

Abstract
Farnesides A and B (1, 2), linear sesquiterpenoids connected by ether links to a ribose dihydrouracil nucleoside, were isolated from a marine-derived Streptomyces sp., strain CNT-372, grown in saline liquid culture. The structures of the new compounds were assigned by comprehensive spectroscopic analysis primarily involving 1D and 2D NMR analysis and by comparison of spectroscopic data to the recently reported ribose nucleoside JBIR-68 (3). The farnesides are only the second example of this exceedingly rare class of microbial terpenoid nucleoside metabolites. Farneside A (1) was found to have modest antimalarial activity against the parasite Plasmodium falciparum.
As part of our continuing interest in exploring the chemical and biological diversity of marine-derived actinobacteria from Fiji,1–4 we cultured a Streptomyces sp., strain CNT-372, from a marine sediment sample collected near Nacula Island in the Yasawa Island chain. Following positive antimalarial testing data from the organic extract, we purified two metabolites, farnesides A and B (1, 2), that were of significant chemical interest due to their rare combination of a sesquiterpenoid ether with a ribose nucleoside. This paper reports the structures of new compounds from this rare biosynthetic class.
Farnesides A and B (1, 2) were isolated from the extract of strain CNT-372 that was identified by 16S rRNA gene sequence analysis as a member of the genus Streptomyces. The strain was cultured in a natural seawater-based medium for seven days, then extracted with Amberlite XAD-16 resin. The resin was collected by filtration and extracted with acetone. The acetone was removed and the remaining material extracted with EtOAc to generate the organic extract. Fractionation of the extract by silica column chromatography, followed by C-18 reversed-phase HPLC purification, led to the purification of farnesides A and B (1, 2) as light yellow amorphous solids.
Farneside A (1) showed [α]25D −15.0 and analyzed for the molecular formula C24H38N2O7 by HR-ESI-TOF-MS. Analysis of 1H, 13C, COSY, HSQC, and HMBC NMR data illustrated that 1 possessed a terpenoid chain and a ribofuranose ring (Table 1 and Supporting Information). A dihydrouracil ring was also recognized by comparison of NMR data to the literature data for the recently published monoterpenoid analog JBIR-68 (3).5 In farneside A, however, a higher homolog sesquiterpenoid chain possessing an epoxide at C-6 – C-7 was confidently defined by analysis of NMR data starting with the terminal quaternary carbon (C-11) at 131.6 ppm, which showed HMBC correlations to the olefinic gem dimethyl protons (H3-12 and H3-13) at δH 1.56, 1.62 ppm and to a methine proton at δH 5.06 ppm (H-10). The methine proton (H-10) showed a COSY correlation to two methylene protons at δH 1.99 ppm (H2-9), which in turn showed COSY correlations to another methylene pair at δH 1.52 and 1.31 ppm (H2-8 by HSQC). These NMR data established a terminal isoprene unit, which was then expanded to a sequential epoxide containing isoprene unit by the observation of HMBC correlations from the quaternary carbon at δC 60.7 ppm to the H2-8 methylene protons, to the protons of a singlet methyl at δH 1.17 ppm (H3-14) and to a methine proton at δH 2.64 ppm (H-6). The shifts of the quaternary carbon at δC 60.7 ppm and the methine carbon (C-6, obtained from an HSQC correlation) at δC 62.7 ppm indicated a direct oxygen linkage to these two carbons. The H-6 methine proton showed a COSY correlation to two methylene protons at δH 1.50 ppm (H2-5), which in turn showed another COSY correlation to the H2-4 methylene protons at δH 3.30 and 2.07 ppm. These NMR data established the second isoprene unit. The remaining isoprenoid fragment was constructed starting from the quaternary carbon at δC 139.3 ppm that showed HMBC correlations to the H2-4 methylene protons, to the protons of an olefinic methyl at δH 1.61 ppm (H3-15) and to an olefin proton at δH 5.28 ppm (H-2). The latter olefinic proton showed a COSY correlation to the methylene protons at δH 4.00 ppm (H2-1). The carbon signal at δC 67.7 ppm, assigned to C-1, is consistent with it bearing an ether linkage. The C-2 - C-3 double bond was assigned an E configuration on the basis of a NOESY correlation between H-2 and H2-4. This is also supported by the 13C chemical shifts of C-15 and C-4 as observed in related terpenoids (δC 16.9 and 36.2 ppm).6
Table 1.
