

Abstract
Four novel, closely related podoviruses, which displayed lytic activity against the gamma-proteobacterium Alteromonas macleodii, have been isolated and sequenced. Alterophages AltAD45-P1 to P4 were obtained from water recovered near a fish farm in the Mediterranean Sea. Their morphology indicates that they belong to the Podoviridae. Their linear and dsDNA genomes are 100–104 kb in size, remarkably larger than any other described podovirus. The four AltAD45-phages share 99% nucleotide sequence identity over 97% of their ORFs, although an insertion was found in AltAD45-P1 and P2 and some regions were slightly more divergent. Despite the high overall sequence similarity among these four phages, the group with the insertion and the group without it, have different host ranges against the A. macleodii strains tested. The AltAD45-P1 to P4 phages have genes for DNA replication and transcription as well as structural genes, which are similar to the N4-like Podoviridae genus that is widespread in proteobacteria. However, in terms of their genomic structure, AltAD45-P1 to P4 differ from that of the N4-like phages. Some distinguishing features include the lack of a large virion encapsidated RNA polymerase gene, very well conserved among all the previously described N4-like phages, a single-stranded DNA binding protein and different tail protein genes. We conclude that the AltAD45 phages characterized in this study constitute a new genus within the Podoviridae.
Keywords: Alteromonas macleodii, Podoviridae, N4-like virus, lytic phage, marine phages
Introduction
Marine phages are the most abundant biological entities in the oceans. They play critical roles in carbon cycling through marine food webs, impel evolutionary change in marine communities by exerting selective pressure and are the agents for frequent lateral gene transfer.1,2 Therefore, they are an important driving force for the genomic diversification and ecological adaptation of a species.3-6 Despite their recognized abundance, most of the marine phages isolated to date are cyanophages preying on Synechococcus or Prochlorococcus species.7-10
Alteromonas macleodii is a globally distributed marine gamma-proteobacterium.11-13 It is a common isolate by standard marine bacteria isolation protocols14 and isolates belonging to this species from different studies have accumulated in individual researchers’ collections.15,16 It has been proven by several independent methodologies that A. macleodii is present worldwide in photic zone depths and is present at all depths in the Mediterranean Sea.13,16,17 Its abundance increased in the particulate fraction and down to a depth of 400 m,16,17A. macleodii was also shown by molecular methods and metatranscriptomic data18,19 to dominate heterotrophic blooms in mesocosms.20,21 In ecological terms, these data and the phenotypic properties, characterize this species as a copiotroph that behaves as an r-strategist in the relatively nutrient depleted regions of the world’s oceans.
Genome sequencing has revealed that there are two separate clades within the species A. macleodii that have average nucleotide identities along the core genome expected from strains belonging to different species (ca. 82%).16,22,23 The surface clade is found in surface waters at temperate or tropical latitudes all around the world while the “deep ecotype” clade has been largely found in the deep Mediterranean. Despite the relevance of A. macleodii in marine environments, no phages from this microbe have been isolated previously.
N4-like bacteriophages are lytic Podoviridae phages. For 40 y, the Escherichia coli bacteriophage N4 was the only described member of this genus. Presently there are 11 known members that infect different Proteobacteria: E. coli (N4,24 EcP1 (unpublished; GenBank ID: NC_019485), IME1125 and vB-EcoP-G7C26), Roseovarius sp (Roseovarius sp 217 phage 1 (unpublished; GenBank ID: FR682616)), Pseudomonas sp (LIT1,PEV2 and LUZ727), Silicibacter sp (DSS3φ2 28), Sulfitobacter sp (EE36ϕ128) and Erwinia sp (vB-EamP-S629). Enterophage N4 is unique in (i) the use of three different DNA-dependent RNA polymerases during its growth cycle,30 (ii) a virion-encapsidated, very large (3500 amino acids) RNA polymerase (N4 vRNAP), that is injected into the host cell upon infection,31-33 (iii) the use of single-stranded DNA binding proteins as transcriptional activators, (iv) the presence of direct repeats varying in length from 390 to 440 bp and 3′ extensions at the ends of the genome,34 and (v) a lysis-inhibited infection cycle.35 All N4-like phages have a proximate size of 70 kb and show significant similarity in key proteins such as the large N4-vRNAP, the DNA polymerase and also similar structural proteins (major capsid protein, tail and tail tip proteins, portal proteins and terminases).
Here we report the morphology, basic biology and genome sequences of four phages, AltAD45-P1 to P4, isolated on A. macleodii AD45 as host, a representative of the surface clade.36 However, they also infected other strains including representatives of the deep clade. The genome organization and some sequence similarity identify these phages as relatives of the N4-like genus of Podoviridae. Nonetheless, they exhibit very distinctive features from all the other known members of the N4-like group and should be considered a new genus within the Podoviridae family.
Results AND Discussion
Morphology and basic biology of AltAD45-P1 to P4
According to the novel universal system of bacteriophage naming,37 the full suggested names of these bacteriophages are vB_AmaP_AD45-P1 to P4. In this work, we use the abbreviated reference AltAD45-P1 to P4. All four phages were isolated from coastal waters near a fish-farm in Altea (Alicante, Spain). Although three strains of A. macleodii were used to detect plaque formation, only one of them (strain AD45) produced visible plaques. Interestingly, this strain was also isolated from waters near a fish farm.38 AltAD45-P1 to P4 were examined by transmission electron microscopy (TEM). Morphologically, they were very similar to each other and have the morphology characteristic of the Podoviridae, with isomorphous icosahedral capsids of ca. 60–70 nm of diameter. A short tail structure could be distinguished in all the Alterophages (Fig. 1).
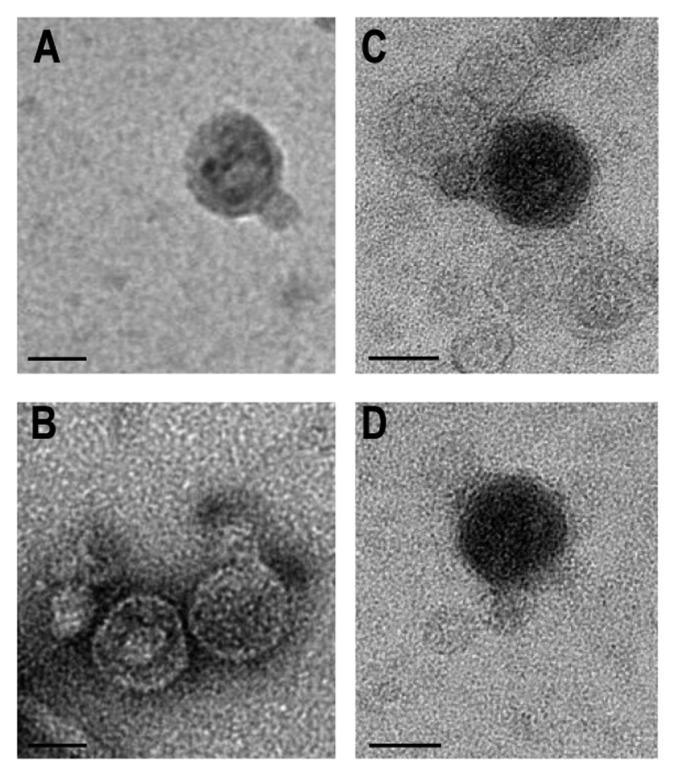
Figure 1. Transmission electron microscopy images of Alterophages stained with Uranil Acetate 2%. (A) AltAD45-P1. (B) AltAD45-P2. (C) AltAD45-P3 and (D) AltAD45-P4. Scale bar: 50 nm.
