

Abstract
The eukaryotic flagellum is a highly conserved organelle serving motility, sensory and transport functions. Although genetic, genomic and proteomic studies have led to the identification of hundreds of flagellar and putative flagellar proteins, precisely how these proteins function individually and collectively to drive flagellum motility and other functions remains to be determined. In this chapter we provide an overview of tools and approaches available for studying flagellum protein function in the protozoan parasite Trypanosoma brucei. We begin by outlining techniques for in vitro cultivation of both T. brucei lifecycle stages, as well as transfection protocols for the delivery of DNA constructs. We then describe specific assays used to assess flagellum function including flagellum preparation and quantitative motility assays. We conclude the chapter with a description of molecular genetic approaches for manipulating gene function. In summary, the availability of potent molecular tools, as well as the health and economic relevance of T. brucei as a pathogen, combine to make the parasite an attractive and integral experimental system for the functional analysis of flagellar proteins.
I. INTRODUCTION
The eukaryotic flagellum, aka cilium1, is a ubiquitous organelle present on most differentiated mammalian cells and on many single-celled microbes. The flagellum performs motility, transport and sensory functions and is essential for normal human development and physiology [1–3]. Flagella are also required for many important human pathogens, including the causative agents of malaria, African sleeping sickness, leishmaniasis, epidemic diarrhea and trichomoniasis [4, 5]. Together, these diseases are responsible for morbidity and mortality in several hundred million people worldwide. Therefore, in addition to providing insight into the operation of molecular motors and principles of cell biology, studies of the mechanisms of flagellum protein function are directly relevant to understanding mechanisms of heritable and infectious human diseases. In this chapter we provide an overview of tools and approaches available for studying flagellum protein function in the protozoan parasite Trypanosoma brucei.
II. RATIONAL
II.A
African trypanosomes, e.g. Trypanosoma brucei2, are protozoan parasites that are the causative agent of African trypanosomiasis, commonly referred to as African sleeping sickness in humans. These parasites are devastating human and animal pathogens that cause significant human mortality and limit economic development in sub-Saharan Africa. Trypanosomes are transmitted between mammalian hosts through the bite of a tsetse fly and parasite motility is important in both hosts [6, 7]. Trypanosome motility is driven by a single flagellum that emerges from the flagellar pocket at the cell posterior and wraps around the cell body as it extends to the cell anterior (Fig. 1) [8]. The propulsive beat is a tractile beat that progresses tipto-base and drives the cell forward with the flagellum tip leading [9]. The flagellum is laterally attached to the cell body along most of its length, causing the entire cell to undulate and rotate in an auger-like fashion as the flagellum beats (supplemental Movie 1). This distinctive motility gives the genus its name, from the Greek trypanon (“auger”) and soma (“body”) [Gruby M. 1843. Recherches et observations sur une nouvelle espèce d’hématozoaire, Trypanosoma sanguinis. C. R. Acad. Sci. Paris 17:1134-336].
Fig. 1. Trypanosoma brucei cell and flagellum structure.
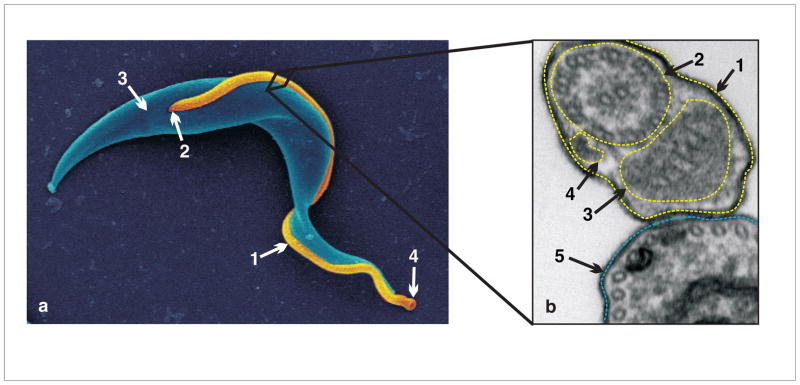
(a) Scanning electron microscopy image of PCF T. brucei. Cell body pseudocolored in blue and flagellum in gold.1 flagellum, 2 flagellar pocket, 3 cell body posterior end, 4 flagellar tip, corresponds to cell anterior. (b) Cross-section of PCF T. brucei cell as imaged by transmission electron microscopy. 1 flagellar membrane, 2 axoneme, 3 paraflagellar rod, 4 Intraflagellar transport particle, 5 cell body membrane. Subpellicular microtubules (not labelled) are visible in cross section in cell body. Adapted from Ralston et al. 2009. Ann. Review. Microbiol, with permission.
The T. brucei flagellum contains a conserved 9+2 axoneme, as well as a unique para-axonemal structure known as the paraflagellar rod (PFR) (Fig. 1). The function of the PFR is not fully understood but it is required for normal motility [10] and is hypothesized to play structural [11] and regulatory roles, since it contains components of calcium and cAMP signalling pathways in addition to proteins with nucleotide transfer domains [12–15]. Trypanosomes are extracellular parasites and thus are presumed to be dependent on their own flagellum-mediated motility to migrate through host tissues. The trypanosome flagellum is also a critical host-parasite interface postulated to provide sensory functions [16, 17] and recent functional studies indicate that the flagellum and/or flagellar motility are required for cell morphogenesis, cell division and evasion of host immune defenses [18–20]. As such, the flagellum and flagellar motility have emerged as attractive drug targets in T. brucei and understanding mechanisms of flagellum protein function in these parasites is critical for exploiting this possibility.
II.B
Genetic screens, biochemical, genomic and proteomic analyses in several organisms have led to the identification of hundreds of flagellar and putative flagellar proteins [20–25]. A key challenge now is to determine where and how these proteins function individually and collectively to drive flagellar motility and other flagellum functions. In addition to its relevance to public health and economic development in some of the poorest places in the world, the molecular tools available in T. brucei make it an excellent experimental system in which to study flagellum protein function. Both lifecycle stages are easily cultured in vitro in semi-defined medium and amenable to biochemical and cell biological analyses. T. brucei also offers facile systems for reverse genetics, including heritable and inducible RNAi, targeted gene knockouts, stable and inducible expression of recombinant proteins, as well as systems for forward genetic screens [26, 27]. Highly efficient homologous recombination allows epitope and tandem affinity purification tags to be readily integrated into any endogenous locus using a single-step PCR approach [28–30]. The genome is sequenced and annotated and the absence of introns makes gene identification simple and reliable [31]. These features combine to make T. brucei a valuable experimental system with tools and approaches for functional analysis of flagellar proteins that are complementary to those available in other organisms.
