. [DOI] [PubMed] [Google Scholar]
- 26.Lauterburg B.H., Adams J.D., Mitchell J.R. Hepatic glutathione homeostasis in the rat: Efflux accounts for glutathione turnover. Hepatology. 1984;4:586–590. doi: 10.1002/hep.1840040402. [DOI] [PubMed] [Google Scholar]
- 27.Cornford E.M., Braun L.D., Crane P.D., Oldendorf W.H. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology. 1978;103:1297–1303. doi: 10.1210/endo-103-4-1297. [DOI] [PubMed] [Google Scholar]
- 28.Lash L.H., Jones D.P. Distribution of oxidized and reduced forms of glutathione and cysteine in rat plasma. Arch. Biochem. Biophys. 1985;240:583–592. doi: 10.1016/0003-9861(85)90065-7. [DOI] [PubMed] [Google Scholar]
- 29.Ammon H.P., Melien M.C., Verspohl E.J. Pharmacokinetics of intravenously administered glutathione in the rat. J. Pharm. Pharmacol. 1986;38:721–725. doi: 10.1111/j.2042-7158.1986.tb04478.x. [DOI] [PubMed] [Google Scholar]
- 30.Olney J.W., Zorumski C., Price M.T., Labruyere J. l-cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science. 1990;248:596–599. doi: 10.1126/science.2185543. [DOI] [PubMed] [Google Scholar]
- 31.Wade L.A., Brady H.M. Cysteine and cystine transport at the blood-brain barrier. J. Neurochem. 1981;37:730–734. doi: 10.1111/j.1471-4159.1982.tb12548.x. [DOI] [PubMed] [Google Scholar]
- 32.Killian D.M., Chikhale P.J. Predominant functional activity of the large, neutral amino acid transporter (LAT1) isoform at the cerebrovasculature. Neurosci. Lett. 2001;306:1–4. doi: 10.1016/s0304-3940(01)01810-9. [DOI] [PubMed] [Google Scholar]
- 33.Johnson W.M., Wilson-Delfosse A.L., Mieyal J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4:1399–1440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato H., Tamba M., Ishii T., Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 35.Pow D.V. Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. doi: 10.1002/glia.1037. [DOI] [PubMed] [Google Scholar]
- 36.Qin S., Colin C., Hinners I., Gervais A., Cheret C., Mallat M. System xc − and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-β peptide 1–40. J. Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dringen R., Kranich O., Loschmann P.A., Hamprecht B. Use of dipeptides for the synthesis of glutathione by astroglia-rich primary cultures. J. Neurochem. 1997;69:868–874. doi: 10.1046/j.1471-4159.1997.69020868.x. [DOI] [PubMed] [Google Scholar]
- 38.Dringen R., Hamprecht B., Broer S. The peptide transporter PepT2 mediates the uptake of the glutathione precursor CysGly in astroglia-rich primary cultures. J. Neurochem. 1998;71:388–393. doi: 10.1046/j.1471-4159.1998.71010388.x. [DOI] [PubMed] [Google Scholar]
- 39.Dringen R., Hamprecht B. Glutathione restoration as indicator for cellular metabolism of astroglial cells. Dev. Neurosci. 1998;20:401–407. doi: 10.1159/000017337. [DOI] [PubMed] [Google Scholar]
- 40.Dringen R., Hirrlinger J. Glutathione pathways in the brain. Biol. Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 41.Dringen R., Gutterer J.M., Gros C., Hirrlinger J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001;66:1003–1008. doi: 10.1002/jnr.10042. [DOI] [PubMed] [Google Scholar]
- 42.Dringen R., Pfeiffer B., Hamprecht B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chance B., Sies H., Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 44.Staniek K., Nohl H. Are mitochondria a permanent source of reactive oxygen species? Biochim. Biophys. Acta. 2000;1460:268–275. doi: 10.1016/s0005-2728(00)00152-3. [DOI] [PubMed] [Google Scholar]
- 45.St-Pierre J., Buckingham J.A., Roebuck S.J., Brand M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 46.Collins Y., Chouchani E.T., James A.M., Menger K.E., Cocheme H.M., Murphy M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012;125:801–806. doi: 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- 47.Girotti A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998;39:1529–1542. [PubMed] [Google Scholar]