The infectivity of the phages was not affected by chloroform treatment (20%), indicating that neither of them is membrane-coated. They were also stable during long-term storage at 4°C. To investigate the host-range, cross-infections were tested against diverse A. macleodii strains AltDE, AltDE1, 673, AD45 and BS11.23,36 All the phages formed clear, round and regular edge plaques (1 mm diameter) infecting more than one strain. The host range results together with the efficiency of plaquing and the adsorption tests are shown in Table 1. Remarkably, all the phages infected AltDE, a strain isolated from the deep Mediterranean. AltDE1, very similar to AltDE and from the same origin,36 could only be infected by AltAD45-P1 and P2. Similarly, BS11, isolated from the Black Sea, was only infected by AltAD45-P1 and P2. Among the strains assayed, only strain 673, isolated from the English Channel, was resistant to all phages.
Table 1. Host ranges and percentage of adsorption of phages AltAD45-P1 to P4 on Alteromonas macleodii strains.
| AltAD45-P1 | AltAD45-P2 | AltAD45-P3 | AltAD45-P4 | |||||
|---|---|---|---|---|---|---|---|---|
| |
EOP* |
A (%)** |
EOP* |
A (%)** |
EOP* |
A (%)** |
EOP* |
A (%)** |
| AD45 |
1 |
91.2 ± 0.6 |
1 |
89.4 ± 14.3 |
1 |
84.4 ± 2.5 |
1 |
76.3 ± 8.9 |
| AltDE |
0.618 |
85.3 ± 9.1 |
0.728 |
94.0 ± 0.6 |
0.708 |
86.4 ± 35.9 |
0.649 |
91.8 ± 5.6 |
| AltDE1 |
0.243 |
97.7 ± 9.4 |
0.321 |
94.8 ± 1.6 |
— |
— |
— |
— |
| BS11 |
0.248 |
74.5 ± 13.6 |
0.329 |
88.0 ± 10.1 |
— |
— |
— |
— |
| 673 | — | — | — | — | — | — | — | — |
EOP: efficiency of plaquing. **A: % adsorption in 15 min. —, not determined, as the phage does not infect the strain.
The latent period of AltAD45-P1 and P2 was about one hour, followed by a gradual increase of released viral particles (Fig. 2). It took about 5 h to reach their growth plateau and this resulted in burst sizes of ca. 600 viral particles per infectious center. These values were very similar to AltAD45-P3 and P4, with 6 h to reach the plateau and burst sizes of near 500 (Fig. 2). Delayed lysis and burst sizes were also characterized in phage N4, which produces ca. 3000 viruses per cell 3 h post infection.39 The longer lysis period of the Alterophages compared with N4 can be partially explained by the nearly four-times slower growth rate of A. macleodii AD45 at 25°C compared with E. coli.

Figure 2. One-step growth curves of AltAD45-P1, P2, P3 and P4 phages in A. macleodii AD45. PFU per ml of AD45 culture at different time points. Each point represents the mean of three experiments.
Overall genome comparison of AltAD45-phages
Alterophages AltAD45-P1 to P4 show a very similar genome size that range between 100–104 kb (Table 2; Fig. 3A). Their genome size is more than 30 kb larger than any other podovirus described till now (genomes of viruses classified as Podoviridae range between 19 and 74 kb). On the other hand, the size is comparable with other marine phages of the Siphoviridae family such us the Cyanophage PSS2 (107 kb), the Vibrio phage pVp-1 (111 kb) or the unclassified Colwellia phage 9A (104 kb). However, our phages do not belong to the Siphoviridae as deduced from the sequence (see below). Their GC-content is very similar to the one of the host (A. macleodii AD45 strain), 44.6% (Table 2).
Table 2. General features of the Alteromonas macleodii phages AltAD45-P1 to P4.
| Phage | Length (bp) | #ORFs | #ORFs N4-like | #ORFs annotated | t-RNA | % GC |
|---|---|---|---|---|---|---|
|
AltAD45-P1 |
103910 |
129 |
18 |
29 |
t-RNA Met |
43.15 |
|
AltAD45-P2 |
104036 |
129 |
18 |
29 |
t-RNA Met |
43.21 |
|
AltAD45-P3 |
101724 |
124 |
18 |
29 |
- |
43.15 |
| AltAD45-P4 | 100619 | 122 | 18 | 29 | - | 43.19 |

Figure 3. (A) Comparative genomic organization of Alterophages AltAD45-P1 to P4. Conserved genomic regions among phages are indicated by gray shaded areas. (B) Map of the single nucleotide polymorphisms (SNP) found in AltAD45-P3 genome when compared with AltAD45-P4. ORFs with non-synonymous substitutions are colored in gray and labeled.
Regions of variability and Single Nucleotide Polymorphisms (SNP)
The comparison of the AltAD45-phage genomes is shown in Figure 3. The similarity among the AltAD45-P1 to P4 genomes was remarkable. Nucleotide-to-nucleotide identity was greater than 98% in more than 88 kb along the genomes with the exception of 9 ORFs (ca. 3 kb) inserted in a region found only in AltAD45-P1 and P2 (Fig. 3A and dash box in Fig. 4). To discard any assembly artifact, the presence of this insertion was confirmed by PCR.

Figure 4. Genomic organization of AltAD45-P1. Conserved ORFs with N4-like viruses are red colored. ORFs with assigned functions are colored in blue and in gray, ORFs without them. Dash box indicate the genomic regions not conserved in AltAD45-P3 and P4. Putative functions of selected genes are indicated. Arrows at the ends indicate the terminal repeats.
AltAD45-P1 and P2 were 99.8% identical and only 13 single-nucleotide-polymorphisms were detected (in the range of the sequencing error). Therefore, AltAD45-P1 and P2 can be considered as different isolates of the same phage species. Although AltAD45-P3 and P4 are also very similar and both possess similar infectivity range, they present a significantly higher number of SNPs (368) plus two extra small ORFs (582 and 264 bp) in AltAD45-P3. When the SNPs were plotted along the AltAD45-P3 genome, it was possible to observe that the point mutations were not uniform (Fig. 3B) and most of them were accumulated in some genes along the nucleotide synthesis and replication module (see below). Unambiguous multiple sequence alignments at the nucleotide level were obtained for individual genes. Only 18 ORFs were found to have more than one non-synonymous change (Fig. 3B). However, for 17 of them, the ratio non-synonymous / synonymous (dN/dS) were < 1, indicating purifying selection40 (Table 3). The tail fiber coded by ORF20 was the only protein that presented significant positive selection. Remarkably the DNA primase had the highest number of polymorphisms in a single gene (128), followed by the DNA helicase (45) (Table 3). In Roseophage SIO1–1989, a tail fiber protein and the DNA primase were also among the most variable areas.41 These results support that, as suggested previously,41 there is an arms race with the host (Red-Queen dynamics) since the tail fiber is the main host target recognition mechanism. The existence of different clonal frames of A. macleodii from a single sample was reported previously.36 Interestingly, a high recombination rate was found within the flexible genomic islands of the A. macleodii genomes coding for the biosynthesis of the O-chain of the lipopolysaccharide LPS, a well-known phage recognition target. Although the isolated Alterophages are very similar among themselves, they showed different polymorphisms and infectivity capacities. As conclusion, these results suggest then that very similar phages co-exist at one single time point together different bacterial clonal frames that co-evolve with them.