III. METHODS
The methods section is organized in three parts. The first part (III.A – III.B) describes basic culture and transfection methodologies. The second part (III.C – III.D) describes specific assays used to assess flagellum function, e.g. flagellum preparation and motility assays. In part three (III.E), we describe molecular genetic approaches for manipulating gene function.
III.A. Culture methods
Insect-form (procyclic culture-form, “PCF”) and bloodstream-form (“BSF”) trypanosomes can be maintained in semi-defined media in suspension culture. Both lifecycle forms can also be maintained on semi-solid agarose plates [32], although the current review is restricted to methods employing suspension cultures.
Procyclic Form (PCF)
Two basic media formulations, SM [33] or SDM-79 [34], are used to cultivate PCF trypanosomes in suspension cultures. The recipe for SM is provided in section IV, while the SDM-79 recipe can be found in [34]. Work in the authors’ laboratory employs SM exclusively and specific growth rates might vary for SDM-79. Cultures are maintained at 27°C with or without 5% CO2. The precise advantage provided by CO2 supplementation is unclear, but is postulated to be via pH effects [http://tryps.rockefeller.edu/] and in our experience generally provides for faster cell doubling and facilitates growth at low cell densities. If desired, phenol red can be added to monitor pH of the medium, though this is not required and should be avoided if sedimentation assays using optical density (III.D) are performed. The most widely used cell line employed for laboratory studies is derived from the 427 strain and engineered to encode a T7 polymerase and Tet repressor for regulated gene expression [35]. This strain has a doubling time of 8–9 hr in SM medium, with logarithmic growth between ~ 1×106–1×107 cells/ml. For routine maintenance, cultures are diluted ~1:100 – 1:300 every 2–3 days. For best results in transfections, cultures should be maintained in log phase for several generations, achieved by daily 1:5 – 1:10 dilution.
Bloodstream Form (BSF)
BSF 427-derived trypanosomes encoding T7 polymerase and Tet repressor for inducible gene expression [35] are most commonly used for laboratory studies. BSF cells are grown in suspension cultures using HMI-9 medium [36] containing 10–15% FBS. The recipe for HMI-9 is provided in section IV. BSF suspension cultures are maintained at 37°C with 5% CO2. Doubling times are 6–8 hr and logarithmic growth is between ~ 2×105 – 3×106 cells/ml. Cultures are diluted as for PCF cultures, though additional care should be taken to avoid overgrowth beyond log phase, because BSF cultures die quickly upon reaching stationary phase.
Freezing and thawing
PCF and BSF cells can be frozen in culture media, including FBS and containing 10% glycerol (“Freezing Medium”), and stored at −80°C for several months or in liquid nitrogen for several years. Trypanosomes should be frozen from a healthy logarithmic phase culture. Cells are harvested by centrifugation at 2500g for 5min. The cell pellet is washed once in pre-warmed culture medium and resuspended gently in pre-warmed freezing medium using 1/2 –1/10 of the initially harvested volume, giving a final density of ~2×107 cells/ml. Freezing medium may be supplemented with 10% conditioned medium and increased FBS concentration to 15%. Conditioned medium is obtained by harvesting a late log phase culture by centrifugation at 2500g for 5 min and filtering the supernatant through a 0.22μm filter. Conditioned medium can be stored for several weeks at 4°C or −20°C. Cells in freezing medium are aliquotted (0.5 – 1.0 ml) into appropriate cryotubes with rubber O-ring, e.g. Sarstedt, 72.694.006, then frozen slowly by placing them into a styrofoam tube rack at −80°C. Stock viability should be checked by thawing a few days after freezing, prior to transfer to permanent storage. Thawing of trypanosomes is done as quickly as possible, e.g. in a waterbath at 27°C for PCF or 37°C for BSF. Thawed cells are diluted 1/10 into fresh, prewarmed culture medium and placed in the incubator. If desired, cells can be washed once in pre-warmed culture medium to wash out the glycerol. Generally, if the culture survives the first 24 hr after thawing, it is viable for future, though should be maintained for an additional week prior to use in experiments.
III.B. Transfection
PCF and BSF cells can be transiently or stably transfected by electroporation with linear and circular plasmid DNA constructs, e.g. [37–39]. PCR products can also be used [28, 29, 40]. Transfection efficiency using standard electroporation systems is 10−3–10−6 for PCF and 10−7–10−8 for BSF [41]. Several labs have reported a sharp increase (70 – 1000-fold) in stable transfection efficiency of BSF trypanosomes when using the Amaxa Nucleofector® system (http://www.lonzabio.com/technology.html) [41], which employs pre-set programs and “cell-type specific” confidential tranfection solutions. Further increases in BSF transfection efficiency have been reported when DNA double strand breaks are introduced in the target cells [42]. These new developments are very promising, particularly for genome-wide analyses in BSF cells. However, the current discussions are restricted to standard electroporation conditions used in the authors’ lab. Alternate protocols and solutions for tranfection may be obtained from online sources, e.g. G. Cross lab (http://tryps.rockefeller.edu/).
DNA preparation
Any standard high-quality plasmid preparation system, e.g. Qiagen®, etc…, is appropriate for transfection. Care should be taken to ensure sterile technique. Miniprepped DNA can be used as well, although care should be taken that it is of good quality. Circular plasmids (25 – 100 ug) are used for transient transfection. Linearized plasmids or DNA constructs (5 – 20 ug) are used for stable transfection.
PCF transfection
Cells in mid log phase, e.g. 4–7×106 cells/ml, are harvested by centrifugation at 2500g for 5min, washed once with room temperature Electroporation Media (“EM”, see recipe in section IV), then resuspended in EM to a final concentration of 5×107 cells/ml. A 0.45ml aliquot of cells (2.25×107 cells) is added to a 0.4 cm BioRad® electroporation cuvette, containing 100μl of DNA in water. 50–100μg of circular DNA is used for transient transfection and 5–20μg of linear DNA for stable transfection. Cells and DNA are mixed by gently pipeting up and down, avoiding air bubbles. Cell-DNA mix is then electroporated with 2 successive pulses, separated by a 10sec interval, in a Gene Pulser II (BioRad®) electroporator using the following settings: 1500V, 25μF. The time constant should be between 0.5–0.7 msec. For transient transfections, 0.3 ml of electroporated cells are transferred to 5 ml fresh medium and monitored 12 – 36 hr post-transfection for phenotype. For stable transfections, following electroporation, cells are immediately transferred into 12 ml pre-warmed culture medium and aliquotted (0.5ml) into each well of a 24-well culture plate. Following an overnight incubation, antibiotic for selection is added as 0.5ml of culture medium containing 2× the amount of required antibiotic to each well. The amount of antibiotic required for selection is summarized in Table 2 in section IV.C. Monitor cells daily and dilute as necessary in presence of drug until mock tranfected samples die out and transfected cells are growing well, generally 1–2 weeks post transfection. Each well represents a “pool” of clones, which can then be isolated by limiting dilution [43]. Alternatively, one can perform limiting dilution immediately after adding drug selection. Clones or pools are tested by Southern blot, northern blot, qRT-PCR, protein expression, etc… as appropriate, to confirm presence of transfected DNA construct.
