

Abstract
Objective:
To estimate the incidence of and to characterize cognitive and imaging findings associated with incident amyloid PET positivity.
Methods:
Cognitively normal (CN) participants in the Mayo Clinic Study of Aging who had 2 or more serial imaging assessments, which included amyloid PET, FDG-PET, and MRI at each time point, were eligible for analysis (n = 207). Twelve subjects with Alzheimer disease dementia were included for comparison.
Results:
Of the 123 CN participants who were amyloid-negative at baseline, 26 met criteria for incident amyloid PET positivity. Compared to the 69 subjects who remained stable amyloid-negative, on average these 26 did not differ on any imaging, demographic, or cognitive variables except amyloid PET (by definition) and task-free functional connectivity, which at baseline was greater in the incident amyloid-positive group. Eleven of the 26 incident amyloid-positive subjects had abnormal hippocampal volume, FDG-PET, or both at baseline.
Conclusions:
The incidence of amyloid PET positivity is approximately 13% per year among CN participants over age 70 sampled from a population-based cohort. In 15/26 (58%), incident amyloid positivity occurred prior to abnormalities in FDG-PET and hippocampal volume. However, 11/26 (42%) incident amyloid-positive subjects had evidence of neurodegeneration prior to incident amyloid positivity. These 11 could be subjects with combinations of preexisting non-Alzheimer pathophysiologies and tau-mediated neurodegeneration who newly entered the amyloid pathway. Our findings suggest that both “amyloid-first” and “neurodegeneration-first” biomarker profile pathways to preclinical AD exist.
Amyloid positivity is associated with higher rates of brain atrophy in cognitively normal (CN) subjects and subjects with mild cognitive impairment (MCI).1,2 CN subjects3–5 and subjects with MCI6,7 who are amyloid-positive experience greater cognitive decline than amyloid-negative individuals. Amyloid positivity must therefore be recognized as a pathologic state.
Like any quantitative biomarker, amyloid PET values exist on a continuous scale. However, defining incident amyloid positivity requires that a precise normal/abnormal threshold be adopted. Categorizing individual subjects as amyloid-positive or -negative is required to implement new diagnostic criteria for preclinical Alzheimer disease (AD) and to integrate amyloid biomarkers into the new diagnostic criteria for MCI and AD dementia.8,9 A precise normal/abnormal threshold is also required to define eligibility for some antiamyloid therapeutic trials.10
A popular model of the temporal evolution of AD biomarkers11,12 proposes that amyloid biomarkers are the first to become abnormal in the pathophysiologic cascade and do so while subjects are still considered CN via neuropsychological and clinical criteria. Thus, characterization of incident amyloid PET positivity should focus on subjects who are CN at baseline. The state of amyloid positivity is clinically important as this defines preclinical AD.9 Therefore, identifying characteristics associated with the transition from amyloid-negative to -positive is likewise important. Our objectives were to estimate the incidence of and to characterize cognitive and imaging findings associated with incident amyloid PET positivity in a cohort of elderly CN subjects drawn from a population-based sample.
METHODS
Participants.
CN participants were drawn from the Mayo Clinic Study of Aging (MCSA). The MCSA is a longitudinal population-based observational study of cognitive aging that was established in Olmsted County, Minnesota, in 2004, initially enrolling subjects age 70–90 years. Continuous replenishment using population-based recruiting results in a continuously active cohort of about 2,000.
Clinical diagnoses of CN or MCI for MCSA participants are rendered through a consensus process that uses information from a mental status examination, a Clinical Dementia Rating, and a psychometric battery containing 9 well-established instruments. A global cognitive summary z score, formed from individual test z scores,13 was used to assess cognitive function in the CN subjects for the present analysis.
To be eligible for inclusion in the current study, subjects must have been classified as CN at the time of baseline imaging and must have had 2 or more multimodal, serial imaging assessments, defined as amyloid PET, FDG-PET, and MRI, all obtained at the same time points. A total of 207 subjects met these criteria. The imaging studies were obtained over the period June 2006–October 2012. The median (interquartile range) for imaging follow-up was 1.3 (1.3, 1.5) years, and for clinical follow-up 2.5 (1.3, 2.7) years.