Table 3. List of genes with highest number of SNP observed between AltAD45-P3 and P4.
| #ORF in AltAD45-P3 | #ORF in AltAD45-P4 | Protein Function | Length (bp) | #SNP | Sd* | Sn** | dN/dS*** |
|---|---|---|---|---|---|---|---|
| ORF101 |
ORF99 |
primase |
2262 |
128 |
118.0 |
10.0 |
0.019 |
| ORF110 |
ORF108 |
helicase |
1428 |
45 |
38.5 |
6.5 |
0.043 |
| ORF104 |
ORF102 |
HP |
807 |
25 |
18.5 |
6.5 |
0.089 |
| ORF121 |
ORF119 |
RNA polymerase |
4230 |
21 |
17.0 |
4.0 |
0.065 |
| ORF102 |
ORF100 |
RIIA |
2088 |
19 |
7.0 |
12.0 |
0.477 |
| ORF105 |
ORF103 |
HP |
462 |
16 |
13.0 |
3.0 |
0.059 |
| ORF52 |
ORF51 |
HP |
2010 |
14 |
8.5 |
5.5 |
0.178 |
| ORF65 |
ORF63 |
transcriptional regulator |
450 |
11 |
9.0 |
2.0 |
0.056 |
| ORF109 |
ORF107 |
HP |
894 |
10 |
4.0 |
6.0 |
0.406 |
| ORF64 |
ORF62 |
HP |
366 |
9 |
6.0 |
3.0 |
0.129 |
| ORF57 |
ORF56 |
HP |
183 |
7 |
5.0 |
2.0 |
0.085 |
| ORF103 |
ORF101 |
HP |
378 |
6 |
4.0 |
2.0 |
0.128 |
| ORF108 |
ORF106 |
HP |
696 |
6 |
4.0 |
2.0 |
0.139 |
| ORF19 |
ORF19 |
tail fiber |
4638 |
5 |
1.0 |
4.0 |
1.1 |
| ORF66 |
ORF64 |
HP |
174 |
4 |
1.0 |
3.0 |
0.801 |
| ORF112 |
ORF110 |
DNA polymerase |
1851 |
3 |
2.0 |
1.0 |
0.145 |
| ORF3 |
ORF3 |
HP |
1911 |
2 |
1.0 |
1.0 |
0.292 |
| ORF111 | ORF109 | exonuclease | 672 | 2 | 1.0 | 1.0 | 0.271 |
Sd: number of synonym substitutions. **Sn: number of non-synonymous substitutions. HP: hypothetical protein. ***dN/dS: non-synonymous vs. synonymous nucleotide substitution ratio
Genome structure and classification
Due to the similarity of all the Alterophages described here, we focus only on the genome of AltAD45-P1 to describe the main genomic features (Fig. 4). A total of 127 open reading frames (ORFs) could be identified, representing 91.6% of the coding capacity of the genome and reflecting a compact genome organization. Since AltAD45-P1 diverged from other available phage genomes, only a limited number (23%) of protein functions could be predicted by similarity searches, highlighting the novelty of this group of phages. Among the identified ORFs, 18 were related to viral genes from the N4-like genus (in red in Fig. 4). However, the ranges of overall similarity were as low as 21–57%, and genomic comparisons with N4-like phages showed that many times only a small part or domain of the ORFs were conserved (a genomic comparison among AltAD45-P1, Escherichia phage N4 and Pseudomonas phage LIT1 and can be found in Fig. S2). The different genomic organization together with the phylogenetic analysis of the DNA polymerase and the terminase large subunit (see ahead), indicates that Alterophages AltAD45-P1 to P4 belong to a new genus within the Podoviridae family.
Despite the low number of annotated ORFs, different modules can be recognized in the AltAD45-P1 genome. Relatively long (437 bp) direct terminal repeats typical of N4-like phages were found at both ends of the genome. The overall architecture of bacteriophage N4 is shared among all the phages of the group.27 Genes for the DNA replication and DNA metabolism are found at the beginning of the genomes, followed by the large vRNAP, and the tail, structural and packaging proteins toward the end. Some of the main differences among them are the existence of extra clusters of small genes located directly upstream from the right or left terminal repeats which code mainly for hypothetical proteins. AltAD45-phages have a different genome organization with the modules following a different order, structural, DNA metabolism, transcription and lysis (Fig. 4). The least conserved region between the Alterophages and N4-like viruses is the large cluster of small genes located in the center of the genomes, both up and downstream of the insertion present in AltAD45-P1 and P2. The AltAD45-P1 and P2 insertion contains a tRNA-Met plus eight ORFs of unknown function (dash box in Fig. 4). Similar to the extra-modules of small genes present in several N4-like viruses, the ORFs of the AltAD45-phages lack any significant similarity to current database entries and any recognizable structural features, so their functions remain unknown. Adjacent to this region, there is one ORF 77% similar to a hypothetical gene of the A. macleodii strain AltDE. The ORF64-like gene in AltDE is placed in one area of the AltDE genome annotated as a putative prophage22 (Fig. S1). This result may suggest that the insertion observed in AltAD45-P1 and P2 could come from another phage preying on A. macleodii. In the following sections, we will describe the major findings on each of the AltAD45-P1 gene-modules.
Tail, structural and packaging module
Sequence-base predictions identified six ORFs as being involved in virion morphogenesis: four phage tail proteins (ORF6, 19, 20 and 22), the major capsid protein (ORF10) and the portal protein (ORF15). The first tail protein is 54% similar to a small region of a large hypothetical protein in the A. macleodii 673 genome (Fig. 4). Similarly, the third tail fiber (ORF20) and the hypothetical protein coded by ORF18 share similarity (71% and 48%, respectively) with genes found in A. macleodii strain BS11 (Fig. 4). Both areas of the genomes of the A. macleodii strains have been described as putative prophages.23 A detailed comparison of these A. macleodii genome areas and AltAD45-P1 showed that no other regions of the phage genome share additional features with the inserted putative prophages (Fig. S2). There is the possibility that some relatives of AltAD45-phages are temperate phages infecting these strains. A similar observation was made in Pseudomonas phage LIT1/PEV2, a member of the N4-like virus, where a cluster of tail genes display significant similarity to a Pseudomonas aeruginosa prophage region.27 The fourth tail protein detected is similar to one found in the IME11 N4-like virus. The other structural components found in this module were similar (49–60%) to Pseudomonas phage LIT1 and Escherichia phage N4, both in the N4-like genus. The podoviral nature of these Alterophages is further supported by the absence of tail baseplate wedge proteins, critical for formation of a contractile tail in myoviruses,7 and tape measure proteins, which acts as scaffold to assemble the phage-tail both in myovirus and siphovirus.
