

Abstract
The rise of obesity and its attendant pathological sequelae, including type 2 diabetes and coronary artery disease, constitute an ongoing public health catastrophe in both the developed and, more recently, the developing world. Although the underlying pathophysiology is complex, chronic low-grade inflammation has emerged as a central driver of both primary metabolic dysfunction and subsequent tissue failure. Importantly, this inflammation has been shown to arise as a consequence of both the disruption of homeostatic tissue resident leukocytes and the recruitment of antagonistic effector cells from the circulation. In this review, we discuss the roles of visceral adipose tissue’s salient leukocyte lineages in the transition to obesity and highlight key points at which this emerging immune axis may be manipulated for therapeutic effect.
Introduction
The obesity epidemic, which started in the industrialized world, has progressed to become a worldwide pandemic in which the number of obese individuals worldwide now exceeds those who are malnourished.1 Unlike other pandemics, however, obesity does not cause disease directly but instead functions as a potent independent risk factor for a number of chronic pathologies, including type 2 diabetes, coronary artery disease, neurodegenerative disorders, and cancer,2 wreaking immense harm in both health and economic terms. Despite such infamy, our understanding of obesity remains inadequate.
The most overt effect of obesity is an increase in fat mass, which results from chronic imbalance between energy intake and energy expenditure.3 In rodents and humans, these excess calories are primarily stored in visceral (also referred to as gonadal in rodents) and subcutaneous white adipose tissues (WATs), the preferred sites for long-term nutrient storage in mammals.4 This observation initially led many to view WAT simply as a depot for excess nutrient storage; however, research over the past 2 decades has revealed that WAT is not an inert organ but rather a dynamic endocrine tissue that secretes adipokines (such as leptin, adiponectin, and resistin) to coordinate changes in nutrient intake, utilization, and storage.4 Importantly, this active coordination of organism-wide metabolism is mediated not only by canonical neuroendocrine control but also by leukocytes that traffic to and reside in metabolic tissues.5 In this review, we review the salient contributions of individual leukocyte lineages to the function and control of adipose tissue and mammalian energy metabolism as a whole.
Obesity, tissue inflammation, and insulin resistance
Our understanding of how leukocytes sense the metabolic state and coordinate environmental adaptation is primarily derived from studies performed in obese animals.5-7 Historically, this stems from the observation that obese visceral WAT expresses inflammatory mediators, such as tumor necrosis factor α (TNF-α), which inhibit insulin action. The inhibitory effects of inflammatory cytokines are primarily mediated via activation of inhibitory nuclear factor κB kinase β8,9 and c-Jun N-terminal kinases,10 which promote serine phosphorylation of insulin receptor substrate proteins to promote insulin resistance (ie, decrease insulin action).11 Moreover, the chronic activation of these stress and inflammatory signaling pathways, as occurs in obesity, decreases insulin action locally in visceral WAT and amplifies the inflammatory response to promote systemic insulin resistance.5
Although initial reports suggested that adipocytes produce TNF-α in obese WAT, subsequent studies demonstrated that infiltrating macrophages are primarily responsible for the inflammatory response in visceral WAT of obese animals.12,13 These observations paved the way for many future studies that examined the mechanisms controlling macrophage recruitment and activation in visceral WAT and the functions performed by these cells in promotion or inhibition of insulin resistance. Furthermore, it is now appreciated that obesity-induced metabolic dysfunction not only alters the repertoire of innate immune cells but also leads to dramatic changes in the composition of the adaptive immune cells taking residence in visceral WAT5 (Figure 1; Sidebox 1). In the subsequent sections, we discuss our current understanding of the mechanisms by which hematopoietic cells orchestrate metabolic adaptations to changes in diet and environment.
Figure 1.