Table 2.
Concentrations of antibiotics most commonly used for selection of PCF and BSF trypanosomes.
| Antibiotic | PCF Final concentration | BSF Final concentration |
|---|---|---|
| G418 | 15μg/ml [1,2,3] | 2.5μg/ml [1] 1–3μg/ml [2] 15μg/ml [3] |
| Phleomycin | 2.5μg/ml [1,2,3] | 2.5μg/ml [1] 1–2.5μg/ml [2] 5μg/ml [3] |
| Hygromycin | 25–50μg/ml [1,2,3] | 5μg/ml [1] 4–5μg/ml [2] 50μg/ml [3] |
| Puromycin | 1μg/ml [2,3] | 0.1μg/ml [2,3] |
| Blasticidin | 10μg/ml [2] | 5μg/ml [2] |
References
Wirtz et al., 1999. MBP
Homepage of Prof. G. Cross (http://tryps.rockefeller.edu/trypsru2)
Hill lab
Note: Concentration may vary dependent on the expression level of the antibiotic resistance marker, e.g. plasmid versus in situ expression, choice of promoter and UTR, etc…
Notes and limitations
Best results are obtained using cells that have been maintained in log-phase for several generations, achieved by daily 1:5 – 1:10 dilutions. Transient transfection efficiencies range from 25 – 80%, depending on the reporter, e.g. tubulin dsRNA constructs provide a more sensitive readout than GFP expression. Stable transfection efficiencies are in the range reported by others [Burkard et al. 2007, MBP], i.e. 10−3 – 10−4. Electroporation with instruments other than the BioRad Gene Pulser II® are also effective, though optimal conditions should be determined empirically.
BSF transfection
Cells in mid log phase (5×105~3×106 cells/ml) are harvested and washed in “EM”, then resuspended at 2×07 cells/ml. A 0.45ml aliquot of cells (0.9×107 cells) is added to a 0.4 cm BioRad® electroporation cuvette, containing 100μl of linearized DNA (20 ug) in water. Cells and DNA are mixed by gently pipeting up and down, avoiding air bubbles. Electroporate with 1 pulse in a Gene Pulser II (BioRad®) electroporator using the following settings: 1500V, 25μF. The time constant should be between 0.5–0.7 msec. Following electroporation, cells are immediately transferred into 12 ml pre-warmed culture medium and aliquotted (0.5ml) into a 24-well culture plate. Following an overnight incubation, antibiotic for selection is added by adding 0.5ml of culture medium containing 2× the amount of the required antibiotic (Table 2) to each well. Monitor cells daily and dilute as necessary in presence of drug until mock tranfected samples die out and transfected cells are growing well, generally 5 to 7 days post transfection. In general, one can expect to see 1–5 wells from a 24-well plate grow out. Each well represents a “pool” of clones, which can then be isolated by limiting dilution. Note that since the stable transfection efficiency is much lower than for PCF cells, cells in each “pool” are quite likely clonal. Clones or pools are tested by Southern blot, northern blot, qRT-PCR, protein expression, etc… as appropriate, to confirm presence of transfected DNA construct. The efficiency for stable transfection using this protocol is 10−6–10−7. We have not had experience with transient tranfection of BSF cells.
III.C. Protocols for flagellum skeleton isolation
The trypanosome flagellum is laterally attached to the cell body along most of its length (Fig. 1), making isolation of intact, i.e. membrane-enclosed, flagella challenging. Progress is being made employing the Fla1 mutant [44] (KH, unpublished) in which the lateral connections are disrupted, but current analyses have been restricted to detergent- and salt-extracted samples lacking the flagellar membrane and soluble matrix [45]. For the current discussion, we refer to such preparations as “flagellum skeletons”, which include the axoneme, PFR, basal body, and some components of the flagellum attachment zone.
A simple and reliable two-step preparation for flagellum skeletons has been published previously [45, 46] and is widely utilized, e.g. [15, 20, 47, 48]. Briefly, this method consists of two basic steps: (i) Non-ionic detergent extraction of whole cells to isolate the cytoskeleton, including flagellum skeleton; followed by (ii) high salt, e.g. 0.5 – 1.0 M NaCl, extraction on ice to depolymerize subpellicular microtubules, leaving the flagellar skeleton intact. In some cases, subpellicular microtubules are depolymerized with 1 mM Ca2+ instead of 1M NaCl [45]. At each step insoluble material, i.e. cytoskeleton or flagellum skeleton, is separated from solubilized material by centrifugation. What remains is the flagellum skeleton, containing extracted axoneme, PFR, basal body and flagellum attachment zone components [45]. Two independent proteomic analyses of such extracted flagella identified a total of 524 potential flagellar proteins [20, 48].
As with other organisms [49] high salt extraction, e.g. 0.5M NaCl and above, perturbs flagellum ultrastructure in T. brucei (Fig. 2a). Commonly outer dyneins are missing and central pair microtubules are compromised. Modification of the procedure to reduce salt concentration and employ a single extraction step yields flagellum skeletons with improved ultrastructure (Fig. 2b). For this one-step preparation, trypanosomes are harvested at 2000g for 5 min, washed in PBS, then resuspended at 2×108 cells/ml in cold PMN buffer (10mM NaPO4 pH7.2, 1mM MgCl2, 150mM NaCl) [45] containing 1% NP-40, DNAse and protease inhibitors. The sample is incubated on ice for 10 min to depolymerize subpellicular microtubules. Addition of Ca2+ [45] sometimes resulted in buffer precipitation and was therefore omitted. Intact flagellum skeletons are harvested by centrifugation at 16,000g for 10 min, the supernatant is aspirated and flagellar skeletons are washed in cold PMN as above. This sample can be used for biochemical, structural, cell biological or proteomic, etc… analyses. Proteomic analysis of the resulting improved flagellum skeleton preparation identified 868 putative flagellar proteins (KH, unpublished), including 76.5% of the proteins identified in earlier analyses.
Fig. 2. T. brucei flagellum skeleton.