Following the structural components, there are genes for the packaging of the DNA. Only the large subunit of the terminase, similar (39%) to the Pseudomonas phage LIT1, could be identified. Phylogenetic analysis of this large subunit terminase, cluster the AltAD45-phages in a separate clade from the N4-like viruses (Fig. 5A). However, the N4-like remain the closest. Although the small terminase subunit could not be identified, the most probable candidate is ORF24, which has the appropriate size and genomic location.

Figure 5. (A) Phylogenetic analysis of DNA-polymerases and (B) large-subunit terminases. Phylogenetic tree constructed using Maximum Likelihood and based on alignments obtained with T-coffee. Numbers in the nodes indicate the bootstrap support (only over 50% is shown). AltAD45-phages cluster is indicated in bold.
The nucleotide synthesis and DNA replication module
These genes concentrated in a region that stretches from ORF90 (transcribed in the opposite direction to the previous genes) to ORF125, just before the RNA-polymerases found at the end of the genome. They are separated from the structural module by a region enriched in small hypothetical proteins which spans about 30 kb. Many N4 early genes that belong to this category, were absent in AltAD45-P1 to P4 and they were replaced by genes with similar functions from other virus or bacteria.
DNA replication
The DNA polymerase (ORF117) present in all AltAD45-phages is 47% similar to the one present in the Escherichia phage vB_EcoP_G7C, a N4-like virus.26 As happens with the large subunit of the terminase, DNA polymerase phylogenetic analysis shows that Alterophages AltAD45-P1 to P4 cluster separated from the N4-genus DNA polymerase and far from the T7 super-group podoviruses (Fig. 5B). All the AltAD45-phages contained a small thioredoxin gene (trx) (ORF105), and the protein coded by this gene shares 37% similarity with the bacterial Trx thioredoxin of A. macleodii AltDE. A host-related thioredoxin has also been found in other T7-like marine phage genomes and in two roseophages from the N4-like group. It has been shown that binding of the host thioredoxin to T7 DNA polymerase, increases its processing speed42 and suggested that the presence of the thioredoxin might be important to phage survival in marine environments.43 The DNA helicase found in the Alterophages also resembles bacterial helicases more closely than the conserved helicases of N4-like viruses.
A DNA primase (ORF106) similar to that of the Enterophage N4 was also identified. In most of the N4-like viruses, a single-stranded DNA binding protein gene, ssb, is located next to the DNA primase. Ssb is involved in DNA replication, DNA recombination44,45 and activation of E. coli RNA polymerase in late N4 transcription.46,47 In AltAD45-phages genomes next to the primase there were genes which coded for proteins that were among the size range of the Ssb protein (200–250 amino acids). However, none of them have any similarity to this protein. The DNA ligase identified here was most closely related that of the Xanthomonas phage Xp15, from the Caudovirales.
Nucleotide synthesis
AltAD45-phages have two rnr genes(ORF119–120), which code for ribonucleotide reductases. It is a key enzyme in DNA synthesis and converts ribonucleosides to deoxynucleotide.48 Several podoviruses isolated from marine environments, such as Roseophages (SIO1, DSS3Φ2, EE36Φ1, P60, P-SSP7 and syn5), and Cyanophages (NATL2A-133 and NATL1A-7)7,49 also contain this gene. Its presence in many marine phages is justified by the nucleotide requirements for phage DNA synthesis in the marine, typically phosphate-deprived, environments.7 Supporting this idea, rnr has been found to be among the most abundant genes in the Sargasso Sea metaviriome analysis.50 Also in common with some Roseophages (DSS3Φ2, EE36Φ1 and SIO-1989), AltAD45-phages have two genes for a thymidylate synthase protein (Thy1) (ORF96, ORF99). These are similar to homologous genes of Vibrio vulnificus MO6–24/O (58%) and Labrenzia alexandrii DFL-11 (54%). Thy1 protein generates thymidine monophosphate (dTMP) which is subsequently phosphorylated to be used in the DNA synthesis and repair. As happens with the ribonucleotide reductases, the viral thymidylate synthases may be involved in scavenging host nucleotides. It is thought that phages carry these genes to alleviate bottlenecks in these key pathways after host transcription has stopped.51
DNA metabolism
Phages can prompt gene recombination via homing endonucleases (HEs) which transfer the genetic elements from the HE-encoding genome to a HE-lacking recipient.52-54 Among the so called “freestanding” endonucleases, two families of proteins are found, the GIY-YIG and HNH homing endonuclease families. Among the AltAD45-phages, three HE were localized, two with domains GIY-YIG (ORF46, ORF98) and one with a HNH domain (ORF97), none of them related with the ones of the N4-like viruses (found in Roseophages DSS3ΦF2 and EE36ΦF1 and the Enterophage N4). Interestingly, these HEs are placed toward the beginning and the end of a ca. 30 kb stretch of small hypothetical genes, within which the insertion affecting AltAD1 and P2 was found. This area of the AltAD45-phages could have been acquired through some recombination event. However, no distinctive genomic features (such as GC-content or oligonucleotide frequency) could be detected. A gene for an endo-deoxyribonuclease, RusA (ORF118), was also found near the end of the DNA replication module. The RusA protein of Escherichia lambdoid phage 82 is an endonuclease that can resolve Holliday intermediates, eliminating branched intermediates that arise from recombinational exchange and DNA repair.55,56 Additional Holliday junction resolvases have been described (T7 endo I, T4 endo VII, Rap and RuvC-like),57 but no similar protein has been found among the N4-like viruses. RusA is also present in other marine phages, such as the cyanosiphoviruses S-CBS1 and S-CBS3 and the recently detected MEDS5 cluster in the Mediterranean Sea.58 The presence in the AltAD45-phages of the RusA protein together with the HE and the large genomic fragment inserted between them might indicate that these phages are highly recombinogenic and do not rely on the host resolution systems.
Transcription module
The most remarkable protein that makes the N4 bacteriophage family unique is the virion RNA polymerase (vRNAP, gp50), which is by far the largest described among all the known phages, ca. 3500 amino acids. The reason for having this single subunit large RNA polymerase is unknown, but its conservation across this phage genus suggests that vRNAP plays an important role in their infection strategy. This protein is packed into the capsid and injected into host cells upon infection.31 It is responsible for the transcription of early and DNA replication genes59 A second RNA-polymerase II gene is also needed for the phage transcription.30,60 None of the AltAD45-phages possess such a large gene and no genes similar to those coding for the second RNA polymerase II were found either. However, two adjacent genes at the end terminus (ORF126 and ORF127) code for two proteins that together will add to the size of vRNAP (1404 and 2481 amino acids respectively). BLASTP comparisons of these proteins showed that they have some similarity to the N and C-terminal part respectively of the N4-gp50 of Pseudomonas phage LIT1 or the Enterophage A427 (Fig. S3). To discard that a stop codon was created artificially due to sequencing errors, the site was PCR amplified and resequenced. In addition, the two polypeptides were identifiable in polyacrylamide gels (sizes of 270 and 175 kDa).