For reference purposes, subjects with AD dementia were drawn from the Mayo Alzheimer's Disease Research Center (n = 11) or incident AD cases in the MCSA (n = 1). All subjects with AD in this analysis were required to be aged 70 years or older to match the age range of the MCSA, and have 2 or more complete multimodal imaging assessments.
Standard protocol approvals, registrations, and patient consents.
These studies were approved by the Mayo Clinic and Olmsted Medical Center institutional review boards. Signed informed consent was obtained from all participants or their surrogates.
Amyloid PET methods.
PET images were acquired using a PET/CT scanner. The 11C Pittsburgh compound B–PET scan, consisting of 4 5-minute dynamic frames, was acquired from 40 to 60 minutes after injection. Image analysis was done with our in-house fully automated pipeline, which uses MRI to guide PET region of interest (ROI) placement.14 An amyloid PET standardized uptake value ratio (SUVR) was formed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus ROIs and dividing this meta ROI by the median uptake over voxels in the cerebellar gray matter ROI of the atlas. Using these methods, we previously found that the relative measurement error (analogous to a coefficient of variation) for serial amyloid PET is about 3%,15 indicating adequate longitudinal measurement precision.
We defined the normal/abnormal threshold for a positive amyloid PET scan to be the 5th percentile (95% sensitivity) in a group of 42 clinically diagnosed subjects with AD described in a prior publication.13 This cutpoint corresponds to an SUVR of 1.4. To qualify for incident amyloid PET positivity, subjects must have changed from SUVR <1.4 to >1.4 over their series of scans and have increased in SUVR by more than 0.04 SUVR (which represents a change greater than noise15).
FDG-PET methods.
FDG-PET images were obtained on the same day 1 hour after the amyloid PET scan. FDG-PET scans were analyzed with the pipeline described above.14 The angular gyrus, posterior cingulate, and inferior temporal cortical ROIs defined an “Alzheimer signature” meta-ROI,16 which was normalized to the pons and cerebellar vermis.
Structural MRI methods.
All subjects underwent MRI scanning at 3T with a standardized protocol that included a 3D magnetization-prepared rapid gradient echo sequence. Hippocampal volume was measured with FreeSurfer. Each subject's raw hippocampal volume was adjusted by total intracranial volume.
Task-free functional MRI methods.
Task-free functional MRI (TF-fMRI) preprocessing methods have been described previously.17 Group independent component analysis of fMRI Toolbox (GIFT) version 2.0d18 was used to extract independent components for each subject. We used the infomax algorithm with ICASSO and the dimensionality for each subject was chosen using minimum description length information theoretic criteria. The posterior default mode network (pDMN) was algorithmically identified and the median z score from the pDMN ROI from a high dimensional functional atlas17 was used as the TF-fMRI metric.
Statistical methods.
We used Poisson regression to estimate the incidence rate for amyloid positivity modeling incident amyloid PET events as an outcome that was scaled to the length of follow-up using an offset term of log (time). Time was calculated as the number of years from the baseline imaging visit to the first imaging visit with incident amyloid PET or to the last follow-up imaging visit in nonincident cases. We compared pairwise group differences between the incident amyloid PET-positive CN group and all other groups in demographics, baseline and annual change in imaging, and baseline and annual change in cognitive performance using Wilcoxon rank-sum tests for continuous variables and χ2 tests for categorical variables.
RESULTS
Incidence of amyloid PET positivity.