1H and 13C NMR Spectroscopic Data for Farnesides A and B (1, 2) in DMSO-d6.
| Farneside A (1) |
Farneside A (2) |
|||||
|---|---|---|---|---|---|---|
| Position | δC, type | δH mul., (J in Hz) | HMBC | δC, type | δH, mul., (J in Hz) | HMBC |
| 1 | 67.7, CH2 | 4.00, m | 2,5”ab | 67.7, CH2 | 3.99, m | 2,5”ab,15 |
| 2 | 121.9, CH | 5.71, d (6) | 1,4ab | 121.1, CH | 5.30, t (7) | 1,4ab,15 |
| 3 | 139.3, C | 1,4ab,15 | 140.7, C | 1,4ab,5ab,15 | ||
| 4a 4b |
36.2, CH2 | 3.32, m 2.12, m |
2,5 | 37.3, CH2 | 2.25, ddd (14, 10, 4) 1.94, m |
2,5ab,6,15 |
| 5 | 27.3, CH2 | 1.58, m | 4ab,6 | 29.7, CH2 | 1.72, m 1.26, dddd |
4ab,6,6-OH |
| 6 | 62.7, CH | 2.69, t (6) | 4ab,5,14 | 76.7, CH | 3.12, dd (10, 7) | 4ab,5ab,6-OH,7-OH,8ab,14 |
| 7 | 60.7, C | 6,8b,9,14 | 73.7, C | 4.39, d (6) 3.99, s |
6,6-OH,7-OH, 8ab,14 | |
| 8a 8b |
39.0, CH2 | 1.58, m 1.35, ddd (14, 9, 8) |
6,9,14 | 39.0, CH2 | 1.46, dt (13, 5) 1.31, dt (13, 5) |
6,7-OH,14,9ab,10 |
| 9 | 24.2, CH2 | 2.04, ddd (8, 8, 7) | 8b | 22.3, CH2 | 1.97, m 2.04, m |
8ab,10 |
| 10 | 124.6, CH | 5.11 t (7) | 9,13 | 126.1, CH | 5.12, t (7) | 8ab,12,13 |
| 11 | 131.6, C | 9,12,13 | 130.6, C | 12,13 | ||
| 12 | 26.6, CH3 | 1.67, s | 26.2, CH3 | 1.67, s | 10,13 | |
| 13 | 18.2, CH3 | 1.60, s | 10 | 18.2, CH3 | 1.60, s | 10,12 |
| 14 | 17.0, CH3 | 1.22, s | 8b | 22.3, CH3 | 0.98, s | 6,7-OH,8ab |
| 15 | 16.9, CH3 | 1.61, s | 4ab | 17.1, CH3 | 1.65, s | 1,2,4ab |
| 2’ | 153.8, C | 1”,6 | 153.8, C | 1”, 6’ | ||
| 3’-NH | 10.29, s | 10.29, s | ||||
| 4’ | 171.7, C | 3’-NH, 5’,6’ | 171.1, C | 3’-NH, 5’,6’ | ||
| 5’ | 31.6, CH2 | 2.51, m | 3’-NH,6’ | 31.6, CH2 | 2.51, m | 3’-NH,6’ |
| 6’ | 36.3, CH2 | 3.35, m | 1”,5’ | 36.3, CH2 | 3.35, m | 1”,5’ |
| 1” | 88.2, CH | 5.71, d (6) | 2”,2”-OH,3” | 88.1, CH | 5.71, d (6) | 3’-NH,6’ |
| 2” | 71.0, CH | 3.99, ddd | 1”,2”-OH,3” | 71.0, CH | 3.99, ddd | 1”,2”-OH,4” |
| 2”-OH | 5.16, d (5) | 5.17, d (6) | ||||
| 3” | 71.5, CH | 3.84, ddd (4, 5, 4) | 2”,3”-OH,4” | 71.5, CH | 3.84, ddd (4, 5, 5) | 1”,4”,5”ab |
| 3”-OH | 5.06, d (4) | 5.07, d (5) | ||||
| 4” | 82.4, CH | 3.81, ddd (3, 4, 4) | 3”-OH,5”ab | 82.4, CH | 3.82, ddd (4, 4, 4) | 3”-OH,5”ab |
| 5”a | 70.7, CH2 | 3.46, dd (10, 5) | 1,3” | 70.7, CH2 | 3.45, dd (11, 5) | 1,3” |
| 5”b | 3.52, dd (10, 4) | 3.51, dd (11, 3) | ||||

The ribofuranose sugar and the dihydrouracil base were defined by analysis of 2D NMR data, (supporting information) and its comparison to NMR data from the recently published metabolite JBIR-68 (3), illustrating that the identical ribofuranose dihydrouracil nucleoside was present in farneside A (1).5
The relative configurations of the asymmetric centers of 1 were assigned by interpretation of NOE correlation data from a 2D NOESY NMR experiment. The stereocenters at the epoxide carbons (C-6 and C-7) of the sesquiterpenoid substituent were defined by the observation of strong NOE interactions between H2-5 and H3-14, and between H-6 and H2-8, demonstrating a C-6R* - C7R* relative configuration. This assignment was further supported by the 13C chemical shifts of C-6 and C-7 (δC 62.7 and 60.7) as seen in related terpenoid epoxides.6 The ribofuranose ring configuration was assigned independently, on the basis of strong NOE enhancements between H-1” and H-4”, and between H-2” to Ha-6’. This assignment is supported by the proton and carbon chemical shifts of the pentose unit that were identical to models of the β-ribofuranose ring.5 The absolute configuration for 1 has not been established.