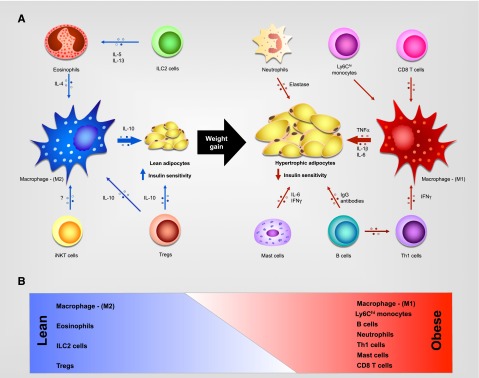
Leukocyte subsets present in WAT. (A) In lean individuals, eosinophils, innate lymphoid type 2 cells (ILC2s), and type I (or invariant) natural killer T (iNKT) cells support activation of alternatively activated (M2) macrophages, which secrete interleukin-10 (IL-10) to promote insulin sensitivity, while regulatory T cells (Treg’s) secrete IL-10, which contributes to M2 activation and directly promotes insulin sensitivity. In obese individuals, CD8+ and CD4+-Th1 T cells secrete proinflammatory cytokines that sustain inflammatory macrophage production of TNF-α, IL-1β, and IL-6 to promote insulin resistance. Neutrophils, mast cells, and B cells also infiltrate the obese adipose tissue and contribute to the inflammatory microenvironment. (B) Schematic representation of weight gain–induced changes in the WAT leukocyte population. IFN-γ, interferon γ; M1, classically activated; Th, T helper.
Innate immune cells
M2 macrophages
Adipose tissues of lean animals contain a resident population of macrophages that express characteristic markers of alternative activation, including arginase 1, CD301, and CD206.14,15 These cells, which are dispersed throughout the WAT of lean animals, promote insulin sensitivity in adipocytes by secreting IL-10 and other as yet unidentified factors14,15 (Figure 1; Sidebox 1). Importantly, impairment in alternative macrophage activation, as in myeloid-specific deletion of peroxisome proliferator activated receptor-γ (PPAR-γ), PPAR-δ, or Klf4, for example, enhances rodents’ susceptibility to diet-induced obesity and insulin resistance.15-18 Conversely, potentiation of alternative macrophage activation, via pharmacologic inhibition or myeloid cell-specific deletion of the mineralocorticoid receptor, for example,19 confers protection from the obesity-related metabolic dysfunction.20 Together, these data implicate M2 macrophages in the maintenance of WAT metabolism and function.
Alternative activation of resident macrophages is mediated by the type 2 cytokines IL-4 and IL-13, which can be secreted by innate or adaptive immune cells. Recent studies, however, demonstrate that eosinophils are the predominant IL-4–producing cells in visceral WAT of lean mice and that their numbers decline with weight gain.21 Congruently, the absence of eosinophils, as in ΔdblGATA mice, abrogates alternative macrophage activation in visceral WAT and renders these animals susceptible to diet-induced obesity and insulin resistance (Figure 1; Sidebox 1). The opposite is observed in IL-5 transgenic mice or parasite-induced models of eosinophilia, which exhibit protection from the deleterious effects of obesogenic diets.21
The production and release of eosinophils from bone marrow and their subsequent recruitment into tissues is largely controlled by IL-5,22 suggesting that IL-5–producing cells might also regulate adipose tissue biology. Interestingly, recent studies have identified ILC2s as the major source of IL-5 and IL-13 in the visceral WAT and that these cells are required for the accumulation of eosinophils and maintenance of M2 macrophages23 (Figure 1; Sidebox 1). Accordingly, mice lacking IL-5 or ILC2s exhibit higher propensity for diet-induced adiposity, insulin resistance, and glucose intolerance.23 In aggregate, these studies have identified a hematopoietic circuit consisting of ILC2s, eosinophils, and M2 macrophages, which promote healthy WAT function and protect against the deleterious effects of excess nutrient intake.