Cross-section of detergent and salt-extracted flagellar skeletons imaged by transmission electron microcopy. (a) Extraction performed using 0.5M NaCl, (b)Extraction performed using 0.15M NaCl.
Notes and limitations
The one-step, low salt preparation above generally yields samples with improved ultrastructure versus high-salt extraction, but there are still samples in the preparation with structural defects. Hence, there is still room for improvement. Coupling flagellum preparation with mutants provides a powerful approach for identification of proteins making up specific flagellum substructures. For example, the protein composition of the PFR was partially determined using an elegant approach that employs quantitative proteomic analysis (iTRAQ and 2D DIGE) of detergent extracted flagella from control cells and mutants lacking the PFR [15].
III.D. Assays for trypanosome motility
A variety of assays are available to assess trypanosome motility, either at the single cell or population level. These assays provide readouts for studying the effects of knockdown, knockout or mutation of flagellar proteins.
Sedimentation Assay
Motility of wild type PCF trypanosomes enables them to remain relatively evenly distributed in suspension cultures over time, while motility mutants sediment (Fig. 3a). This provides for a convenient motility assay simply by measuring the optical density at 600 nm (OD600) over time [19, 50, 51]. For sedimentation assays, a culture grown to a density of 5×106 cells/ml is aliquotted into four cuvettes and incubated under standard growth conditions. We standardly use 1ml size BioRad® disposable cuvettes (BioRad® part # 223-9955). The optical density at 600nm (OD600) is then measured every 2 hours for 12 hours. In order to distinguish a motility defect from a growth defect, two cuvettes are left undisturbed, while the other two cuvettes are resuspended at each timepoint. The ΔOD600 is calculated by subtracting the OD600 of the resuspended samples from that of the undisturbed samples and plotted versus time. This ΔOD600 plot is then compared to the control population. An example of the assay is given in Fig. 3b [adapted from [19]]. Sedimentation can also be done reiteratively to enrich for motility mutants (III.E.3).
Fig. 3. Assays for trypanosome motility.


(a) Cartoon illustration depicting sedimentation assays. Cells with wild type motility remain relatively evenly suspended (on left), where as motility mutants sediment (on right). Degree of sedimentation is quantitated using OD600 (arrow). (b) Example dataset derived from a sedimentation assay (adapted from Ralston et al. 2006). Depletion of RSP3 by Tet-inducible RNAi (RSP3 + Tet) cause cells to sediment, while uninduced cells, expressing wt level of RSP3 (RSP3-Tet) remain suspended over time. Adapted from Ralston et al. 2006, with permission. (c) Illustration depicting a motility chamber. (d) Example dataset derived from a motility trace assay (adapted from Hutchings et al 2002). Open circles represent cell position at 1 second intervals, arrowheads indicate ending position. Uninduced cells, expressing wt level of Trypanin (-Tet) exhibit robust motility, whereas Trypain depleted cells (+Tet) fail to exhibit productive forward translocation. Respecitive western blots (WB) against Trypanin or tubulin as a loading control show protein levels. Adapted from Hutchings et al. 2002. JBC, with permission.
Notes and limitations
Care should be taken to keep test and control samples at equivalent cell densities, because motility can change as a function of cell density. In our hands, BSF cells do not generally remain evenly suspended, so sedimentation assays have not worked well for assessing motility of this life cycle stage. Note that the sedimentation assay provides a convenient and quantitative assessment of motility, but applies only populations and is “low-resolution”. Mutants with sedimentation phenotypes are therefore followed through with more detailed analyses by video microscopy (see below).
Motility Traces
A second method for assessing motility is to track movement of individual cells in a population [20, 50, 52–54]. This method employs a motility chamber for digital video microscopy analysis (Fig. 3c) [54]. Each motility chamber is comprised of a glass slide and glass coverslip held together by 50-μm-thick (approximately two cell lengths) double sided tape. To prevent cell adherence, the slide and coverslip are pre-coated in 0.25% poly-L-glutamate [53] as described here: 1) Clean slide and coverslip with 6N Nitric Acid for 10 minutes. 2) Rinse thoroughly with double-distilled water, rinse with ethanol (abs.) and dry using compressed gas duster. 3) Coat slide and coverslip with poly-L-glutamic acid (poly-E) sodium salt (cat. no. P4886, Sigma) in PBS. Incubate for 20 minutes. 4) Rinse off excess poly-E with double-distilled water, rinse with ethanol (abs.) and dry using compressed gas duster. To mimimize capillary flow, the edges of the cover slip are sealed with Vaseline® following the application of cells.
Cells are grown to a density of 5 × 106 cells/ml and resuspended into fresh, pre-warmed medium at a final concentration of 1 × 106 cells/ml. Next, the cells are applied to motility chamber and viewed under dark-field illumination, using a 10× objective. Generally, 30 seconds of digital video from separate regions on each slide are captured for PCF and 2min for BSF. For a camera, typically any good quality hand-held video camera will suffice and can be mounted directly to the microscope light path using appropriate adaptors available from the microscope vendor. Digital video is captured directly to the computer hard drive using 3rd party video capture software, e.g. Adobe Photoshop Premier Elements®. Digital videos, either as avi files or as image stacks, are then analyzed using motility tracking software. We have used Metoamorph® from Molecular Devices, CA, USA [54], which offers a robust suite of programs that can be taylored to individual preferences for motility analyses. Cell movements can be readily quantitated and traces generated automatically (Fig. 3d, adapted from [52]). Cell movements are analyzed for average velocity, burst velocity, distance migrated, and directionality of each cell within a video, etc…
Video microscopy analysis of flagellar beat
To characterize subtleties of flagellar beating and more rigorously describe motility phenotypes, video-microscopy of individual cells at high magnification is employed. Cells are placed in a motility chamber (see above) and imaged using 63× or 100× oil-immersion objectives. The use of differential-interference contrast (DIC) optics aids in viewing of the flagellum. In some cases, it can be useful to image at high resolution using dark field optics, which enhances outline features of the cell. Alternatively, cells can be imaged directly in the tissue culture flask using a 60× long distance objective. Depending on the optics of the microscope used, this approach can give very high resolution and high-contrast videos even with out oil-immersion objectives or DIC optics (see for example supplementary movie 1). Flagellum movements can further be quantified via video microscopy [19, 51]. This approach provides a high-resolution means whereby the beat defects of individual mutants can be quantitatively assessed. Additionally, through the use of this approach, one can distinguish the severity in paralysis between different motility mutants, and can also describe phenotypes characteristic to more severe motility defects.