Farneside B (2) was isolated as a glassy light yellow solid that showed [α]26D −0.4, and analyzed for the molecular formula C24H40N2O8 by HRMS. As in 1, the structure of 2 was defined by analysis of NMR spectroscopic data, including COSY, HSQC and HMBC data, as summarized in Table 1. Comparison of the overall NMR data for 2 with that from 1, revealed that farneside B differed in the degree of oxygenation on the side chain. The molecular formula for 2 indicated an addition of a water molecule to 1. The proton NMR spectrum showed the presence of two additional exchangeable protons signals (δH 3.99 and 4.39 ppm). In addition, the chemical shifts of C-6 (δC 76.7 ppm), H-6 (δC 3.12 ppm) and C-7 (δC 73.7 ppm) were shifted to lower field indicating that 2 was the corresponding diol derivative produced by a presumed hydrolytic ring opening of the epoxide in 1. In order to determine the relative configuration of the asymmetric centers C-6 and C-7, the diol was converted to the corresponding diacetonide 4 by treatment of 2 with 2,2-dimethoxypropane under standard conditions.6 2D ROESY NMR data from 4 allowed the relative configuration of the side chain acetonide to be assigned. The H-6 proton (δH 3.63) showed two NOE correlations to H3-14 (δH 1.13) and to the acetonide methyl H3-B (δH 1.24) demonstrating a C-6S* - C-7R* relative configuration. The absolute configuration of 2 was also not assigned. The transformation of epoxide 1 to the diol 2 is results in an inversion of configuration at C-6 as seen in the terpenoid epoxide and diol observed in the nitropyrrolins A and B.6 This reactivity is consistent with the role of epoxide hydrolases (EHs) that catalyze epoxide hydrolysis yielding the corresponding vicinal diols.7 The EHs have been isolated from wide variety of sources, including actinobacteria.8
The bioactivities of farnesides A and B were examined in a cytotoxicity assay against the human colon adenocarcinoma cell line, HCT-116, in an antibacterial screen against methicillin-resistant Staphylococcus aureus (MRSA), and in an antiviral screen against influenza type A virus. In addition, the farnesides 1 and 2 were screened in an antimalarial assay against Plasmodium falciparum. The farnesides were not appreciably cytotoxic and did not show significant antibacterial properties. While the analogous metabolite JBIR-68 (3) was reported to possess activity against influenza in plaque inhibition assays, examination of the farnesides showed they possessed negligible activity. In antimalarial testing, however, farneside A (1) demonstrated a modest antimalarial effect, showing IC50 = 69.3 3M against native Plasmodium falciparum.