- 48.Trepanier G., Furling D., Puymirat J., Mirault M.E. Immunocytochemical localization of seleno-glutathione peroxidase in the adult mouse brain. Neuroscience. 1996;75:231–243. doi: 10.1016/0306-4522(96)00222-9. [DOI] [PubMed] [Google Scholar]
- 49.Arthur J.R. The glutathione peroxidases. Cell. Mol. Life Sci. 2000;57:1825–1835. doi: 10.1007/PL00000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Power J.H., Blumbergs P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:63–73. doi: 10.1007/s00401-008-0438-3. [DOI] [PubMed] [Google Scholar]
- 51.Yang Y., Sharma R., Sharma A., Awasthi S., Awasthi Y.C. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Pol. 2003;50:319–336. [PubMed] [Google Scholar]
- 52.Winterbourn C.C., Metodiewa D. The reaction of superoxide with reduced glutathione. Arch. Biochem. Biophys. 1994;314:284–290. doi: 10.1006/abbi.1994.1444. [DOI] [PubMed] [Google Scholar]
- 53.Hogg N., Singh R.J., Kalyanaraman B. The role of glutathione in the transport and catabolism of nitric oxide. FEBS Lett. 1996;382:223–228. doi: 10.1016/0014-5793(96)00086-5. [DOI] [PubMed] [Google Scholar]
- 54.Dringen R., Gutterer J.M. Glutathione reductase from bovine brain. Methods Enzymol. 2002;348:281–288. doi: 10.1016/s0076-6879(02)48646-6. [DOI] [PubMed] [Google Scholar]
- 55.Dringen R., Kussmaul L., Gutterer J.M., Hirrlinger J., Hamprecht B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999;72:2523–2530. doi: 10.1046/j.1471-4159.1999.0722523.x. [DOI] [PubMed] [Google Scholar]
- 56.Hayes J.D., Flanagan J.U., Jowsey I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 57.Leier I., Jedlitschky G., Buchholz U., Center M., Cole S.P., Deeley R.G., Keppler D. ATP-dependent glutathione disulphide transport mediated by the MRP gene-encoded conjugate export pump. Biochem. J. 1996;314:433–437. doi: 10.1042/bj3140433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ballatori N., Krance S.M., Marchan R., Hammond C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 2009;30:13–28. doi: 10.1016/j.mam.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho Y.S., Magnenat J.L., Bronson R.T., Cao J., Gargano M., Sugawara M., Funk C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 60.Malinski T., Bailey F., Zhang Z.G., Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 61.Cherian L., Goodman J.C., Robertson C.S. Brain nitric oxide changes after controlled cortical impact injury in rats. J. Neurophysiol. 2000;83:2171–2178. doi: 10.1152/jn.2000.83.4.2171. [DOI] [PubMed] [Google Scholar]
- 62.Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szabo C., Ischiropoulos H., Radi R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 64.Beckman J.S. Peroxynitrite versus hydroxyl radical: The role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci. 1994;738:69–75. doi: 10.1111/j.1749-6632.1994.tb21791.x. [DOI] [PubMed] [Google Scholar]
- 65.Koppenol W.H. The basic chemistry of nitrogen monoxide and peroxynitrite. Free Radic. Biol. Med. 1998;25:385–391. doi: 10.1016/s0891-5849(98)00093-8. [DOI] [PubMed] [Google Scholar]
- 66.Yamakura F., Taka H., Fujimura T., Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- 67.Aoyama K., Watabe M., Nakaki T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008;108:227–238. doi: 10.1254/jphs.08r01cr. [DOI] [PubMed] [Google Scholar]
- 68.Halliwell B., Gutteridge J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sies H. Strategies of antioxidant defense. Eur. J. Biochem. 1993;215:213–219. doi: 10.1111/j.1432-1033.1993.tb18025.x. [DOI] [PubMed] [Google Scholar]