Notes and limitations
Although the above-mentioned strategies offer a way by which to characterize motility defects at an individual cell level, they are not without limitations. To begin with, although video microscopy captured at conventional speeds (30 frames per second) can effectively yield information about the gross number of beats carried out by a flagellum, the specific mechanics of that beat remain unappreciated in T. brucei, owing to highly erratic and three-dimensional beating of the flagellum, together with the challenges posed by lateral attachment of the flagellum to the cell body. In other kinetoplastids, the use of high-speed imaging has yielded novel data sets that have been used to more rigorously describe the mechanisms driving biological functions [53, 55]. A second concern is the fact that the flagellar beat in T. brucei is three-dimensional and bending can occur independently in the distal and proximal portions of the flagellum, it becomes impossible to reliably measure the frequency and amplitude of a single waveform along the length of the entire flagellum. Lastly, when conducting motility assays, one should take care to be aware of cell density, as cells swim faster when entering stationary phase, and appear to be slower when in cytokinesis.
Examples of application and related references
See references in text
III.E. Genetic approaches to study individual components of the T. brucei flagellum
Molecular genetic tools based on homologous recombination, controllable promoters, RNAi, and a suite of plasmid systems combine to provide complementary approaches for investigating flagellum protein function in T. brucei. Here we outline these approaches, which employ the simple and straight-forward methodologies presented in earlier sections. Where relevant, we list a few published examples, though this is not a comprehensive listing.
1. Genetic tools based on homologous recombination
African trypanosomes exhibit highly efficient homologous recombination and this is exploited to target DNA constructs to specific sites in the parasite genome [38, 39, 56, 57]. The approach can be used to “knock-out” a target gene, or to introduce an altered transgene at its endogenous locus, i.e.“knock-in”. The altered transgene carries specific point mutations or an epitope tag. Recombination efficiency varies depending on the length of the homology region and is generally higher in procyclic PCF form versus BSF cells. Homology regions can be as short as 40–80 base pairs [28, 29, 40], although the length required varies from locus to locus and a few hundred base pairs are generally used for increased reliability.
1.1. Gene disruption by homologous recombination
Constructs for gene deletion are constructed by flanking a selectable marker in a plasmid vector with sequences corresponding to the 5′ untranslated region (UTR) and 3′ UTR of the targeted gene. This construct is than excised from the plasmid backbone by restriction digest and stably transfected into trypanosomes (section III.B). The integrated construct employs endogenous expression signals, e.g. splice acceptor and poly-adenylation. In PCF trypanosomes, where stable transfection efficiency is highest, constructs for gene disruption can be generated by a fast, one-step PCR-based approach using long (~100-bp) primers containing the homology region in their 5′ ends [40]. Efficiency of integration into the targeted locus is reduced relative to employing longer flanking sequences and the choice of using the PCR-based or classical, cloning-based approach is decided on a case-by-case basis. Disruption of multiple genes by homologous recombination is limited to the available resistance markers. The recent development of Cre-lox system for trypanosomes opens new possibilities for the generation of novel selection markers and multiple gene deletions in the same cell line [58].
Examples of application and related references
[40] One step PCR-based disruption of genes
[17, 59] Multiple gene disruption in the same cell line
[58] Cre-lox system for T. brucei
1.2 In situ tagging of genes by a “knock-in” approach
Homologous recombination is also exploited to introduce small epitope or fluorescent protein tags at endogenous chromosomal loci, a procedure referred to as “in situ tagging” (Fig. 4) [28–30, 60]. The epitope or fluorescent protein tag can be introduced at either end of the target gene by choosing the appropriate “tagging cassette”, consisting of a tag, a spacer DNA element (intergenic region, igr) and an antibiotic resistance marker for selection (Fig. 4 and S1). The tagging cassette is flanked by homology regions, for homologous recombination into the target gene locus, using one of two approaches: either a two-step cloning procedure or a single-step PCR amplification (Fig. 4). Both approaches employ the same set of pMOTag tagging plasmids [29], which contain a tag, selectable marker, intergenic sequences to direct RNA processing and multicloning sites (Fig. 4). For C-terminal tagging, the 3′ end of the target open reading frame (without stop codon) is fused in-frame to the 5′ end of the tag and the target gene’s 3′UTR is cloned downstream of the drug resistance marker (Fig. 4). For N-terminal tagging, the target gene’s 5′UTR is fused upstream of the drug resistance marker and the 5′ end of the target open reading frame is fused in frame to the 3′ end of the tag [28, 30, 60] (Suppl. Fig. 1). The tagging cassette, either excised from the plasmid backbone using restriction enzymes or obtained as a PCR product, is stably-transfected into recipient cells as outlined in section III.B. Resulting tagged lines are analyzed as desired. Generally, a single copy of the tagging cassette integrates into one allele of the target gene. If desired, tagging of both alleles or a different gene is easily accomplished by employing an additional selectable drug marker and selecting under both drugs [61]. The genomic landscape at the site of integration and the number of integration events can be verified by PCR and Southern blot analysis.
Fig. 4. Schematic Illustration outlining procedure for C-terminal in situ tagging of T. brucei genes via “knock-in” approach using the pMOTag vector series (Oberholzer et al. 2006).

The tagging-integration construct is generated using PCR with chimeric primers including target gnee ORF (blue) or 3′UTR (gray) at the 5′ end and pMOTag sequences at the three prime end. Alternitively, the integration construct is generated by cloning ORF and UTR sequences into the pMOTag multicloning sites (MCS). The tagging-integration construct is integrated at the endogenous chromosomal locus by homologous recombination. Analogous pMOTag vectors are used for tagging at theN-terminus (Supplemental Fig. 1).
A wide variety of epitope or fluorescent protein tags are available (Table 1), including tags for protein visualization in fixed or live cells, tandem affinity purification or analysis of protein-protein interaction. DNA elements encoding different epitopes have been synthesized for optimum condon usage in trypanosomes (HA, c-Myc, Flag [29]). The most widely used epitope tags are hemagglutinin HA, c-Myc and TY1 tag for protein localization and PTP-tag for affinity purification of protein complexes (Table 1). Most of the antibodies used for the detection of these tags are commercially available, although some cross-react with endogenous trypanosome proteins in western blotting and immunofluorescence microcopy (see Table 1). The highest signal intensity with minimal cross-reactivity has been observed when using the 3xc-Myc tag in combination with the monoclonal antibody 9E10 (Santa Cruz) ([29], Cross lab, unpublished]. Besides small epitope tags, various fluorescent proteins (GFP, eYFP, mCherry,…) are widely used for in situ tagging of genes in trypanosomes [60, 62, 63]. A list of published and unpublished vectors for in situ tagging of genes via the use of various epitope and fluorescent protein tags is provided in the supplemental Material (Suppl. Table 1).