Inflammatory macrophages
In contrast to the protection conferred by resident M2 macrophages, inflammatory Ly6Chi monocytes recruited into obese visceral WAT contribute to its overall inflammatory timbre and to the development of local and systemic insulin resistance24,25 (Figure 1; Sidebox 1). In obese visceral WAT, these recruited Ly6Chi monocytes differentiate into macrophages and can be distinguished by their location and cell surface markers. Unlike resident M2 macrophages, recruited macrophages express CD11c and often circumscribe dying adipocytes to form characteristic “crownlike” structures.26 Furthermore, CD11c+ cells express high levels of TNF-α, Nos2, and IL-6 and thus participate in the development of adipose tissue inflammation.14 In support of this, genetic perturbations that enhance the recruitment of Ccr2+Ly6Chi inflammatory monocytes into WAT promote adipose tissue inflammation and insulin resistance,25 whereas inhibition of Ccr2+Ly6Chi inflammatory monocyte trafficking or their inflammatory activation results in the converse.8,24 Finally, the antagonism of macrophage inflammatory activation by Gpr120 agonists and parasite-derived products21,27 supports macrophage-targeted interventions as a potential therapeutic strategy for obesity-induced inflammation and metabolic disease.28
In addition to their putative roles in propagating adipose tissue inflammation, CD11c+ cells in obese adipose tissue have been suggested to function as dendritic cells, the classical antigen-presenting cells. For instance, purified CD11c+ cells from obese mice express major histocompatibility complex (MHC) class II and costimulatory molecules and are capable of presenting antigens to naïve CD4+ T cells.29,30 Furthermore, Flt3l−/− mice, which lack mature dendritic cells, are protected from diet-induced obesity and insulin resistance.30 These results suggest that the M1/M2 dichotomy might be overly simplistic, and in the future, it will be important to identify better markers to distinguish inflammatory M1 macrophages from the CD11c+ dendritic cells in obese adipose tissue.
Mast cells
Although infiltration of mast cells into obese visceral WAT was first documented 50 years ago, their functions in metabolic disease remained controversial. A recent report using genetic and pharmacologic approaches implicates these innate cells in the pathogenesis of diet-induced metabolic disease.31 Mast cell–deficient mice (KitW-sh/W-sh and KitWv/W) were protected from obesity-induced metabolic syndrome, which was in part mediated by mast cell–derived IL-6 and IFN-γ. Congruent with this observation, stabilization of mast cells by disodium cromoglycate or ketotifen decreases weight gain and improves metabolic function in dietary models of obesity.31 However, these results must be interpreted cautiously because these mice also lack interstitial cells of Cajal in the gut and have bile acid reflux into the stomach, both of which may contribute to lipid malabsorption and intestinal dysbiosis.32
Neutrophils
Unlike macrophages, neutrophils are not present in visceral WAT of lean mice; however, increased energy intake and storage results in their rapid recruitment into visceral WAT.33 Although neutrophils in obese WAT are fewer in number than macrophages, they nonetheless potently augment adipose tissue inflammation and insulin resistance via secretion of elastase.33 Thus, like mast cells, the functions of neutrophils seem to be primarily restricted to the pathological state of obesity.
Adaptive immune cells
T cells
Visceral adipose tissue of lean mice contains a relatively large number of Treg’s, whose frequency exceeds that in other lymphoid tissues, such as the spleen or lymph nodes34 (Figure 1; Sidebox 1). Moreover, obesity is accompanied by a precipitous decline in the proportion of Treg’s relative to other T cells in WAT, which contributes to the observed increase in WAT inflammation.34 Congruently, depletion of Treg’s worsens insulin resistance in lean animals, whereas their in situ expansion leads to partial protection from the metabolic sequelae of obesity.34 Interestingly, unlike splenic Treg’s, WAT Treg’s express the adipogenic transcription factor PPARγ, which is required for their anti-inflammatory functions in obese WAT.35 Accordingly, deletion of PPARγ in Treg’s decreases their numbers in WAT but not the spleen and exacerbates adipose tissue inflammation and insulin resistance.