Of the 207 CN participants in the MCSA who were eligible for analysis, 123 (59%) were amyloid PET-negative at baseline and thus represented the group at risk for incident amyloid PET positivity (figure 1). Twenty-six subjects with 198 person-years of imaging follow-up met criterion for incident amyloid PET positivity for an annual incidence of 13%. Sixty-nine CN subjects remained amyloid-negative over their entire series of scans and did not increase by more than 0.04 SUVR (labeled stable amyloid-negative). Twenty-five subjects were amyloid-negative at baseline and follow-up but over time their SUVR value increased by more than 0.04 (labeled nonstable amyloid-negative). Eighty-four subjects were amyloid-positive at baseline (labeled prevalent amyloid-positive). Three subjects were amyloid-negative at baseline and over time changed to SUVR values greater than 1.4, but the change was less than the noise value of 0.04 so they did not meet the incident amyloid PET definition. These 3 were not included in the analysis. Spaghetti plots of SUVR vs time are presented in figure 2 for the 5 groups of subjects we focus on: incident amyloid-positive, stable amyloid-negative, nonstable amyloid-negative, prevalent amyloid-positive, and AD.
Figure 1. Flowchart.
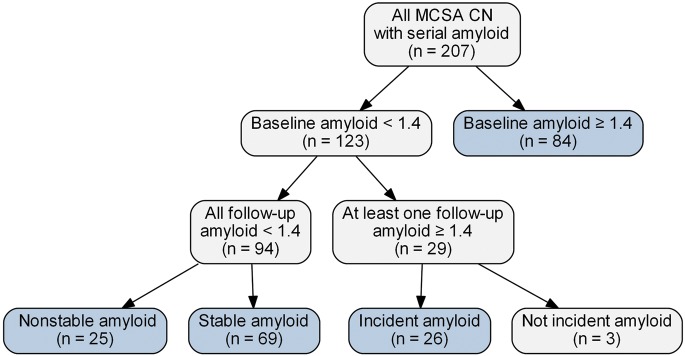
The cognitively normal (CN) groups in blue are the focus of this article. MCSA = Mayo Clinic Study of Aging.
Figure 2. Trajectory plots of amyloid by age.
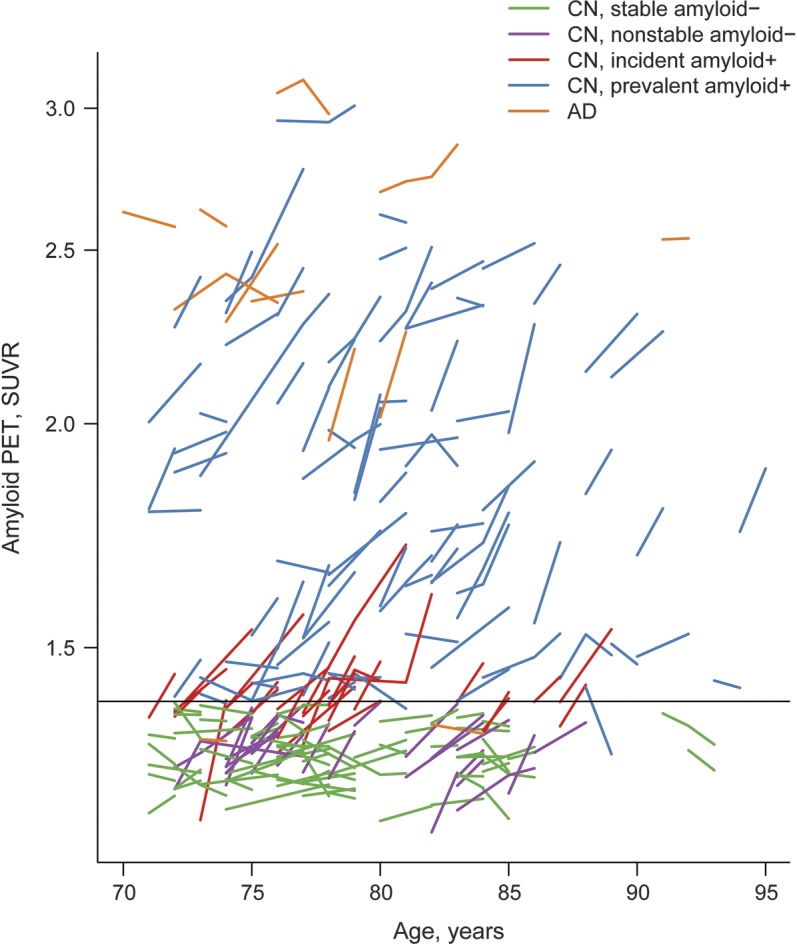
Stable amyloid PET-negative cognitively normal (CN) participants are represented with green lines, nonstable amyloid-negative CN with purple lines, incident amyloid-positive CN with red lines, prevalent amyloid-positive CN with blue lines, and Alzheimer disease (AD) with orange lines. Of the 84 amyloid-positive participants at baseline, 2 became amyloid-negative (<1.4) and decreased by >0.04 over time and thus could be considered to have reverted from amyloid-positive to -negative. We attribute this infrequent phenomenon (2/84 = 2%) to measurement noise in subjects who lie at the boundary of amyloid positivity. SUVR = standardized uptake value ratio.