- 70.Ghezzi P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta. 2013;1830:3165–3172. doi: 10.1016/j.bbagen.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 71.Klatt P., Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 72.Dalle-Donne I., Aldini G., Carini M., Colombo R., Rossi R., Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giustarini D., Rossi R., Milzani A., Colombo R., Dalle-Donne I. S-Glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004;8:201–212. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arner E.S., Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 75.Berndt C., Lillig C.H., Holmgren A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim. Biophys. Acta. 2008;1783:641–650. doi: 10.1016/j.bbamcr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 76.Gilbert H.F. Redox control of enzyme activities by thiol/disulfide exchange. Methods Enzymol. 1984;107:330–351. doi: 10.1016/0076-6879(84)07022-1. [DOI] [PubMed] [Google Scholar]
- 77.Gilbert H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 78.Arrigo A.P. Gene expression and the thiol redox state. Free Radic. Biol. Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 79.Klatt P., Molina E.P., de Lacoba M.G., Padilla C.A., Martinez-Galesteo E., Barcena J.A., Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 80.Pineda-Molina E., Klatt P., Vazquez J., Marina A., Garcia de Lacoba M., Perez-Sala D., Lamas S. Glutathionylation of the p50 subunit of NF-κB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 81.Fratelli M., Goodwin L.O., Orom U.A., Lombardi S., Tonelli R., Mengozzi M., Ghezzi P. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc. Natl. Acad. Sci. USA. 2005;102:13998–14003. doi: 10.1073/pnas.0504398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Poot M., Teubert H., Rabinovitch P.S., Kavanagh T.J. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J. Cell. Physiol. 1995;163:555–560. doi: 10.1002/jcp.1041630316. [DOI] [PubMed] [Google Scholar]
- 83.Markovic J., Borras C., Ortega A., Sastre J., Vina J., Pallardo F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007;282:20416–20424. doi: 10.1074/jbc.M609582200. [DOI] [PubMed] [Google Scholar]
- 84.Voehringer D.W. BCL-2 and glutathione: Alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic. Biol. Med. 1999;27:945–950. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 85.Filomeni G., Rotilio G., Ciriolo M.R. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 2003;17:64–66. doi: 10.1096/fj.02-0105fje. [DOI] [PubMed] [Google Scholar]
- 86.Sagara J.I., Miura K., Bannai S. Maintenance of neuronal glutathione by glial cells. J. Neurochem. 1993;61:1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- 87.Kranich O., Hamprecht B., Dringen R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996;219:211–214. doi: 10.1016/s0304-3940(96)13217-1. [DOI] [PubMed] [Google Scholar]
- 88.Danbolt N.C. Glutamate uptake. Prog. Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 89.Shanker G., Allen J.W., Mutkus L.A., Aschner M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001;902:156–163. doi: 10.1016/s0006-8993(01)02342-3. [DOI] [PubMed] [Google Scholar]
- 90.Kanai Y., Hediger M.A. The glutamate and neutral amino acid transporter family: Physiological and pharmacological implications. Eur. J. Pharmacol. 2003;479:237–247. doi: 10.1016/j.ejphar.2003.08.073. [DOI] [PubMed] [Google Scholar]
- 91.Had-Aissouni L. Maintenance of antioxidant defenses of brain cells: Plasma membrane glutamate transporters and beyond. Amino Acids. 2012;42:159–161. doi: 10.1007/s00726-011-0860-z. [DOI] [PubMed] [Google Scholar]
- 92.Had-Aissouni L. Toward a new role for plasma membrane sodium-dependent glutamate transporters of astrocytes: Maintenance of antioxidant defenses beyond extracellular glutamate clearance. Amino Acids. 2012;42:181–197. doi: 10.1007/s00726-011-0863-9. [DOI] [PubMed] [Google Scholar]