In addition to Treg’s, diet-induced obesity dynamically alters the landscape of other visceral WAT T-cell subsets as well. For example, visceral WAT of lean animals is populated with Th2 cells and iNKT cells, which actively maintain the tolerogenic immune environment and promote insulin sensitivity.36 During obesity, however, the recruitment and antigen-specific expansion of Th1 and CD8+ T cells overwhelms the tolerogenic effect of Th2 and Treg’s.37 Consequently, obese animals develop insulin resistance, which can be rescued by selective restoration of Treg’s in visceral WAT either through Treg-specific supplementation or wholesale T-cell depletion, which resets the Treg-Th1/CD8+ balance.37
B cells
Lastly, diet-induced obesity results in both the accumulation of B cells within visceral WAT and the appearance of autoreactive IgG antibodies that collect around crownlike structures.38 Interestingly, salient disease phenotypes including insulin resistance can be recapitulated in lean animals by either the transfer of serum IgG or MHC class II–expressing B cells from obese animals, suggesting that both autoantibody production and T-cell antigen presentation contribute to a low-grade autoimmunity within visceral WAT that potentiates metabolic dysfunction.38
Conclusion
Although the field of immunometabolism is only a decade old, it has grown at an unprecedented rate and has provided major insights into how the immune system mediates metabolic adaptations to changes in dietary intake. A general theme that has emerged from this collective work is that excess nutrient storage results in metabolic dysfunction in adipocytes, which in turn activates innate and adaptive inflammation in the enlarging WAT. The net effect of this inflammatory response is to promote insulin resistance and limit nutrient storage, presumably in an effort to relieve metabolic stress in the lipid-engorged adipocytes. In this setting, inflammation-induced insulin resistance is thus an adaptation that allows organisms to mitigate the deleterious effects of excess nutrient intake.5 With chronic imbalance between nutrient intake and expenditure, however, this inflammatory response becomes maladaptive, resulting in a decreased metabolic flexibility and the development of obesity-induced metabolic disease.
Although the majority of these initial studies have focused on the pathological state of obesity, more recent work has begun to uncover important roles for leukocytes in tissue function under conditions of physiological normalcy as well. For example, the contribution of the ILC2-eosinophil-macrophage axis described previously is critical in basal WAT function, whereas a similar M2 macrophage–driven axis has been shown to support brown adipose tissue’s thermogenic functionality.39 Indeed, some experts now believe that such leukocyte-parenchyma collaboration in tissue function may be the rule more than the exception, although the details remain yet undescribed for most tissues.
Despite remarkable progress in explicating immunity’s interaction with both metabolic function and dysfunction in the mouse, our understanding of these processes in humans remains incomplete. Nevertheless, animal model–driven research has already opened previously unappreciated therapeutic avenues to combating obesity and its attendant pathology that are currently under aggressive pursuit in human studies, including interventions directed against pathogenic cell populations (eg, CD3- and CD20-targeted antibodies), mediators (eg, canakinumab and infliximab), and signal transduction apparatuses (eg, amlexanox and salsalate).5 Although yet investigational, these studies clearly demonstrate that immunity is inextricably linked to human metabolism and its related pathology.
Sidebox 1.
Macrophages
Macrophages are innate immune sentinels that participate in various physiological and pathological states by elaborating cytokines, chemokines, growth factors, lipid mediators, and reactive oxygen/nitrogen species. In response to cytokines and pathogens, macrophages can undergo distinct activation programs. For instance, IFN-γ and lipopolysaccharide drive activation of M1 macrophages, whereas IL-4 and IL-13 promote the maturation of M2 macrophages.
Monocytes
There are 2 circulating subsets of monocytes in mice. Ly6Chi monocytes, also known as the inflammatory monocytes, are recruited to sites of infection, inflammation, or damage via the CC chemokine receptor 2, which can bind and be activated by CC chemokine ligand (CCL) 2, CCL5, and CCL8. Upon infiltration into tissues, Ly6Chi monocytes are thought to give rise to M1 macrophages. Ly6Clow cells, in contrast, patrol the local vascular environment using the CX3CR1 chemokine receptor and are termed the “patrolling” monocytes. Although Ly6Clow monocytes have been suggested to give rise to M2 macrophages, this relationship has not been firmly established.
Type 2 innate lymphoid cells
ILC2s belong to a group of novel cells termed innate lymphoid cells (ILCs), which have a lymphoid morphology but lack antigen-specific receptors encoded by rearranged genes, such as the T-cell receptors (TCRs). Unlike other lymphoid cells, ILCs form a first-line defense against microbes and are an important source of cytokines that are later elaborated by effector T cells. ILC2s secrete type 2 cytokines IL-5 and IL-13.