Characteristics of incident amyloid-positive CN subjects.
Compared to the stable amyloid-negative CN group, the incident amyloid-positive CN group had greater baseline amyloid SUVR values (median 1.38 vs 1.30, p < 0.001), greater baseline pDMN connectivity (median 1.52 vs 1.35, p = 0.03), and (by definition) greater longitudinal increase in amyloid (median 0.06 vs 0, p < 0.001). The 2 groups did not differ on any other baseline or longitudinal imaging measures, demographic characteristics, or baseline or longitudinal change in cognition (table and figures 3 and 4).
Table.
Characteristics of participantsa
Figure 3. Boxplot of baseline imaging and cognitive performance by diagnosis and amyloid group.

APOE ε4-negative participants are represented with yellow circles and APOE ε4-positive participants are represented with blue plus signs. (A) Boxplot of baseline amyloid by diagnosis and amyloid group. The horizontal line represents a cutpoint of 1.40, which was used in the definition of incident amyloid. This cutpoint has a sensitivity of approximately 95% in 42 subjects with clinically diagnosed Alzheimer disease (AD). (B) Boxplot of baseline FDG-PET by diagnosis and amyloid group. The horizontal line represents a cutpoint of 1.38, which corresponds to a sensitivity of approximately 95% in 42 subjects with clinically diagnosed AD. (C) Boxplot of baseline adjusted hippocampal volume by diagnosis and amyloid group. The horizontal line represents a cutpoint of −0.34, which corresponds to a sensitivity of approximately 95% in 42 subjects with clinically diagnosed AD. (D) Boxplot of baseline posterior default mode network (pDMN) connectivity by diagnosis and amyloid group. (E) Boxplot of baseline global z score by amyloid group among cognitively normal (CN) participants. SUVR = standardized uptake value ratio.
Figure 4. Boxplot of annual change in imaging and cognitive performance by diagnosis and amyloid group.

APOE ε4-negative participants are represented with yellow circles and APOE ε4-positive participants are represented with blue plus signs. (A) Boxplot of annual change in amyloid PET by diagnosis and amyloid group. (B) Boxplot of annual change in FDG-PET by diagnosis and amyloid group. (C) Boxplot of annual % change in hippocampal volume by diagnosis and amyloid group. (D) Boxplot of annual change in posterior default mode network (pDMN) connectivity by diagnosis and amyloid group. (E) Boxplot of annual change in global z score by amyloid group among cognitively normal (CN) participants. AD = Alzheimer disease; SUVR = standardized uptake value ratio.
Compared to the nonstable amyloid-negative CN group, the incident amyloid-positive CN group had greater baseline amyloid SUVR (median 1.38 vs 1.29, p < 0.001) and marginally lower hippocampal volumes (median −0.05 vs 0.40, p = 0.06), greater pDMN connectivity (median 1.52 vs 1.28, p = 0.09), and greater longitudinal increase in amyloid (median 0.06 vs 0.04, p = 0.05).
Compared to the prevalent amyloid-positive CN group, the incident amyloid PET-positive CN group were less often APOE ε4 carriers (19% vs 45%, p = 0.02) and had marginally younger age (median 77 vs 80, p = 0.07). They also had greater baseline FDG uptake (median 1.47 vs 1.39, p = 0.003) and (by definition) lower baseline amyloid SUVR (median 1.38 vs 1.74, p < 0.001).