Table 1.
Most commonly used epitope tags for in situ tagging of T. brucei genes
| Tag | Tag sequence (* C-terminal stop) | Antibody (vendor) | Antibody specificity | Selected references |
|---|---|---|---|---|
| HA | YPYDVPDYA* | HA.11 monoclonal mouse (Convance) | Highly specific in WB and IF | Oberholzer et al. FASEB, 2007 |
| 3xHA | YPYDVPDYAYPYDVPDYAYPYDVPDYA* | HAY-11 polyclonal rabbit (Santa Cruz) | Background in WB and IF. Needs pre- absorbtion | Siegel et al. Genes and Development, 2009 Zamudio et al. 2009, Mol.Cell.Biol. |
| HA monoclonal mouse 12CA5 (Roche) | Background in WB and IF | Umeyama et. al, 2008, Eucary. Cell | ||
|
| ||||
| c-Myc | EQKLISEEDL* | Anti-c-Myc monoclonal mouse | Highly specific in WB and IF | Zhao et al. EMBO, 2008 |
| 3xc-Myc | GGRSRSEEQKLISEEDLLRSEQKLISEDLLRSEEQKLISEEDLL* | 9E10 (Santa Cruz Biotechnology) |
Oberholzer et al. FASEB, 2007 Kunz et al. 2009 |
|
|
| ||||
| TY1 | EVHTNQDPLD* | BB2, monoclonal mouse (Prof. K. Gull, Oxford) | Highly specific in WB and IF | Bastin et al. 1996 Portman et al. JBC, 2008 Lowell et al. 2004 |
|
| ||||
| PTP | ProtC-TEV-ProtA-ProtA |
Schimanski et al. Eucaryotic cell, 2005 Zamudio et al. 2009, Mol.Cell.Biol. Nguyen et al. 2007, MBP |
||
Other in situ tagging systems
The in situ tagging system developed by the Tschudi lab [28]as well as well as the pMOTag vectors [29] discussed above, employ two regions of homology to direct integration at a specific locus. An alternate in situ tagging strategy uses a single region of homology to direct integration [30, 60]. The advantage of this second system is that only one homology region has to be cloned, thus simplifying the cloning approach. The disadvantages are: (i) a truncated version of the tagged gene is left behind in the genome and (ii) the plasmid backbone also integrates into the genome. This approach thus generates cell lines with one wildtype allele, one tagged allele and a third truncated allele, which might cause unforeseen complications. Nonetheless, this tagging system has been extremely successful and has been utilized in several instances [15, 30, 62, 64], particularly for incorporating tags that enable tandem affinity purification.
Considerations and Limitations
In situ tagging has two main advantages over traditional, plasmid-based tagging methods. First, because the tagged copy is under the control of the endogenous promoter, one avoids several of the potential risks associated with expressing an extra copy of a gene from a heterologous promoter. Second, large quantities of the tagging construct can be generated via a single-step PCR, thus circumventing multiple sublconing steps and expediting the tagging process. Replacement of the target gene’s endogenous 3′ UTR by the intergenic region of the α-β tubulin locus might alter expression level relative to endogenous levels and this should be taken into consideration. Gene expression through the life-cycle of trypanosomes is regulated largely at the post-transcriptional level [e.g. [65], therefore care should be taken to understand the developmentally-regulated expression profile of the gene targeted for in situ tagging. Next-generation tagging cassettes may overcome this limitation by taking advantage of the CRE recombinase-based system developed for T. brucei [58, 66]. In this case, loxP sites flanking the igr and selectable marker of the tagging cassette allow these sequences to be excised, placing the target gene back under control of the endogenous UTR. Such a in situ tagging strategy would introduce a tag at the 3′ end of a gene leaving only a LoxP-site, a 34bp “scar” between the stop codon and the endogenous 3′UTR. The CRE recombinase system was integrated into the pMOTag in situ tagging vectors series by the research group of Prof. G. Cross but its use remains to be tested (Cross lab homepage for vector details, unpublished]).
Examples of applications and related references
[28, 29, 60] Vectors for in situ tagging of T. brucei genes
[13, 15, 61, 63] In situ tagging of T. brucei genes with epitope tags
[62, 63] In situ tagging of T. brucei genes with fluorescent protein tags
[30, 64, 67] In situ tagging of T. brucei genes with PTP tag for affinity purification of protein complexes
2. Genetic tools based on controllable promoters
T. brucei offers a variety of integratable plasmids for constitutive and tetracycline-inducible expression of genes and RNAi constructs [35, 44, 68, 69]. Tet-inducible systems are particularly useful for studying protein localization as a function of cell cycle, analyzing proteins harboring specific point mutations (see section IIIE.4), and for investigating function of essential genes [13, 19, 20, 44]. The key for the Tet regulated system is a modified cell line, which expresses a Tet repressor and a T7 polymerase [35]. Construction of this cell line was keystone development for functional analysis in T. brucei, with the initial publication [35] being cited more 382 times in the Pubmed database as of July 2009.
2.1. Plasmids for inducible expression
Plasmid details vary, but they all have three key features: 1) a Tet operator sequence to control the transgene promoter; 2) a drug resistance marker to enable selection of stable transfectants; 3) sequences homologous to a silent chromosomal region to allow stable integration. The selectable marker is either located downstream from the inducible promoter and transcribed with the transgene (tandem arrangement), or oriented divergently from the Tet-inducible transgene and transcribed by a separate, constitutive promoter (Fig. 5) [Wirtz et al. 1999, MBP]. Divergent promoters are generally preferred, as this arrangement enables more complete silencing of the transgene in the absence of Tetracycline. For protein expression, the plasmid backbone includes 5′ and 3′ untranslated regions to direct post-transciptional processing and translation. The constructs are targeted to transcriptionally silent loci to ensure no transgene expression in the absence of tetracycline. Most frequently the spacer region of the rDNA locus or the silent regions of the minichromosomes are used [35, 70]. Two widely used vectors that served as templates, physically and conceptually, for the various inducible systems are pLEW100 and pLEW82 [35]. Their overall structure and modules serve as an example for Tet inducible expression systems (Fig. 5). Sequences of the vectors are accessible on the homepage of the research group of Prof. G. Cross (http://tryps.rockefeller.edu/).
Fig. 5. Plasmids for inducible gene expression.