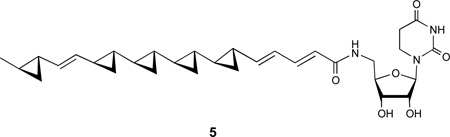
The farnesides are hybrid isoprenoids composed of a linear sesquiterpenoid chain coupled via an ether linkage with a dihydrouracil ribofuranose nucleoside. There are numerous natural products that contain nucleic acids, but the ribofuranose dihydrouracil composition is rare. An example of this class is the antifungal metabolite FR-900848 (5), which is composed of a fatty acid amide linkage at C-5” of the ribofuranose.9 The compound JBIR-68 (3) is the only compound known with a linear isoprenoid dihydrouracil ribofuranoside ether composition.5
Experimental Section
General Experimental Procedures
Optical rotations were measured using a Rudolph Research Autopol III polarimeter with a 10-cm cell. UV spectroscopic data were recorded in a Varian Cary UV-visible spectrophotometer with a path length of 1 cm. IR data were recorded on a Perkin-Elmer 1600 FT-IR spectrometer. 1H and 2D NMR spectroscopic data were recorded at 500 MHz in DMSO-d6 and CDCl3 solutions containing Me4Si as internal standard (at 300 and 500 MHz) on Varian Inova spectrometers. 13C NMR spectroscopic data were acquired at 125 MHz on a Varian Inova spectrometer. High-resolution ESI-TOF mass spectroscopic data were recorded on an Agilent 6530 Accurate – Mass QTOF LC-MS. Low-resolution LC-MS data were measured using a Hewlett-Packard series 1100 LC-MS system with a reversed-phase C18 (Phenomenex Luna, 4.6 mm × 100 mm, 5 mm) column at a flow rate of 1.0 mL/min.
Collection and Phylogenetic Analysis of Strain CNT-372
Strain CNT-372 was isolated from a sediment sample collected in July 2008 at a depth of 5 m off Nacula Island in the Yasawa Island chain, Fiji. The sample was air dried in a laminar flow hood and stamped onto medium A1 (10 g soluble starch, 4 g yeast extract, 2 g peptone, 18 g agar, 1 L natural seawater) using previously described methods.10 The strain has been identified as a Streptomyces sp. based on 16S rRNA gene sequence analysis (Genbank accession number KC699532).
Cultivation and Extraction
Streptomyces sp. (strain CNT-372) was cultured in 20 Fernbach flasks (2.8 L) each containing 1 L of a natural seawater-based medium (10 g starch, 4 g yeast extract, 2 g peptone, 1 g CaCO3, 40 mg Fe2(SO4)3 4H2O, 100 mg KBr) and shaken at 230 rpm at 27 ° C. After seven days of cultivation, sterilized XAD-16 resin (20 g/L) was added to adsorb the organic products, and the culture and resin were shaken at 215 rpm for 2 h. The resin was filtered through cheesecloth, washed with deionized water, and eluted with acetone. The acetone was removed under reduced pressure and the resulting aqueous layer extracted with EtOAc (2 × 300 mL). The EtOAc soluble fraction was dried under vacuum to yield 1.3 g of organic extract from a 20 L culture.