In bacteria, RNA-polymerases (RNAP) are large molecules constituted by five core-subunits (ca. 400 kDa), from which the β and β′ subunits are the ones where the active center rely.61 Only in a few cases, RNAP are synthetized in one single polypeptide (some Helicobacter and Campylobacter species62). N4 vRNAP’s size is comparable with the combined sizes of the eubacterial RNAPs β and β′ subunits, but it appears to be a evolutionarily highly divergent member of this class of enzymes.63 Although vRNAP consists of a single 3500 amino acid polypeptide, Kazmierczak et al. (2002) described that an 1106 amino acid domain, called “mini-vRNAP,” possess the same transcriptional properties than the full-length version of the protein. Also, they suggested that the other parts of the vRNAP have other functional purposes, the C-terminus comprises a stable domain that is required for polymerase encapsidation, while the N-terminal domain is required for genome injection. The equivalent part of the mini-vRNAP in the Alterophage AltAD45-P1 is conserved at the N-terminus of the protein coded by ORF127, while a region similar to the C-terminus of vRNAP is found in the next ORF126. Importantly, vRNAP contains no cystein residues, a requirement for the enzyme to pass through the tail into the cell and/or through the host periplasm, which contains proteins that catalyze disulfide bond formation.63 The protein coded by ORF127 contains two cysteins, which suggests that this ORF will not be injected into the host cell. In conclusion, it appears that AltAD45-phages have a RNAP synthesized as two independent polypeptides. Single-subunit RNAPs share blocks of conserved sequence and they appear to have evolved from a single subunit common ancestor.63 Therefore, the most likely scenario to explain the RNAP configuration of AltAD45-P1 to P4 is that they suffered a fission event from a single polypeptide ancestor.
Lysis genes
A key step of the phage infecting process is the release of new virions at the end of the lytic cycle. Among the N4-viruses, an N-acetyl-muramidase and an endolysin have been described as involved in host cell lysis.26,35 Marine roseophages DSS3ΦF2 and EE36ΦF1 are both lacking any N4-like endolysin gene. In AltAD45-phages, the hypothetical protein coded by ORF128 (next to the RNAP) is likely to act as a lysis gene as HHpred predictions showed a remote similarity to the lysozyme hydrolase found in bacteriophage N4.
Conclusions
AltAD45-P1 to P4 are the first phage isolates that infect the abundant marine gamma-proteobacteria A. macleodii. They belong to the Podoviridiae family, with an isomorphous icosahedral head of about 60–70 nm and a short non-contractile tail. Remarkably, their genomes sizes range from 100 to 104 kb, which are 30 kb larger than any other described podovirus. Genomic comparisons showed that the four AltAD45-phages are very similar, with very little differences in gene content. AltAD45-P1 and P2 had an insertion of ca. 3 kb that could be involved in the different host-infections capacities showed by these phages.
Although the AltAD45-phages described here have the N4-like phages as closest relatives, they have enough differences to be considered a separate genus. The size and genome organization are already significant but probably the lack of a cysteine-free N4-like vRNAP divided in two independent polypeptides, indicates major differences in the replication mechanism of these phages. Also the lack of a protein similar to the single-stranded DNA binding protein, and the absent of an extra DNA-dependent RNA-polymerase suggests a different growth style. Supporting a separate classification, phylogenetic analysis of the DNA polymerase and the large subunit terminase, indicates that they are phylogenetically distant from the N4 and T7 phages. Therefore, we propose the creation of a new phage genus inside the Podoviridae, the “AltAD45-like viruses.”
The ever-increasing number of complete phage genome sequences has greatly improved our knowledge regarding phage diversity. A comprehensive appreciation of phage diversity and evolution, however, will require both the sequencing of additional phage genomes and extensive molecular research to relate those genomic sequences to biological functions. The characterization of AltAD45 phages will help to determine the molecular background of ecologically meaningful phenotypic differences identifiable in genetically related phage isolates.
Materials and Methods
Collection of samples and isolation of the AltAD45-phages
A seawater sample (5 ml) was collected near a fish farm in Altea, Spain, on November, 2011. Following filtration through 0.22 µm filter (Millipore, Westborough, MA, USA), A. macleodii strains AltDE1, AD45 and 673 were assayed for infection. A lawn of bacterial cells was produced using the agar overlay technique. For the base layer, marine agar (Pronadisa, Madrid, Spain) was used and marine soft agar 0.5% for the top layer. Exponentially growing A. macleodii cells (100 µl) were mixed with the filtered sample (100 µl) and incubated 20 min at room temperature. Then, 4 mL of soft agar previously cooled to 45°C was added to this mixture and spread in the marine agar plate. The plates were incubated at room temperature in darkness until plaques appeared, approximately 12 h later. Phages were isolated by picking individual plaques and resuspended in 500 µl of marine media (sea salts 3.5% (Sigma), 0.5% peptone (AppliChem) and 0.1% of yeast extract (AppliChem)). To ensure that each plaque was derived from a single phage, three additional rounds of purification on top agar were performed.
Purification of phage DNA
To obtain a stock of the isolated viruses, they were grown in confluent plaques and the soft agar was recovered for 50 plates and mix with marine liquid media (2 ml/plate), incubated two hours and then centrifuged to obtain the supernatant. The stocks were filtered using 0.22 µm filters (Millipore) and concentrated to a final volume of 1 ml with a 100 kDa amicon (Millipore). Next, a treatment with DNase I (2U/ml) (Invitrogen) was done during 30 min at 37°C. Next, SDS (final concentration of 0.4%) and protease K (20 mg/ml, AppliChem) was added and incubated for 1 h at 56°C. DNA was extracted using a phenol-chloroform isolation and precipitated with ethanol 100% and ammonium acetate 3M. The phage DNA was finally resuspended in miliQ water and quantified by Quant-iT® PicoGreen ® dsDNA Reagent (Invitrogen).
Electron microscopy
A 10 μl sample from each viral suspension was fixed with formaldehyde (4%, Sigma) for 30 min at 4°C and stained for 5 min with uranyl acetate (0.5%, Electron Microscopy Sciences) on Formvar-coated carbon grids (Electron Microscopy Sciences). Virus-like particles were observed in a Jeol JEM-2010 transmission electron microscope operating at 200 kV.
Host specifity studies
To assess the host range, the Alterophage isolates AltAD45-P1 to P4 were screened against A. macleodii AD45 (the original host), AltDE, AltDE1, 673, and BS11. A volume of 5 μl of 10−2 and 10−4 dilutions of a high-titer lysate (109 PFU ml−1) were spotted on top of agar containing cells of one of the five Alteromonas strains tested (see Table 1). To study the phage adsorption process, A. macleodii cultures were grown in marine media to an optical density at 600 nm of 0.6 to 0.8, after which they were in contact with the Alterophages at a final concentration of 103 PFU ml−1. The phage-containing cultures were incubated at room temperature for 15 min to allow adsorption of the phages to the cells. The suspension was centrifuged for 2 min at 12100 g, and the phages present in the supernatant were enumerated by plaque assay.64 This assay was repeated three times (n = 3). To determine the presence of active natural defense mechanisms against any of the Alterophages, the efficiency of plaquing was calculated by dividing the phage titer on the test A. macleodii strain by the titer of the phage on the phage-sensitive host strain A. macleodii AD45. For the one step growth curves of Figure 2, exponentially growing cultures of A. macleodii AD45 (20 ml) were inoculated with the AltAD45-P1 to P4 phages at a multiplicity of infection of 0.1–0.01. Fifteen minutes after infection, non-adsorbed phage particles were eliminated from the culture by pelleting the cells. The AD45 cells were resuspended in new fresh media (20 ml). The number of infectious centers was determined using plaquing serial dilutions with AD45 cells. An aliquot of the cell suspension was collected from each culture every hour for a period of 20 h. The number of phages was determined plaquing serial dilutions.