(a) Illustration of two vectors (pLEW100 and pLEW82), commonly used for inducible expression in T. brucei. Adapted from Wirtz et al. 1999, MBP, with permission. (b) Example of a PCF cell line expressing a GFP-tagged flagellum protein (PFR2) through the use of pLEW100 (Fig. 5a). Top panels, fluorescence image of a Tet induced, live cell. On bottom, series of images showing the movement of the GFP-PFR2 expressing cell line under fluorescence microscopy (see supplementary movie 2. Cells are co-stained with a live DNA dye, CFSE to visualize nucleus and kinetoplast. Movie is recorded using a 63× objective (recorded and played back at 30fps).
Considerations and Limitations
Several factors contribute to the expression level of a transgene from ectopic sites.
The design of the vector used for expression, including promoter identity, promoter orientation and nature of the UTR sequences flanking the transgene. Any given construct might also be subject to life cycle stage-specific regulation and hence, this should be considered when choosing the recipient cell line.
The nature of the protein expressed from the construct can also influence the final expression levels. Namely, in our experience cells are able to more readily accommodate high expression levels of proteins that are endogenously highly expressed. For example cells expressing GFP-tagged paraflagellar rod 2 (PFR2) [71] can be readily visualized in live fluorescent video microscopy (Fig. 5b and suppl. Movie 2, MO unpublished), while less abundant proteins, such as trypanin-GFP or CMF19-GFP (two flagellar proteins) require exposure times in excess of 1 second [72], despite using the same expression vector pLEW100 and same induction conditions.
A limitation of Tet regulated expression system can be leakiness of the vector in uninduced state. The extent leakiness varies and has not been systematically studied for a large number of genes, though it would primarily only be problematic for expression toxic gene products or dsRNA.
Examples of application and related references
[35] Cell line expressing Tet repressor and T7 polymerase. Descibing of pLEW100 and pLEW82 vectors [62, 72–75] Selected applications of pLEW100 and pLEW82
2.2 Plasmids for inducible RNA interference (RNAi)
T. brucei is exceptionally well-suited to analyze gene function using RNA interference (RNAi), as first demonstrated by the research group of Prof. E. Ullu [76] and subsequently used to tremendous success, e.g. [69, 77–79]. RNAi in T. brucei is potent, specific and can be achieved with long or short dsRNA triggers [76]. The T. brucei genome is sequenced, lack of introns makes gene identification simple and reliable, and web-based algorithms provide for automated identification and design of primers to amplify the appropriate non-redundant sequences in any target gene [80]. These features, coupled with the tightly regulated Tet-inducible promoter systems described above, allow for facile construction of cell lines in which target gene expression is ablated by heritable and inducible RNAi [44, 69, 81–83].
RNAi knockdown of an endogenous mRNA requires production of dsRNA homologous to a portion of the target mRNA [84]. In T. brucei, this is commonly achieved by placing a single copy of the target between opposing, “head-to-head”, promoters (Fig. 6a) [69, 81]. Alternatively, inverted copies of the target sequence are placed downstream of a single promoter [13, 52, 82]. It is also possible to simultaneously knockdown two or more genes by cloning the target sequences in tandem between opposing promoters, thereby allowing for assessment of genetic interactions [19]. Details on individual systems for generating dsRNA to induce RNAi in trypanosomes are reviewed elsewhere [79, 85]. The head-to-head system is used most commonly, owing to simplicity of cloning. The vectors used most commonly for head-to-head systems are p2T7 or pZJM [44, 69]. Each contains a multicloning site between opposing, tetracycline-inducible promoters, together with a selectable marker and sequences to direct homologous integration at the silent rRNA spacer locus. Appropriate target sequence and primers are identified using the online web-based RNAit application [80], then the target sequence is PCR-amplified and inserted into the multicloning site using standard methods. The resulting plasmid is linearized within the rRNA homology sequence and transfected into PCF or BSF trypanosomes as described above.
Fig. 6. Inducible RNA interference (RNAi) in T. brucei.

(a) Illustration of a widely used inducible RNAi vector (p2T7). Adapted from LaCount et al. 2002, with permission. (b) Illustration of a forward genetic screen through the use of a Tet inducible RNAi library. Adapted from Wang et al. 2000, with permission. See text for details.
Considerations and Limitations
Tet-inducible RNAi in T. brucei is robust and powerful and has been utilized successfully in hundreds of applications. A potential limitation can be that the RNAi construct may be “leaky” in the uniduced state, therefore expressing a small amount of dsRNA without induction by Tet. This can be problematic when targeting an essential gene, although it has not posed a significant limitation in practice, and selective pressures enrich for more completely silenced constructs. RNAi over extended time periods, e.g. more than a week, should be monitored closely, as this can select for cells that down-regulate the RNAi machinery. In large part, this limitation is overcome by utilizing inducible rather than constitutive promoters to drive expression of the RNAi construct. By definition, RNAi phenotypes result from loss of the target protein. Hence, if the target protein is part of a complex, as are most flagellar proteins, one must consider whether the entire complex might be destabilized and, if so, whether the phenotype results from loss of the target protein per se, or loss of the entire complex. Recent developments (see section III.E.4) provide approaches to overcome this limitation through heterologous expression of the RNAi target gene harboring point mutations, thereby disrupting function without altering protein expression and assembly.
Examples of application and related references
[44, 69, 81] Description of p2T7 and pZJM RNAi vectors
[13, 61, 86–88] Selected applications of inducible RNAi
3. Forward genetic screens using RNAi libraries
Forward genetic screens are technically challenging to perform in trypanosomes, owing primarily to the fact that experimentally accessible life cycle stages are diploid. Recently, the research group of Paul Englund described a novel method for conducting forward genetic screens in T. brucei [27, 89]. This method employs a tetracycline-inducible RNAi library that contains random T. brucei DNA fragments between two opposing, tetracycline-inducible promoters in a plasmid that can be stably integrated into the genome (Fig. 6b). The library was introduced into PCF 29-13 cells to provide approximately five-fold genome coverage [27]. Induction with Tet produces a distinct dsRNA in each cell that down-regulates expression of the target gene. From this heterogeneous population of “mutated” (tetracycline induced) cells, mutants are isolated by screening for the desired phenotype, e.g. [27]. Alternatively, the library can be sorted and individual clones induced and screened independently, e.g. [61]. The target gene is identified by PCR-amplification using primers flanking the DNA insert in the RNAi plasmid, and then tested independently. The dominant nature of RNAi circumvents limitations posed by diploidy in trypanosomes and the RNAi plasmid provides a simple and rapid means of identifying the target gene. The approach has been successfully used to identify genes regulating surface protein expression, targets of the trypanosome drug tubericidine, proteins linking the kinetoplast to the flagellum basal body and regulators of the cell-cycle [27, 61, 90, 91]. Although RNAi library screening has not yet been applied for isolation of motility mutants, the approach is readily adaptable to motility mutant screens, e.g. through reiterative rounds of sedimentation (section III.D) to enrich for motility mutants. Forward genetic screens employing transposon mutagenesis have also been established [26]. While promising, this methodology has yet to be implemented widely and is not discussed here.