Compared to subjects with AD, incident amyloid-positive CN subjects were less often APOE ε4 carriers (19% vs 75%, p < 0.001) and had lower baseline amyloid SUVR (median 1.38 vs 2.33, p < 0.001), greater baseline pDMN connectivity (median 1.52 vs 1.04, p = 0.001), higher baseline FDG uptake (median 1.47 vs 1.12, p < 0.001), and higher adjusted hippocampal volumes (median −0.05 vs −1.35, p < 0.001). They also had a greater annual increase in amyloid (median 0.06 vs 0, p = 0.03), less annual decrease in FDG uptake (median −0.01 vs −0.08, p = 0.002), and less annual hippocampal volume loss (median −0.8 vs −4.0, p < 0.001).
A cutpoint of 1.40 for amyloid PET had a sensitivity of approximately 95% in a group of 42 clinically diagnosed subjects with AD in a prior publication.13 The same sensitivity corresponded to a cutpoint of 1.38 for FDG and −0.34 for adjusted hippocampal volume in this same group of subjects with AD.13 Using these thresholds, 11 of our 26 incident amyloid PET-positive subjects had abnormal hippocampal volume (n = 4), FDG (n = 2), or both (n = 5) at baseline (figure 3 and figure e-1 on the Neurology® Web site at www.neurology.org). These 11 therefore had abnormal neurodegenerative biomarkers (FDG-PET or hippocampal volume) with normal amyloid PET at baseline, but later become amyloid-positive. Nine of these 11 (82%) subjects were not APOE ε4 carriers. We did not have TF-fMRI data in all of the 42 subjects with AD13 and therefore were not able to generate cutpoints for TF-fMRI analogous to those used for the other 3 imaging modalities.
DISCUSSION
Using our criteria, the annual incidence of amyloid PET positivity was approximately 13% among CN participants over age 70 drawn from a population-based sample. A different group reported an annual amyloid PET positivity incidence of 3.1% based on following 125 subjects for a mean of 2.6 years.19 The difference from our observed rate of 13% could be explained by the younger age of the sample in reference 19 (mean 66 years vs 78 years in our sample).
The unexpectedly low APOE ε4 frequency of 19% in our incident amyloid-positive group may be expected from the age range in our study. Subjects had to remain amyloid-negative until at least age 70, which was the lower limit in the MCSA, to be at risk for incident amyloid positivity.20 A higher prevalence of APOE ε4 would be expected in incident amyloid-positive subjects in a younger sample.21
While the threshold of 1.4 SUVR is not a universal standard, amyloid positivity cannot be defined without adopting a precise normal/abnormal threshold. A threshold of SUVR 1.4 has been used by others in analyses of CN subjects.1,22 Slightly higher thresholds such as SUVR 1.5 are often used to define amyloid PET positivity in the context of identifying the etiology of a dementia.14 However, when the objective is to ascertain the earliest evidence of abnormality in CN subjects, a lower threshold seems justified.23 Here we used the SUVR value (1.4) that corresponds to the fifth percentile of subjects with clinically diagnosed AD at our institution.13 This cutpoint satisfies the criterion of leniency in that false-negatives among subjects with AD are controlled at a rate of 5%.