- 93.Zerangue N., Kavanaugh M.P. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 94.Rothstein J.D., Martin L., Levey A.I., Dykes-Hoberg M., Jin L., Wu D., Nash N., Kuncl R.W. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 95.Coco S., Verderio C., Trotti D., Rothstein J.D., Volterra A., Matteoli M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur. J. Neurosci. 1997;9:1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x. [DOI] [PubMed] [Google Scholar]
- 96.Shashidharan P., Huntley G.W., Murray J.M., Buku A., Moran T., Walsh M.J., Morrison J.H., Plaitakis A. Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res. 1997;773:139–148. doi: 10.1016/s0006-8993(97)00921-9. [DOI] [PubMed] [Google Scholar]
- 97.Holmseth S., Dehnes Y., Huang Y.H., Follin-Arbelet V.V., Grutle N.J., Mylonakou M.N., Plachez C., Zhou Y., Furness D.N., Bergles D.E., et al. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci. 2012;32:6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tanaka K., Watase K., Manabe T., Yamada K., Watanabe M., Takahashi K., Iwama H., Nishikawa T., Ichihara N., Kikuchi T., et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 99.Grewer C., Rauen T. Electrogenic glutamate transporters in the CNS: Molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J. Membr. Biol. 2005;203:1–20. doi: 10.1007/s00232-004-0731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zerangue N., Kavanaugh M.P. Interaction of l-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996;493:419–423. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Himi T., Ikeda M., Yasuhara T., Nishida M., Morita I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003;110:1337–1348. doi: 10.1007/s00702-003-0049-z. [DOI] [PubMed] [Google Scholar]
- 102.Aoyama K., Suh S.W., Hamby A.M., Liu J., Chan W.Y., Chen Y., Swanson R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 103.Berman A.E., Chan W.Y., Brennan A.M., Reyes R.C., Adler B.L., Suh S.W., Kauppinen T.M., Edling Y., Swanson R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1−/− mouse. Ann. Neurol. 2011;69:509–520. doi: 10.1002/ana.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fournier K.M., Gonzalez M.I., Robinson M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem. 2004;279:34505–34513. doi: 10.1074/jbc.M404032200. [DOI] [PubMed] [Google Scholar]
- 105.Ruggiero A.M., Liu Y., Vidensky S., Maier S., Jung E., Farhan H., Robinson M.B., Sitte H., Rothstein J.D. The ER exit of glutamate transporter is regulated by the inducible mammalian Yip6b/GTRAP3–18 protein. J. Biol. Chem. 2008;283:6175–6183. doi: 10.1074/jbc.M701008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Watabe M., Aoyama K., Nakaki T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3–18 (GTRAP3–18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol. 2007;72:1103–1110. doi: 10.1124/mol.107.039461. [DOI] [PubMed] [Google Scholar]
- 107.Watabe M., Aoyama K., Nakaki T. A dominant role of GTRAP3–18 in neuronal glutathione synthesis. J. Neurosci. 2008;28:9404–9413. doi: 10.1523/JNEUROSCI.3351-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aoyama K., Nakaki T. Inhibition of GTRAP3–18 may increase neuroprotective glutathione (GSH) synthesis. Int. J. Mol. Sci. 2012;13:12017–12035. doi: 10.3390/ijms130912017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aoyama K., Nakaki T. Neuroprotective properties of the excitatory amino acid carrier 1 (EAAC1) Amino Acids. 2013;45:133–142. doi: 10.1007/s00726-013-1481-5. [DOI] [PubMed] [Google Scholar]