Isolation of Farnesides A and B (1, 2)
The organic extract (1.3 g) from strain CNT-372 was subjected to silica chromatography using sequential mixtures of CH2Cl2 and MeOH as eluents (elution order: 5, 7, 10, 20% MeOH in CH2Cl2, and 100% MeOH). Guided by LC-MS analyses, the 7% and 10% MeOH fractions containing 1 (436.7 mg), and the 20% and 50% MeOH fractions containing 2 were combined (186.1 mg). Each fraction was subsequently purified by C-18 reversed-phase semipreparative HPLC using a Phenomenex Luna C18(2) column (250 × 10 mm, 10 µm) with a gradient of 20 to 80% CH3CN in H2O for 20 min, flow rate of 3 mL / min, to yield 26.3 mg and 7.1 mg of farnesides A and B (1, 2), respectively.
Farneside A (1)
glassy yellow solid; [α]25D −15 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 210 nm (3.90) nm; IR (CHCl3) νmax 3390, 3243, 2947, 2919, 2850, 1689, 1484, 1442, 1377, 1207, 1118, 1018 cm−1; see Table 1 for 1H NMR and 13C NMR data; ESI-TOF-MS m/z 489.2589 [M+Na], (calcd for C24H38N2O7Na, 489.2571, Δ 4.18 ppm).
Farneside B (2)
glassy yellow solid; [α]26D −0.4 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 210 nm (3.90) nm; IR (CHCl3) νmax 3390, 3243, 2947, 2919, 2850, 1689, 1484, 1442, 1377, 1207, 1118, 1018 cm−1; see Table 1 for 1H NMR and 13C NMR data; ESI-TOF-MS m/z 507.2678 [M+Na], (calcd for C24H40N2O8Na, 507.2677, Δ 0.61 ppm).
Preparation of Acetonide 4
Farneside B (2, 2.0 mg) was dissolved in 2,2-dimethoxypropane (1 mL) and methanol (0.2 mL), and pyridinium-p-toluenesulfonate (2 mg) was added. The reaction was allowed to stir for 2 d at 0 °C and 8 h at rt, then the reaction mixture was purified by C-18 RP HPLC (isocratic, 70% CH3CN in H2O for 10 min, followed by a gradient from 70% to % 95 CH3CN in H2O for 10 min, followed by 5 min of initial conditions) to provided compound 4 (1.1 mg, ret. time 20.8 min). UV (MeOH) λmax (log ε) 210 nm (3.90) nm; ESI-TOF-MS m/z 587.3299 [M+Na], (calcd for C30H48N2O8Na, 564.3408, Y 0.55 ppm); 1H NMR (500 MHz, DMSO-d6): δ 1.13 (3H, s, H-14), 1.24 (3H, s, H-B), 1.27 (3H, s, H-D), 1.29 (3H, s, H-A), 1.46 (3H, s, H-C), 1.55 (3H, s, H-12), 1.62 (6H, s, H-13, H-15), 3.48 (2H, m, H-5”), 3.63 (1H, dd, J = 9.5, 3.7 Hz, H-6), 3.94 (1H, q, J = 4.2 Hz, H-4”), 3.96 (2H, d, J = 6.6 Hz, H-1), 4.57 (1H, dd, J = 6.8, 4.6 Hz, H-3”), 4.79 (1H, dd, J = 7.1, 2.9 Hz, H-2”), 5.11 (1H, t, J = 7.3 Hz, H-10), 5.28 (1H, t, J = 6.7 Hz, H-2), 5.74 (1H, dd, J = 3.2, 0.5 Hz, H-1”), 10.32 (1H, s, H-1’).