We were not able to generate cutpoints for TF-fMRI analogous to those used for the other 3 imaging modalities. Consequently, the TF-fMRI results are discussed separately here. On average, baseline pDMN connectivity and amyloid SUVR were both greater in the incident amyloid-positive than the stable amyloid-negative group. Baseline amyloid and (marginally) pDMN connectivity were greater in the incident amyloid-positive than the nonstable amyloid-negative group. Baseline amyloid was less while pDMN connectivity was greater in incident amyloid-positive subjects than in subjects with AD. This is consistent with prior studies indicating increased connectivity in CN subjects with higher amyloid deposition24,25 followed later by a decrease in connectivity in subjects with dementia.26 Interestingly, incident amyloid-positive subjects also had greater FDG uptake than prevalent amyloid PET-positive subjects, which could be spurious or could represent a cerebral glucose metabolism analogue of early compensatory TF-fMRI hyperconnectivity. Because both amyloid SUVR and pDMN connectivity differed between incident amyloid-positive, nonstable amyloid-negative, and stable negative CN subjects at baseline, our data do not imply that one of these 2 imaging modalities becomes abnormal before the other. Neither FDG-PET nor hippocampal volume differed on average between the incident amyloid-positive and the stable amyloid-negative CN groups, however, which suggests that in at least some individuals TF-fMRI becomes abnormal prior to FDG-PET or hippocampal volume. Rates of change in pDMN connectivity, however, did not differ between the incident amyloid-positive amyloid group and any other, including AD. The most logical explanation is that, in its current form, TF-fMRI is a noisy measure compared to the other 3 imaging modalities examined.
In terms of identifying defining features of incident amyloid positivity, this group did not differ notably on demographic, FDG-PET, hippocampal volume, or cognitive features from the 2 groups that remained amyloid-negative (stable and nonstable) throughout. There were some expected exceptions to this generality. Rates of amyloid accumulation were greater in incident amyloid-positive than in the stable amyloid-negative group by definition. Baseline amyloid levels were also greater, which is logical as slightly elevated (but subthreshold) amyloid at baseline might be expected in subjects who later became amyloid-positive. The incident amyloid-positive group also had marginally lower hippocampal volumes than the nonstable amyloid-negative group. However, the most notable feature of incident amyloid positivity vs those who remained amyloid-negative was the lack of obvious differentiating features other than baseline and change in amyloid itself.
Fifteen of our 26 incident amyloid-positive subjects (58%) had normal hippocampal volume, FDG-PET, and amyloid (by definition) at baseline, and later became amyloid-positive. In 2010, several of us proposed a model11 of the temporal evolution of AD biomarkers in which amyloid biomarkers become abnormal first, followed by biomarkers of neurodegeneration, followed by clinical symptoms. These 15 fit this amyloid-first model of biomarker evolution. This 2010 biomarker model11 would order the groups of subjects studied here from earliest to latest in the disease process as follows: nonstable amyloid-negative, incident amyloid-positive CN, prevalent amyloid-positive CN, then AD. Stable amyloid-negative subjects have not entered the AD pathway and serve as a reference group at the normal end of the pathway as subjects with AD do at the more abnormal end of the pathway. We found that rates of amyloid accumulation were greater in incident amyloid-positive CN than in AD, which may seem counterintuitive. However, we and others15,27–29 have recently shown that rates of amyloid accumulation slow at high baseline levels. As predicted by our model,11 our current findings support the concept that amyloid is an early changing biomarker whose rate of change slows as subjects develop dementia. In contrast, FDG and structural MRI continue to change at high rates in early dementia.2,30