- 110.Dalton T.P., Dieter M.Z., Yang Y., Shertzer H.G., Nebert D.W. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: Embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun. 2000;279:324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- 111.Yang Y., Dieter M.Z., Chen Y., Shertzer H.G., Nebert D.W., Dalton T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 112.Chen Y., Johansson E., Fan Y., Shertzer H.G., Vasiliou V., Nebert D.W., Dalton T.P. Early onset senescence occurs when fibroblasts lack the glutamate-cysteine ligase modifier subunit. Free Radic. Biol. Med. 2009;47:410–418. doi: 10.1016/j.freeradbiomed.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ristoff E., Larsson A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007;2:16. doi: 10.1186/1750-1172-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Winkler A., Njalsson R., Carlsson K., Elgadi A., Rozell B., Abraham L., Ercal N., Shi Z.Z., Lieberman M.W., Larsson A., et al. Glutathione is essential for early embryogenesis—Analysis of a glutathione synthetase knockout mouse. Biochem. Biophys. Res. Commun. 2011;412:121–126. doi: 10.1016/j.bbrc.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 115.Choi I.Y., Gruetter R. Dynamic or inert metabolism? Turnover of N-acetyl aspartate and glutathione from d-[1-13C]glucose in the rat brain in vivo. J. Neurochem. 2004;91:778–787. doi: 10.1111/j.1471-4159.2004.02716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jenner P. Oxidative damage in neurodegenerative disease. Lancet. 1994;344:796–798. doi: 10.1016/s0140-6736(94)92347-7. [DOI] [PubMed] [Google Scholar]
- 117.Jenner P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003;53:S26–S36. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 118.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 119.Beckman K.B., Ames B.N. The free radical theory of aging matures. Physiol. Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 120.Jain A., Martensson J., Stole E., Auld P.A., Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc. Natl. Acad. Sci. USA. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dringen R., Gutterer J.M., Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 122.Schulz J.B., Lindenau J., Seyfried J., Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 123.Bains J.S., Shaw C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 124.Andersen J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004;10:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 125.Mandal P.K., Tripathi M., Sugunan S. Brain oxidative stress: Detection and mapping of anti-oxidant marker “Glutathione” in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 2012;417:43–48. doi: 10.1016/j.bbrc.2011.11.047. [DOI] [PubMed] [Google Scholar]
- 126.Metcalfe T., Bowen D.M., Muller D.P. Vitamin E concentrations in human brain of patients with Alzheimer’s disease, fetuses with Down’s syndrome, centenarians, and controls. Neurochem. Res. 1989;14:1209–1212. doi: 10.1007/BF00965511. [DOI] [PubMed] [Google Scholar]
- 127.Paraskevas G.P., Kapaki E., Libitaki G., Zournas C., Segditsa I., Papageorgiou C. Ascorbate in healthy subjects, amyotrophic lateral sclerosis and Alzheimer’s disease. Acta Neurol. Scand. 1997;96:88–90. doi: 10.1111/j.1600-0404.1997.tb00245.x. [DOI] [PubMed] [Google Scholar]
- 128.Lovell M.A., Xie C., Markesbery W.R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology. 1998;51:1562–1566. doi: 10.1212/wnl.51.6.1562. [DOI] [PubMed] [Google Scholar]
- 129.Casado A., Encarnacion Lopez-Fernandez M., Concepcion Casado M., de la Torre R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem. Res. 2008;33:450–458. doi: 10.1007/s11064-007-9453-3. [DOI] [PubMed] [Google Scholar]
- 130.Spalletta G., Bernardini S., Bellincampi L., Federici G., Trequattrini A., Ciappi F., Bria P., Caltagirone C., Bossu P. Glutathione S-transferase P1 and T1 gene polymorphisms predict longitudinal course and age at onset of Alzheimer disease. Am. J. Geriatr. Psychiatry. 2007;15:879–887. doi: 10.1097/JGP.0b013e3180547076. [DOI] [PubMed] [Google Scholar]
- 131.Paz-y-Mino C., Carrera C., Lopez-Cortes A., Munoz M.J., Cumbal N., Castro B., Cabrera A., Sanchez M.E. Genetic polymorphisms in apolipoprotein E and glutathione peroxidase 1 genes in the Ecuadorian population affected with Alzheimer’s disease. Am. J. Med. Sci. 2010;340:373–377. doi: 10.1097/MAJ.0b013e3181e93475. [DOI] [PubMed] [Google Scholar]