Sequencing and assembly
The four Alterophages AltAD45-P1 to P4 were sequenced individually by 454 GS-FLX-system (Roche 454 GS-FLX system, CSIPS, Valencia, Spain) at high coverage (55–111 fold). Sequencing details can be found in Supplementary Table 1. Low quality regions were clipped using sff_extract (http://bioinf.comav.upv.es/sff_extract/). Sequences were assembled by two different assemblers: Geneious v. 5.6.0 (http://www.geneious.com/) (using the default parameters) and MIRA (http://www.chevreux.org/projects_mira.html)) to check the consistency of the sequences assembled. All the AltAD45-phages were assembled in one single contig by both assemblers.
PCR amplification
Different PCR amplifications were done in order to corroborate the existence of the inserted region observed in AltAD45-P1 and P2 and absent in AltAD45-P3 and P4. Several oligonucleotides were designed to amplify the complete inserted region in AltAD45-P1 and P2 (4400 pb) or a smaller genomic fragment from AltAD45-P3 and P4 (1950 pb). Also, PCRs were performed to verify the existence of a stop codon in the vRNAP-like gene present in all the Alterophages. PCR was performed in 50-μL volumes and contained 1.5 mm/L MgCl2, 200 μm/L dNTPs, 1 U of Expand High Fidelity DNA polymerase (Roche), 500 nm/L of each of the primers and 100 ng of genomic DNA. Following an initial denaturation step of 4 min at 94°C, 35 PCR cycles were performed (94°C for 2 min, 58°C for 60 sec, 72°C for 2 min) followed by a final extension step at 72°C for 10 min. All PCRs yielded only specific products, that is, single bands as judged by electrophoresis of the PCR products on agarose gels. PCR products were sequenced in both directions.
Analysis of the sequences
Gene prediction was done using Prodigal,65 and the predicted protein sequences were compared with the NCBI-nr database using BLASTP (E-value < 1e−4). Domain predictions in the sequences were performed using the HMMER package66 (against the Pfam database), the HHpred server (http://toolkit.tuebingen.mpg.de)67 and the NCBI Conserved Domains Database.68 Phage annotation was manually inspected, especially those initially annotated as hypothetical proteins. All-vs-all comparisons of the AltAD45-P1 with other N4-like viruses (Enterobacter phage N4 and Pseudomonas phage LIT1, obtained from NCBI Viral RefSeq) and AltAD45-P1 with specific areas of the A. macleodii genomes were performed using reciprocal BLASTN and TBLASTX searches and visualized using custom perl scripts and the ACT Artemis Comparation tool v.9.69 Only homologous genomic regions bigger than 50 bp are shown in the figures. GC-content was calculated using the EMBOSS tool ‘geecee’70. BioEdit software was used to manipulate the sequences.71 tRNAs were identified using tRNAscan-SE.72
Single Nucleotide Polymorphism detection
‘Nucmer’ and ‘show-snp’ of the MUMmer3+ package was used to calculate the single nucleotide polymorphisms between the AltAD45-P3 and AltAD45-P4. Non-synonymous vs. synonymous nucleotide substitution ratio (dN/dS) was used as an indicator of selective pressure acting on a protein-coding gene. Orthologous protein sequence pairs were aligned using MUSCLE73 and the protein alignments imposed upon the nucleotide sequences using the program pal2nal.74 Protein-encoding sequences with a high dN/dS ratio (> 1) were considered to exhibit diversifying selection (positive selection for variability at some sites), whereas a low dN/dS ratio (< 1) indicates purifying (negative) selection.
DNA polymerase and large subunit terminase phylograms
Phylogenetic analysis of the DNA polymerase and large subunit terminase were performed. For making the phylogenetic trees, alignments were created using T-Coffee,75 manually inspected and trimmed when necessary. Maximum likelihood trees were made using the program FastTree2.76 Bootstrapping was performed using the seqboot program in the Phylip package.77 MEGA 5.05 sofware was used to represent the tree.78
Accession Numbers
The sequences have been deposited in GenBank under the Bioproject number PRJNA191132.
Supplementary Material
Acknowledgments
This work was supported by projects MAGYK (BIO2008-02444), MICROGEN Programa CONSOLIDER-INGENIO 2010 (CDS2009-00006), (CGL2009-12651-C02-01) from the Spanish Ministerio de Ciencia e Innovación, DIMEGEN (PROMETEO/2010/089), ACOPM/2012/146 from the Generalitat Valenciana, BIO2011-12879-E from Spanish Ministerio de Ciencia e Innovación and MaCuMBA from EU (P. no. 311975). FEDER funds supported this project. Critical reading of the manuscript by Nikole Kimes is appreciated.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/bacteriophage/article/24766
Footnotes
Previously published online: www.landesbioscience.com/journals/bacteriophage/article/24766
REFERENCES
- 1.Canchaya C, Fournous G, Chibani-Chennoufi S, Dillmann ML, Brüssow H. Phage as agents of lateral gene transfer. Curr Opin Microbiol. 2003;6:417–24. doi: 10.1016/S1369-5274(03)00086-9. [DOI] [PubMed] [Google Scholar]
- 2.Fuhrman JA. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399:541–8. doi: 10.1038/21119. [DOI] [PubMed] [Google Scholar]
- 3.Breitbart M, Thompson LR, Suttle CA, Sullivan MB. Exploring the vast diversity of marine viruses. Oceanography (Wash DC) 2007;20:135–9. doi: 10.5670/oceanog.2007.58. [DOI] [Google Scholar]
- 4.Paul JH. Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008;2:579–89. doi: 10.1038/ismej.2008.35. [DOI] [PubMed] [Google Scholar]
- 5.Paul JH, Sullivan MB, Segall AM, Rohwer F. Marine phage genomics. Comp Biochem Physiol B Biochem Mol Biol. 2002;133:463–76. doi: 10.1016/S1096-4959(02)00168-9. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Valera F, Martin-Cuadrado AB, Rodriguez-Brito B, Pasić L, Thingstad TF, Rohwer F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–36. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 2005;3:e144. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12:3035–56. doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan MB, Waterbury JB, Chisholm SW. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;424:1047–51. doi: 10.1038/nature01929. [DOI] [PubMed] [Google Scholar]
- 10.Labrie SJ, Frois-Moniz K, Osburne MS, Kelly L, Roggensack SE, Sullivan MB, et al. Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ Microbiol. 2013;15:1356–76. doi: 10.1111/1462-2920.12053. [DOI] [PubMed] [Google Scholar]
- 11.García-Martínez J, Acinas SG, Massana R, Rodríguez-Valera F. Prevalence and microdiversity of Alteromonas macleodii-like microorganisms in different oceanic regions. Environ Microbiol. 2002;4:42–50. doi: 10.1046/j.1462-2920.2002.00255.x. [DOI] [PubMed] [Google Scholar]