Regulatory T cells
Treg’s are a subpopulation of T cells that express the transcription factor FOXP3 and participate in immune homeostasis. Treg’s secrete IL-10 to suppress immune activation and play an important role in tolerance to self-antigens and in the abrogation of autoimmunity.
Type I natural killer T cells
iNKTs are a unique T-cell lineage that shares characteristics with T and NK cells. They are MHC class I and CD1d restricted and have a limited TCR diversity. Importantly, iNKT cells are important producers of both type 1 (eg, IFN-γ) and type 2 (eg, IL-4) cytokines.
CD8+ T cells
CD8+ T cells play an important role in killing cancerous, virally infected, or damaged cells in an antigen-specific manner and can participate in otherwise inflammatory responses.
CD4+ T cells
CD4+ T cells are helper T cells that give rise to effector, memory, and regulatory T cells. Upon activation in an antigen-specific manner, Th1 and Th2 effector T cells produce type 1 (eg, IFN-γ) and type 2 (eg, IL-4, IL-5, and IL-13) cytokines to orchestrate cellular and humoral immunity, respectively. IL-17–producing Th17 cells and other more recently discovered CD4+ T-cell lineages are of unclear significance in obesity at this time.
Acknowledgments
We thank Justin I. Odegaard for suggestions on this manuscript.
This work was supported by grants from National Institutes of Health: National Heart, Lung, and Blood Institute (HL076746) and National Institute of Diabetes and Digestive and Kidney Diseases (DK094641) (A.C.), a Larry L. Hillblom Foundation Network Grant (A.C.), the Diabetes Family Fund (University of California, San Francisco) (A.C.), the American Heart Association Innovative Award (12PILT11840038) (A.C.), the National Institutes of Health Director’s Pioneer Award (DP1AR064158) (A.C.), a grant from Conselho Nacional de Desenvolvimento Científico e Tecnológico (J.B.C.C.), and a postdoctoral fellowship from the American Heart Association (Y.Q.).
Authorship
Contribution: J.B.C.C. and Y.Q. wrote the initial drafts of the manuscript; A.C. revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ajay Chawla, University of California, San Francisco, 555 Mission Bay Blvd South, Room 252W, P.O. Box 589001, San Francisco, CA 94158-9001; e-mail: ajay.chawla@ucsf.edu.
References
- 1.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world—a growing challenge. N Engl J Med. 2007;356(3):213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 2.Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA. 2013;309(1):71–82. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444(7121):847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21(12):1443–1455. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- 5.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 7.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 9.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 11.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 12.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao X, Sharma N, Kapadia F, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121(7):2736–2749. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7(6):496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7(6):485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Usher MG, Duan SZ, Ivaschenko CY, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120(9):3350–3364. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150(11):776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu D, Molofsky AB, Liang HE, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13(1):9–22. doi: 10.1038/nri3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molofsky AB, Nussbaum JC, Liang HE, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–549. doi: 10.1084/jem.20121964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamei N, Tobe K, Suzuki R, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281(36):26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- 26.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46(11):2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Bhargava P, Li C, Stanya KJ, et al. Immunomodulatory glycan LNFPIII alleviates hepatosteatosis and insulin resistance through direct and indirect control of metabolic pathways. Nat Med. 2012;18(11):1665–1672. doi: 10.1038/nm.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertola A, Ciucci T, Rousseau D, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61(9):2238–2247. doi: 10.2337/db11-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefanovic-Racic M, Yang X, Turner MS, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. 2012;61(9):2330–2339. doi: 10.2337/db11-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15(8):940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167(3):835–848. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talukdar S, Oh DY, Bandyopadhyay G, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18(9):1407–1412. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cipolletta D, Feuerer M, Li A, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486(7404):549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch L, Nowak M, Varghese B, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. 2012;37(3):574–587. doi: 10.1016/j.immuni.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen KD, Qiu Y, Cui X, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480(7375):104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]