- 132.Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., Pericak-Vance M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 133.Ramassamy C., Averill D., Beffert U., Theroux L., Lussier-Cacan S., Cohn J.S., Christen Y., Schoofs A., Davignon J., Poirier J. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol. Dis. 2000;7:23–37. doi: 10.1006/nbdi.1999.0273. [DOI] [PubMed] [Google Scholar]
- 134.Petersen R.C. Mild cognitive impairment: Transition between aging and Alzheimer’s disease. Neurologia. 2000;15:93–101. [PubMed] [Google Scholar]
- 135.Sultana R., Piroddi M., Galli F., Butterfield D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res. 2008;33:2540–2546. doi: 10.1007/s11064-008-9593-0. [DOI] [PubMed] [Google Scholar]
- 136.Mattson M.P. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 138.Hodgson N., Trivedi M., Muratore C., Li S., Deth R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013;36:197–209. doi: 10.3233/JAD-130101. [DOI] [PubMed] [Google Scholar]
- 139.Duerson K., Woltjer R.L., Mookherjee P., Leverenz J.B., Montine T.J., Bird T.D., Pow D.V., Rauen T., Cook D.G. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 2009;19:267–278. doi: 10.1111/j.1750-3639.2008.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ansari M.A., Scheff S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frank C., Pari G., Rossiter J.P. Approach to diagnosis of Parkinson disease. Can. Fam. Physician. 2006;52:862–868. [PMC free article] [PubMed] [Google Scholar]
- 142.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 143.Thomas B., Beal M.F. Parkinson’s disease. Hum. Mol. Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 144.Khandhar S.M., Marks W.J. Epidemiology of Parkinson’s disease. Dis. Mon. 2007;53:200–205. doi: 10.1016/j.disamonth.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 145.Schapira A.H., Jenner P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011;26:1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- 146.Sian J., Dexter D.T., Lees A.J., Daniel S., Agid Y., Javoy-Agid F., Jenner P., Marsden C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 147.Riederer P., Sofic E., Rausch W.D., Schmidt B., Reynolds G.P., Jellinger K., Youdim M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 148.Dexter D.T., Ward R.J., Wells F.R., Daniel S.E., Lees A.J., Peters T.J., Jenner P., Marsden C.D. α-Tocopherol levels in brain are not altered in Parkinson’s disease. Ann. Neurol. 1992;32:591–593. doi: 10.1002/ana.410320420. [DOI] [PubMed] [Google Scholar]
- 149.Dexter D.T., Sian J., Rose S., Hindmarsh J.G., Mann V.M., Cooper J.M., Wells F.R., Daniel S.E., Lees A.J., Schapira A.H., et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann. Neurol. 1994;35:38–44. doi: 10.1002/ana.410350107. [DOI] [PubMed] [Google Scholar]
- 150.Paik S.R., Lee D., Cho H.J., Lee E.N., Chang C.S. Oxidized glutathione stimulated the amyloid formation of α-synuclein. FEBS Lett. 2003;537:63–67. doi: 10.1016/s0014-5793(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 151.Klein C., Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee M.K., Stirling W., Xu Y., Xu X., Qui D., Mandir A.S., Dawson T.M., Copeland N.G., Jenkins N.A., Price D.L. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53→Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vali S., Chinta S.J., Peng J., Sultana Z., Singh N., Sharma P., Sharada S., Andersen J.K., Bharath M.M. Insights into the effects of α-synuclein expression and proteasome inhibition on glutathione metabolism through a dynamic in silico model of Parkinson’s disease: Validation by cell culture data. Free Radic. Biol. Med. 2008;45:1290–1301. doi: 10.1016/j.freeradbiomed.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 155.Bossy-Wetzel E., Schwarzenbacher R., Lipton S.A. Molecular pathways to neurodegeneration. Nat. Med. 2004;10:S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 156.Wong E.S., Tan J.M., Wang C., Zhang Z., Tay S.P., Zaiden N., Ko H.S., Dawson V.L., Dawson T.M., Lim K.L. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J. Biol. Chem. 2007;282:12310–12318. doi: 10.1074/jbc.M609466200. [DOI] [PubMed] [Google Scholar]