- 12.Ivars-Martínez E, D’Auria G, Rodríguez-Valera F, Sânchez-Porro C, Ventosa A, Joint I, et al. Biogeography of the ubiquitous marine bacterium Alteromonas macleodii determined by multilocus sequence analysis. Mol Ecol. 2008;17:4092–106. doi: 10.1111/j.1365-294X.2008.03883.x. [DOI] [PubMed] [Google Scholar]
- 13.Sass AM, Sass H, Coolen MJ, Cypionka H, Overmann J. Microbial communities in the chemocline of hypersaline deep-sea basin (Uranian Basin- Mediterranean Sea) Appl Environ Microbiol. 2001;67:3083–91. doi: 10.1128/AEM.67.12.5392-5402.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Floyd MM, Tang J, Kane M, Emerson D. Captured diversity in a culture collection: case study of the geographic and habitat distributions of environmental isolates held at the american type culture collection. Appl Environ Microbiol. 2005;71:2813–23. doi: 10.1128/AEM.71.6.2813-2823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivars-Martínez E, D’Auria G, Rodríguez-Valera F, Sânchez-Porro C, Ventosa A, Joint I, et al. Biogeography of the ubiquitous marine bacterium Alteromonas macleodii determined by multilocus sequence analysis. Mol Ecol. 2008;17:4092–106. doi: 10.1111/j.1365-294X.2008.03883.x. [DOI] [PubMed] [Google Scholar]
- 16.López-López A, Bartual SG, Stal L, Onyshchenko O, Rodríguez-Valera F. Genetic analysis of housekeeping genes reveals a deep-sea ecotype of Alteromonas macleodii in the Mediterranean Sea. Environ Microbiol. 2005;7:649–59. doi: 10.1111/j.1462-2920.2005.00733.x. [DOI] [PubMed] [Google Scholar]
- 17.Acinas SG, Antón J, Rodríguez-Valera F. Diversity of free-living and attached bacteria in offshore Western Mediterranean waters as depicted by analysis of genes encoding 16S rRNA. Appl Environ Microbiol. 1999;65:514–22. doi: 10.1128/aem.65.2.514-522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, McCarren J, DeLong EF. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment. Environ Microbiol. 2012;14:191–206. doi: 10.1111/j.1462-2920.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- 19.McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR, et al. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci U S A. 2010;107:16420–7. doi: 10.1073/pnas.1010732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pukall R, Buntefuss D, Frühling A, Rohde M, Kroppenstedt RM, Burghardt J, et al. Sulfitobacter mediterraneus sp. nov., a new sulfite-oxidizing member of the alpha-Proteobacteria. Int J Syst Bacteriol. 1999;49:513–9. doi: 10.1099/00207713-49-2-513. [DOI] [PubMed] [Google Scholar]
- 21.Schäfer H, Servais P, Muyzer G. Successional changes in the genetic diversity of a marine bacterial assemblage during confinement. Arch Microbiol. 2000;173:138–45. doi: 10.1007/s002039900121. [DOI] [PubMed] [Google Scholar]
- 22.Ivars-Martinez E, Martin-Cuadrado AB, D’Auria G, Mira A, Ferriera S, Johnson J, et al. Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2008;2:1194–212. doi: 10.1038/ismej.2008.74. [DOI] [PubMed] [Google Scholar]
- 23.López-Pérez M, Gonzaga A, Martin-Cuadrado AB, Onyshchenko O, Ghavidel A, Ghai R, et al. Genomes of surface isolates of Alteromonas macleodii: the life of a widespread marine opportunistic copiotroph. Sci Rep. 2012;2:696. doi: 10.1038/srep00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schito GC. Intracellular crystallization of the DNA coliphage N4. Virology. 1967;32:723–5. doi: 10.1016/0042-6822(67)90050-5. [DOI] [PubMed] [Google Scholar]
- 25.Fan H, Fan H, An X, Huang Y, Zhang Z, Mi Z, et al. Complete genome sequence of IME11, a new N4-like bacteriophage. J Virol. 2012;86:13861. doi: 10.1128/JVI.02684-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kulikov E, Kropinski AM, Golomidova A, Lingohr E, Govorun V, Serebryakova M, et al. Isolation and characterization of a novel indigenous intestinal N4-related coliphage vB_EcoP_G7C. Virology. 2012;426:93–9. doi: 10.1016/j.virol.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Ceyssens PJ, Brabban A, Rogge L, Lewis MS, Pickard D, Goulding D, et al. Molecular and physiological analysis of three Pseudomonas aeruginosa phages belonging to the “N4-like viruses”. Virology. 2010;405:26–30. doi: 10.1016/j.virol.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Wang K, Jiao N, Chen F. Genome sequences of two novel phages infecting marine roseobacters. Environ Microbiol. 2009;11:2055–64. doi: 10.1111/j.1462-2920.2009.01927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Born Y, Fieseler L, Marazzi J, Lurz R, Duffy B, Loessner MJ. Novel virulent and broad-host-range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and a relationship to Enterobacteriaceae phages. Appl Environ Microbiol. 2011;77:5945–54. doi: 10.1128/AEM.03022-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willis SH, Kazmierczak KM, Carter RH, Rothman-Denes LB. N4 RNA polymerase II, a heterodimeric RNA polymerase with homology to the single-subunit family of RNA polymerases. J Bacteriol. 2002;184:4952–61. doi: 10.1128/JB.184.18.4952-4961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falco SC, Zehring W, Rothman-Denes LB. DNA-dependent RNA polymerase from bacteriophage N4 virions. Purification and characterization. J Biol Chem. 1980;255:4339–47. [PubMed] [Google Scholar]
- 32.Pesce A, Casoli C, Schito GC. Selectivity of transcription and structure of coliphage N4 virion-associated RNA polymerase. Biochem Biophys Res Commun. 1978;82:1040–8. doi: 10.1016/0006-291X(78)90888-4. [DOI] [PubMed] [Google Scholar]
- 33.Davydova EK, Kazmierczak KM, Rothman-Denes LB. Bacteriophage N4-coded, virion-encapsulated DNA-dependent RNA polymerase. Methods Enzymol. 2003;370:83–94. doi: 10.1016/S0076-6879(03)70008-1. [DOI] [PubMed] [Google Scholar]
- 34.Ohmori H, Haynes LL, Rothman-Denes LB. Structure of the ends of the coliphage N4 genome. J Mol Biol. 1988;202:1–10. doi: 10.1016/0022-2836(88)90512-8. [DOI] [PubMed] [Google Scholar]
- 35.Stojković EA, Rothman-Denes LB. Coliphage N4 N-acetylmuramidase defines a new family of murein hydrolases. J Mol Biol. 2007;366:406–19. doi: 10.1016/j.jmb.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 36.Gonzaga A, Martin-Cuadrado AB, López-Pérez M, Megumi Mizuno C, García-Heredia I, Kimes NE, et al. Polyclonality of concurrent natural populations of Alteromonas macleodii. Genome Biol Evol. 2012;4:1360–74. doi: 10.1093/gbe/evs112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kropinski AM, Prangishvili D, Lavigne R. Position paper: the creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ Microbiol. 2009;11:2775–7. doi: 10.1111/j.1462-2920.2009.01970.x. [DOI] [PubMed] [Google Scholar]