While 15 of our 26 incident amyloid-positive subjects follow an amyloid-first biomarker profile pathway, 11/26 (42%) had an abnormal neurodegenerative biomarker study at baseline. This phenomenon may be explained in at least 3 ways. First, this could just be subject classification error at the boundaries of biomarker thresholds, although this seems unlikely to be the explanation in all 11 of these subjects. Second, this could be downstream neurodegeneration in response to β-amyloid dysmetabolism prior to amyloid plaque formation. Third, these 11 subjects may have had pathophysiologies such as Lewy body disease, vascular disease, TDP43, argyrophilic grain disease, or hippocampal sclerosis present at baseline and then independently later developed fibrillar amyloid deposits. In prior publications,13,31,32 we have labeled CN subjects with normal amyloid PET studies but abnormal neurodegenerative biomarker studies suspected non-AD pathophysiology (SNAP). We include medial temporal lobe tauopathy in the definition of SNAP.13,31,32 While entorhinal and hippocampal neurofibrillary tangles are commonly found at autopsy in the absence of amyloid plaques in middle-aged and older CN subjects,33–35 neurodegeneration in most elderly subjects is most commonly due to a mixture of pathologies rather than a single isolated pathophysiologic process.36
Frequent abnormal neurodegenerative biomarker studies at baseline were not limited to our incident amyloid-positive group but rather were pervasive among all elderly CN groups (table). According to Braak and Braak,33 by the late 70s, 97% of individuals have some neurofibrillary tangles at autopsy, most often confined to the medial temporal lobe. Therefore, some portion of the imaging evidence of neurodegeneration seen in most of our 11 incident amyloid-positive subjects with abnormal neurodegeneration biomarkers at baseline must be attributable to tauopathy. Whether or not or not this represents a pre-amyloid AD biomarker abnormality is a matter of debate. However, this debate gets at the heart of the definition of late-onset AD (not autosomal dominant AD or perhaps AD in APOE ε4 homozygotes) and its pathogenesis. Tau-related neurodegeneration before amyloid is not consistent with the amyloid cascade hypothesis, which proposes a series of causal molecular events with amyloid dysmetabolism preceding tauopathy.37 Alternative hypotheses propose that tauopathy and β-amyloid arise independently,38,39 or that amyloid arises independently on a background of preexisting medial temporal tauopathy.12,34,40 Our current data cannot resolve this controversy about disease pathogenesis. However, our data do show that both amyloid-first and neurodegeneration-first biomarker profiles characterize incident amyloid positivity. Amyloid positivity defines preclinical AD9; therefore, both amyloid-first and neurodegeneration-first biomarker profile pathways to preclinical AD exist.
Supplementary Material
GLOSSARY
- AD
Alzheimer disease
- CN
cognitively normal
- MCI
mild cognitive impairment
- MCSA
Mayo Clinic Study of Aging
- pDMN
posterior default mode network
- TF-fMRI
task-free functional MRI
- ROI
region of interest
- SNAP
suspected non-AD pathophysiology
- SUVR
standardized uptake value ratio
Footnotes
Editorial, page 1728
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Clifford Jack: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, statistical analysis, study supervision, obtaining funding. Heather J. Wiste: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, statistical analysis. Stephen D. Weigand: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, statistical analysis. David S. Knopman: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval. Val Lowe: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, acquisition of data, study supervision, obtaining funding. Prashanthi Vemuri: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval. Michelle M. Mielke: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. David T. Jones: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, statistical analysis. Matthew L. Senjem: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, contribution of vital reagents/tools/patients, statistical analysis. Jeffrey L. Gunter: analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data, statistical analysis. Brian E. Gregg: analysis or interpretation of data, accepts responsibility for conduct of research and final approval, acquisition of data. Vernon S. Pankratz: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and final approval, statistical analysis. Ronald C. Petersen: drafting/revising the manuscript, accepts responsibility for conduct of research and final approval, obtaining funding.
STUDY FUNDING
Supported by The Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation, USA, and the Robert H. and Clarice Smith Alzheimer's Disease Research Program of the Mayo Foundation, USA; and by the NIH/National Institute on Aging (R01 AG011378, RO1 AG041851, U01 AG006786, P50 AG16574, C06 RR018898).