- 157.Meng F., Yao D., Shi Y., Kabakoff J., Wu W., Reicher J., Ma Y., Moosmann B., Masliah E., Lipton S.A., et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011;6:34. doi: 10.1186/1750-1326-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Casarejos M.J., Solano R.M., Rodriguez-Navarro J.A., Gomez A., Perucho J., Castano J.G., Garcia de Yebenes J., Mena M.A. Parkin deficiency increases the resistance of midbrain neurons and glia to mild proteasome inhibition: The role of autophagy and glutathione homeostasis. J. Neurochem. 2009;110:1523–1537. doi: 10.1111/j.1471-4159.2009.06248.x. [DOI] [PubMed] [Google Scholar]
- 159.Zhou W., Zhu M., Wilson M.A., Petsko G.A., Fink A.L. The oxidation state of DJ-1 regulates its chaperone activity toward α-synuclein. J. Mol. Biol. 2006;356:1036–1048. doi: 10.1016/j.jmb.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 160.Zhou W., Freed C.R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T α-synuclein toxicity. J. Biol. Chem. 2005;280:43150–43158. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- 161.Choi J., Sullards M.C., Olzmann J.A., Rees H.D., Weintraub S.T., Bostwick D.E., Gearing M., Levey A.I., Chin L.S., Li L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006;281:10816–10824. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Plaitakis A., Shashidharan P. Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: Implications for the pathogenesis of Parkinson’s disease. J. Neurol. 2000;247:II25–II35. doi: 10.1007/pl00007757. [DOI] [PubMed] [Google Scholar]
- 163.Trotti D., Danbolt N.C., Volterra A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998;19:328–334. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- 164.Nafia I., Re D.B., Masmejean F., Melon C., Kachidian P., Kerkerian-Le Goff L., Nieoullon A., Had-Aissouni L. Preferential vulnerability of mesencephalic dopamine neurons to glutamate transporter dysfunction. J. Neurochem. 2008;105:484–496. doi: 10.1111/j.1471-4159.2007.05146.x. [DOI] [PubMed] [Google Scholar]
- 165.Aoyama K., Matsumura N., Watabe M., Nakaki T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J. Neurosci. 2008;27:20–30. doi: 10.1111/j.1460-9568.2007.05979.x. [DOI] [PubMed] [Google Scholar]
- 166.Chi L., Ke Y., Luo C., Gozal D., Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007;144:991–1003. doi: 10.1016/j.neuroscience.2006.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Babu G.N., Kumar A., Chandra R., Puri S.K., Singh R.L., Kalita J., Misra U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008;52:1284–1289. doi: 10.1016/j.neuint.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 168.Lanius R.A., Krieger C., Wagey R., Shaw C.A. Increased [35S]glutathione binding sites in spinal cords from patients with sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 1993;163:89–92. doi: 10.1016/0304-3940(93)90236-e. [DOI] [PubMed] [Google Scholar]
- 169.Usarek E., Gajewska B., Kazmierczak B., Kuzma M., Dziewulska D., Baranczyk-Kuzma A. A study of glutathione S-transferase pi expression in central nervous system of subjects with amyotrophic lateral sclerosis using RNA extraction from formalin-fixed, paraffin-embedded material. Neurochem. Res. 2005;30:1003–1007. doi: 10.1007/s11064-005-6771-1. [DOI] [PubMed] [Google Scholar]