Anti-Malarial Bioassay
The anti-malarial activity of the farnesides A and B (1, 2) were evaluated using a SYBR Green based parasite proliferation assay.11,12 Plasmodium falciparum parasites (3D7 strain, MRA-102, MR4/ATCC, Manassas, VA) were cultured in human O+ erythrocytes as previously described.13 The farnesides were diluted in complete medium and 40 µL transferred to 96-well assay plates (Costar #3904). To this 80 µL of complete media with 3D7 infected erythrocytes were dispensed in order to obtain a 2.5% hematocrit and 0.5% parasitemia in the assay. Uninfected erythrocytes were dispensed into the background wells at the same final hematocrit. Plates were incubated for 72 h in a low oxygen environment (96% N2, 3% CO2, 1% O2) in a modular incubation chamber (Billups-Rothenberg). The plates were sealed and placed in a −80 ° C freezer overnight then thawed and 120 µL of lysis buffer (20 mM Tris-HCl, pH 7.5, 5mM EDTA, 0.08% Triton X-100, (Promega), 0.008% saponin (Acros)) with 0.2 µL/mL Sybr Green I (Invitrogen) was dispensed into each well and incubated at 37 ° C in the dark for 6 h to achieve maximum signal to noise ratio. The plates were read with a Molecular Devices SpectraMAX Gemini EM at ex: 495 nm, em: 525 nm with 515 nm cut-off.
Supplementary Material
Acknowledgment
This work was supported by the U.S. National Institutes of Health’s International Cooperative Biodiversity Groups program (Grant No. U01 TW007401). The malaria parasite, Plasmodium falciparum 3D7, MRA-102, deposited by D. J. Carucci, was obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH. We thank James Noah, from the Southern Research Institute and Amy E. Krafft from NIAID/NIH for providing the influenza bioassay.
Footnotes
Supporting Information Available:
Spectroscopic data, 1D and 2D NMR, HRMS and IR spectra for 1 and 2, are available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Sun P, Maloney KN, Nam SJ, Haste NM, Raju R, Aalbersberg W, Jensen PR, Nizet V, Hensler ME, Fenical W. Bioorg. Med. Chem. 2011;19:6–11. doi: 10.1016/j.bmc.2011.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nam SJ, Gaudencio SP, Kauffman CA, Jensen PR, Kondratyuk TP, Marler LE, Pezzuto JM, Fenical W. J. Nat. Prod. 2010;73:1080–1086. doi: 10.1021/np100087c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haste NM, Perera VR, Maloney KN, Tran DN, Jensen PR, Fenical W, Nizet V, Hensler ME. J. Antibiot. 2010;63:219–224. doi: 10.1038/ja.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freel KC, Edlund A, Jensen PR. Envir. Microbiol. 2012;14:480–493. doi: 10.1111/j.1462-2920.2011.02641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takagi M, Motohashi K, Nagai A, Izumikawa M, Tanaka M, Fuse S, Doi T, Iwase K, Kawaguchi A, Nagata K, Takahashi T, Shin-ya K. Org. Lett. 2010;12:4664–4666. doi: 10.1021/ol102007d. [DOI] [PubMed] [Google Scholar]
- 6.Kwon C, Espindola A-P, Park J-S, Prieto-Davo A, Rose M, Jensen P, Fenical W. J. Nat. Prod. 2010;73:2047–2052. doi: 10.1021/np1006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang JH, Jung HW, Sung GK, Young OH, Sang JK. J. Microbiol. Biotechnol. 2008;18:1445–1452. [PubMed] [Google Scholar]
- 8.Steinreiber A, Farber K. Curr. Opin. Biotechnol. 2001;12:552–558. doi: 10.1016/s0958-1669(01)00262-2. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida M, Ezaki M, Hashmoto M, Yamashita M, Shigematsu N, Okuhara M, Kohsaka M, Horikoshi K. J. Antibiot. 1990;28:748–754. doi: 10.7164/antibiotics.43.748. [DOI] [PubMed] [Google Scholar]
- 10.Jensen PR, Gontang E, Mafnas C, Mincer TJ, Fenical W. Environ. Microbiol. 2005;7:1039–1048. doi: 10.1111/j.1462-2920.2005.00785.x. [DOI] [PubMed] [Google Scholar]
- 11.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar A, Davidson E, Roepe P. Antimicrob. Agents Chemother. 2004;48:1807–1810. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trager W, Jensen JB. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.