DISCLOSURE
R. Petersen reports receiving consulting fees from Elan Pharmaceuticals and GE Healthcare, receiving royalties from Oxford University Press, and serving as chair of data monitoring committees for Pfizer and Janssen Alzheimer Immunotherapy; and receives research support from the NIH/NIA. C. Jack serves as a consultant for Siemens and receives research funding from the NIH (R01-AG011378, RO1-AG037551, U01-HL096917, U01-AG032438, U01-AG024904) and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation Family. H. Wiste and S. Weigand report no disclosures. D. Knopman serves as Deputy Editor for Neurology®, serves on a Data Safety Monitoring Board for Lilly Pharmaceuticals, is an investigator in clinical trials sponsored by Janssen Pharmaceuticals, and receives research support from the NIH. V. Lowe serves on scientific advisory boards for Bayer Schering Pharma and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, the NIH (NIA, NCI), the MN Partnership for Biotechnology and Medical Genomics, and the Leukemia & Lymphoma Society. P. Vemuri, M. Mielke, D. Jones, M. Senjem, J. Gunter, and B. Gregg report no disclosures. V. Pankratz is funded by the NIH (R01AG040042, U01AG06786, Mayo Clinic Alzheimer's Disease Research Center/Core C P50AG16574/Core C, and R01AG32990). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Chetelat G, Villemagne VL, Villain N, et al. Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition. Neurology 2012;78:477–484 [DOI] [PubMed] [Google Scholar]
- 2.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PiB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol 2011;69:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 2009;66:1469–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, Johnson KA, Doraiswamy PM, et al. Amyloid deposition detected with florbetapir F 18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging 2013;34:822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR, Jr, Wiste HJ, Vemuri P, et al. Brain beta-amyloid measure and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain 2010;133:3336–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordberg A, Carter SF, Rinne J, et al. A European multicentre PET study of fibrillar amyloid in Alzheimer's disease. Eur J Nucl Med Mol Imaging 2013;40:104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKhann G. Revision of the NINCDS-ADRDA diagnostic criteria. Presented at Alzheimer's Association International Conference on Alzheimer's Disease; July 13, 2010; Honolulu, Hawaii
- 9.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sperling RA, Jack CR, Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med 2011;3:111cm33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jack CR, Jr, Knopman DS, Weigand SD, et al. An operational approach to NIA-AA criteria for preclinical Alzheimer's disease. Ann Neurol 2012;71:765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Jr, Wiste HJ, Lesnick TG, et al. Brain beta-amyloid load approaches a plateau. Neurology 2013;80:890–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010;75:230–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DT, Vemuri P, Murphy MC, et al. Non-stationarity in the “resting brain's” modular architecture. PLoS One 2012;7:e39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calhoun VD, Adali T, Pearlson GD, Pekar JJ. A method for making group inferences from functional MRI data using independent component analysis. Hum Brain Mapp 2001;14:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vlassenko AG, Mintun MA, Xiong C, et al. Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C]Pittsburgh compound B data. Ann Neurol 2011;70:857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleisher AS, Chen K, Liu X, et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging 2013;34:1–12 [DOI] [PubMed] [Google Scholar]
- 21.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283 [DOI] [PubMed] [Google Scholar]
- 23.Mormino EC, Brandel MG, Madison CM, et al. Not quite PiB-positive, not quite PiB-negative: slight PiB elevations in elderly normal control subjects are biologically relevant. Neuroimage 2012;59:1152–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sperling RA, Laviolette PS, O'Keefe K, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009;63:178–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheline YI, Raichle ME, Snyder AZ, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry 2010;67:584–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci USA 2004;101:4637–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jack CR, Jr, Vemuri P, Wiste HJ, et al. Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch Neurol 2012;69:856–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villain N, Chetelat G, Grassiot B, et al. Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer's disease: a voxelwise PiB-PET longitudinal study. Brain 2012;135:2126–2139 [DOI] [PubMed] [Google Scholar]
- 29.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 2013;12:357–367 [DOI] [PubMed] [Google Scholar]
- 30.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology 2012;78:1576–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Brain injury biomarkers are not dependent on β- amyloid in normal elderly. Ann Neurol Epub 2012 Nov 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:351–357 [DOI] [PubMed] [Google Scholar]
- 34.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 1999;45:358–368 [DOI] [PubMed] [Google Scholar]
- 35.Haroutunian V, Purohit DP, Perl DP, et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol 1999;56:713–718 [DOI] [PubMed] [Google Scholar]
- 36.Sonnen JA, Santa Cruz K, Hemmy LS, et al. Ecology of the aging human brain. Arch Neurol 2011;68:1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356 [DOI] [PubMed] [Google Scholar]
- 38.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly with β-amyloidosis. JAMA Neurol 2013;70:1030–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron 2008;60:534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musiek ES, Holtzman DM. Origins of Alzheimer's disease: reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-beta and tau involvement. Curr Opin Neurol 2012;25:715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}