- 170.Rothstein J.D., van Kammen M., Levey A.I., Martin L.J., Kuncl R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 171.Bristol L.A., Rothstein J.D. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol. 1996;39:676–679. doi: 10.1002/ana.410390519. [DOI] [PubMed] [Google Scholar]
- 172.Barber S.C., Mead R.J., Shaw P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 173.Azbill R.D., Mu X., Springer J.E. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 174.Frizzo M.E., Dall’Onder L.P., Dalcin K.B., Souza D.O. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell. Mol. Neurobiol. 2004;24:123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Deng Y., Xu Z.F., Liu W., Xu B., Yang H.B., Wei Y.G. Riluzole-triggered GSH synthesis via activation of glutamate transporters to antagonize methylmercury-induced oxidative stress in rat cerebral cortex. Oxid. Med. Cell. Longev. 2012;2012:534705. doi: 10.1155/2012/534705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fitzmaurice P.S., Ang L., Guttman M., Rajput A.H., Furukawa Y., Kish S.J. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov. Disord. 2003;18:969–976. doi: 10.1002/mds.10486. [DOI] [PubMed] [Google Scholar]
- 177.Friguet B., Stadtman E.R., Szweda L.I. Modification of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Formation of cross-linked protein that inhibits the multicatalytic protease. J. Biol. Chem. 1994;269:21639–21643. [PubMed] [Google Scholar]
- 178.Kinter M., Roberts R.J. Glutathione consumption and glutathione peroxidase inactivation in fibroblast cell lines by 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 1996;21:457–462. doi: 10.1016/0891-5849(96)00128-1. [DOI] [PubMed] [Google Scholar]
- 179.Aoyama K., Matsubara K., Kobayashi S. Aging and oxidative stress in progressive supranuclear palsy. Eur. J. Neurol. 2006;13:89–92. doi: 10.1111/j.1468-1331.2006.01139.x. [DOI] [PubMed] [Google Scholar]
- 180.Albers D.S., Augood S.J. New insights into progressive supranuclear palsy. Trends Neurosci. 2001;24:347–353. doi: 10.1016/s0166-2236(00)01794-x. [DOI] [PubMed] [Google Scholar]
- 181.Klepac N., Relja M., Klepac R., Hecimovic S., Babic T., Trkulja V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007;254:1676–1683. doi: 10.1007/s00415-007-0611-y. [DOI] [PubMed] [Google Scholar]
- 182.Li X., Valencia A., Sapp E., Masso N., Alexander J., Reeves P., Kegel K.B., Aronin N., Difiglia M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010;30:4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Trapp B.D., Nave K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 184.Dutta R., Trapp B.D. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol. 2011;93:1–12. doi: 10.1016/j.pneurobio.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Srinivasan R., Ratiney H., Hammond-Rosenbluth K.E., Pelletier D., Nelson S.J. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn. Reson. Imaging. 2010;28:163–170. doi: 10.1016/j.mri.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 186.Choi I.Y., Lee S.P., Denney D.R., Lynch S.G. Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult. Scler. 2011;17:289–296. doi: 10.1177/1352458510384010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gilgun-Sherki Y., Melamed E., Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: The need for effective antioxidant therapy. J. Neurol. 2004;251:261–268. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- 188.Wendel A., Cikryt P. The level and half-life of glutathione in human plasma. FEBS Lett. 1980;120:209–211. doi: 10.1016/0014-5793(80)80299-7. [DOI] [PubMed] [Google Scholar]
- 189.Wendel A., Jaeschke H. Drug-induced lipid peroxidation in mice-III. Glutathione content of liver, kidney and spleen after intravenous administration of free and liposomally entrapped glutathione. Biochem. Pharmacol. 1982;31:3607–3611. doi: 10.1016/0006-2952(82)90583-4. [DOI] [PubMed] [Google Scholar]