- 38.Pujalte MJ, Sitjà-Bobadilla A, Alvarez-Pellitero P, Garay E. Carriage of potentially fish-pathogenic bacteria in Sparus aurata cultured in Mediterranean fish farms. Dis Aquat Organ. 2003;54:119–26. doi: 10.3354/dao054119. [DOI] [PubMed] [Google Scholar]
- 39.Schito GC. Dvelopment of coliphage N4: ultrastructural studies. J Virol. 1974;13:186–96. doi: 10.1128/jvi.13.1.186-196.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angly F, Youle M, Nosrat B, Srinagesh S, Rodriguez-Brito B, McNairnie P, et al. Genomic analysis of multiple Roseophage SIO1 strains. Environ Microbiol. 2009;11:2863–73. doi: 10.1111/j.1462-2920.2009.02021.x. [DOI] [PubMed] [Google Scholar]
- 42.Huber HE, Tabor S, Richardson CC. Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J Biol Chem. 1987;262:16224–32. [PubMed] [Google Scholar]
- 43.Hardies SC, Comeau AM, Serwer P, Suttle CA. The complete sequence of marine bacteriophage VpV262 infecting vibrio parahaemolyticus indicates that an ancestral component of a T7 viral supergroup is widespread in the marine environment. Virology. 2003;310:359–71. doi: 10.1016/S0042-6822(03)00172-7. [DOI] [PubMed] [Google Scholar]
- 44.Lindberg G, Kowalczykowski SC, Rist JK, Sugino A, Rothman-Denes LB. Purification and characterization of the coliphage N4-coded single-stranded DNA binding protein. J Biol Chem. 1989;264:12700–8. [PubMed] [Google Scholar]
- 45.Choi M, Miller A, Cho NY, Rothman-Denes LB. Identification, cloning, and characterization of the bacteriophage N4 gene encoding the single-stranded DNA-binding protein. A protein required for phage replication, recombination, and late transcription. J Biol Chem. 1995;270:22541–7. doi: 10.1074/jbc.270.38.22541. [DOI] [PubMed] [Google Scholar]
- 46.Cho NY, Choi M, Rothman-Denes LB. The bacteriophage N4-coded single-stranded DNA-binding protein (N4SSB) is the transcriptional activator of Escherichia coli RNA polymerase at N4 late promoters. J Mol Biol. 1995;246:461–71. doi: 10.1006/jmbi.1994.0098. [DOI] [PubMed] [Google Scholar]
- 47.Carter RH, Demidenko AA, Hattingh-Willis S, Rothman-Denes LB. Phage N4 RNA polymerase II recruitment to DNA by a single-stranded DNA-binding protein. Genes Dev. 2003;17:2334–45. doi: 10.1101/gad.1121403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 49.Pope WH, Weigele PR, Chang J, Pedulla ML, Ford ME, Houtz JM, et al. Genome sequence, structural proteins, and capsid organization of the cyanophage Syn5: a “horned” bacteriophage of marine synechococcus. J Mol Biol. 2007;368:966–81. doi: 10.1016/j.jmb.2007.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, et al. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson LR, Zeng Q, Kelly L, Huang KH, Singer AU, Stubbe J, et al. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc Natl Acad Sci U S A. 2011;108:E757–64. doi: 10.1073/pnas.1102164108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleinstiver BP, Bérubé-Janzen W, Fernandes AD, Edgell DR. Divalent metal ion differentially regulates the sequential nicking reactions of the GIY-YIG homing endonuclease I-BmoI. PLoS One. 2011;6:e23804. doi: 10.1371/journal.pone.0023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgell DR, Gibb EA, Belfort M. Mobile DNA elements in T4 and related phages. Virol J. 2010;7:290. doi: 10.1186/1743-422X-7-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burt A, Koufopanou V. Homing endonuclease genes: the rise and fall and rise again of a selfish element. Curr Opin Genet Dev. 2004;14:609–15. doi: 10.1016/j.gde.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 55.Mahdi AA, Sharples GJ, Mandal TN, Lloyd RG. Holliday junction resolvases encoded by homologous rusA genes in Escherichia coli K-12 and phage 82. J Mol Biol. 1996;257:561–73. doi: 10.1006/jmbi.1996.0185. [DOI] [PubMed] [Google Scholar]
- 56.Sharples GJ, Chan SN, Mahdi AA, Whitby MC, Lloyd RG. Processing of intermediates in recombination and DNA repair: identification of a new endonuclease that specifically cleaves Holliday junctions. EMBO J. 1994;13:6133–42. doi: 10.1002/j.1460-2075.1994.tb06960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharples GJ. The X philes: structure-specific endonucleases that resolve Holliday junctions. Mol Microbiol. 2001;39:823–34. doi: 10.1046/j.1365-2958.2001.02284.x. [DOI] [PubMed] [Google Scholar]
- 58.Mizuno CM, Rodriguez-Valera F, Garcia-Heredia I, Martin-Cuadrado AB, Ghai R. Reconstruction of novel cyanobacterial siphovirus genomes from Mediterranean metagenomic fosmids. Appl Environ Microbiol. 2013;79:688–95. doi: 10.1128/AEM.02742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tunitskaya VL, Kochetkov SN. Structural-functional analysis of bacteriophage T7 RNA polymerase. Biochemistry (Mosc) 2002;67:1124–35. doi: 10.1023/A:1020911223250. [DOI] [PubMed] [Google Scholar]
- 60.Choi KH, McPartland J, Kaganman I, Bowman VD, Rothman-Denes LB, Rossmann MG. Insight into DNA and protein transport in double-stranded DNA viruses: the structure of bacteriophage N4. J Mol Biol. 2008;378:726–36. doi: 10.1016/j.jmb.2008.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ebright RH. RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J Mol Biol. 2000;304:687–98. doi: 10.1006/jmbi.2000.4309. [DOI] [PubMed] [Google Scholar]
- 62.Zakharova N, Hoffman PS, Berg DE, Severinov K. The largest subunits of RNA polymerase from gastric helicobacters are tethered. J Biol Chem. 1998;273:19371–4. doi: 10.1074/jbc.273.31.19371. [DOI] [PubMed] [Google Scholar]
- 63.Kazmierczak KM, Davydova EK, Mustaev AA, Rothman-Denes LB. The phage N4 virion RNA polymerase catalytic domain is related to single-subunit RNA polymerases. EMBO J. 2002;21:5815–23. doi: 10.1093/emboj/cdf584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foley S, Lucchini S, Zwahlen MC, Brüssow H. A short noncoding viral DNA element showing characteristics of a replication origin confers bacteriophage resistance to Streptococcus thermophilus. Virology. 1998;250:377–87. doi: 10.1006/viro.1998.9387. [DOI] [PubMed] [Google Scholar]
- 65.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 67.Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server issue):W244-8. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32(Web Server issue):W327-31. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 70.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 71.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–8. [Google Scholar]
- 72.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(Web Server issue):W609-12. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 76.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- 78.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.