Considerations and Limitations
Currently, the RNAi library approach has only been used in the PCF life cycle stage. Until recently, BSF trypanosome transfection efficiencies were too low to enable construction of a library with sufficient coverage. However, the recent development of a novel high-efficiency transfection protocol for BSF trypanosomes [41], see section III.B, now make it feasible to generate RNAi libraries in BSF cells. As always, coverage of the library needs to be considered and large or multicopy genes are biasedly screened over smaller, single copy genes. This is due to the fact that larger genes, as well as multicopy genes, are represented with greater frequency as a consequence of the methodology employed to generate the RNAi library. As the approach becomes more widely utilized, new and improved libraries will evolve. As with any library-based approach, the extent of genome coverage is based on the quality of the library and there is also a risk of loosing library complexity the more it is amplified. Likewise, as with any mutant screen, care must be put into the development and implementation of the screening strategy, as this largely determines what one ultimately gets out of the screen.
A potential limitation of the approach is that the RNAi vector can integrate in a genomic locus other than the intended rDNA locus, leading to unexpected mis-regulation of flanking genes [92], particularly if the target fragment happens to contain a site for the restriction enzyme used to linearize the plasmid. For this reason, each target gene identified should be tested independently to confirm the requirement for motility. Note that even in the event of apparent mis-integration, the mis-regulated gene(s) responsible for the phenotype can be identified by careful analysis of the mutant through restriction mapping and PCR to identify the site of vector integration, combined with northern blotting or qRTPCR to assess flanking gene expression levels [92].
Examples of application and related references
[27, 61, 90, 91] Description and applications of the pZJM RNAi library
4. Combination of RNAi and heterologous expression for structure-function studies
An inherent limitation to simple RNAi or gene knockout approaches to assess gene function is that the phenotypes observed reflect loss of the target protein. If the protein is part of a complex, as is generally the case for flagellar proteins, this can cause loss of other subunits or the entire complex, thereby confounding or limiting interpretation. Such cases provide validation of flagellum protein identification and offer information about proteins required for assembly of flagellum sub-structures, but offer little or no information about specific functions of the target protein. Indeed, despite the expansion of inventories of flagellar proteins through genetic, genomic and proteomic studies, knowledge about how these proteins function and details on key amino acids and domains is severely limited. In order to fill this gap in knowledge of flagellum biology, we recently developed an inducible RNAi-based system for structure-function analysis of T. brucei flagellar proteins (Ralston and Hill, unpublished). Details on the system and its application will be published elsewhere, so we provide an outline of the approach here.
The system employs dual vectors to allow for inducible knockdown of an endogenous gene, with simultaneous induction of a heterologous copy harboring specific point mutations (Fig. 7). The “knockdown vector” targets the target gene 3′ UTR so that the coding sequence can still be expressed in the RNAi background. The heterologous copy of the target protein carries an epitope tag to distinguish it from the endogenous protein. Using this system, we identified amino acids necessary for function of the outer arm dynein light chain 1 (LC1) protein (Ralston and Hill, unpublished).
Fig. 7. Combination of RNAi and heterologous expression for structure-function studies.

In the example, knock-down of the target gene is achieved by RNAi against the gene’s 3′UTR. A tagged copy of the target gene, WT or carrying directed point mutations is expressed simultaneously from a different vector.
Considerations and Limitations
Important for the success of this approach is that the heterologous copy of the target gene be expressed at a level that is reasonable, compared to wild type levels. If antibodies are available, this can be tested empirically. In any case, a wild-type copy of the target gene should be expressed using the system, in order to validate that functional rescue of the knockdown is achieved, before testing site mutants. The system also requires that the 3′ UTR is effective at ablating expression of the endogenous target gene, without significantly affecting the gene downstream of the target. This is tested using quantitative, real-time reverse transcriptase PCR. In our experience we have found both criteria to be met for all genes so far tested. As with any epitope tagging analysis, care must be taken to ensure that the tag does not interfere with protein localization, assembly or function. Heterologous expression of a non-mutated, tagged copy of the target to test for rescue is done to address this question.
Examples of application
Ralston et al. in preparation
IV. MATERIALS
IV. A. Culture Medium
1. Cunningham’s culture media (“SM” medium) for cultivating PCF trypanosomes
Reference: [33]
| Inorganic Salts: | 4 Liters |
|---|---|
| I. NaH2PO4 • 1 H2O (Fisher #S369. FW=137.99) | 2,120 mg |
| II. A. MgCl2 • 6 H2O (Fisher #M33. FW=203.31) | 12,160 mg |
| B. MgSO4 • 7 H2O (Fisher #M63. FW=246.48) | 14,800 mg |
| C. KCl (anhydrous) (Fisher. #P217. FW=74.56 | 11,920 mg |
| III. CaCl2 • 2 H2O (BDH #10070. FW=147.02) | 600 mg |
| Sugars: | ||
|---|---|---|
| D-Glucose | (Fisher. #D16. FW=180.16) | 2,800 mg |
| D-Fructose | (Fisher #L95 FW=180.16) | 1,600 mg |
| Sucrose | (B.Mann. #100168 FW=342.3) | 1,600 mg |
| Organic Acids: | 4 Liters | |
|---|---|---|
| L-Malic | 2,680 mg | (M-1000 FW=134.1) |
| a-Ketoglutaric | 1,480 mg | (K-1128 FW=146.1 @ 4C) |
| Fumaric | 220 mg | (Fisher #A120 FW=116.07) |
| Succinic | 240 mg | (MCB Rgts. #SX1040 FW=118.09) |
| cis-Aconitate (3 mM Final) | 2,088 | (A-3412 FW=174.1 @-20C) |
| Na-Pyruvate (1 mM Final) | 40 ml (100 mM Stock) | (Gibco MEM #06304 @4C in dark) |
| Vitamins: | ||
|---|---|---|
| BME Vitamins (100X Stock) | 8 ml | (B-6891 stored @ −20C) |
| Pen-Strep (100X Stock) | 40 ml | (Gibco #15140-122 stored @ −20C) |
| Phenol Red (0.5% Stock)* | 16 ml | (Sigma, βP-0290, *optional) |
| Amino Acids: | Cat. Sigma #: | F.W.: (g/mol) | 4 Liters |
|---|---|---|---|
| 1. B-Alanine | A-7752 | 89.09 | 8,000 mg |
| 2. DL-Alanine | A-7502 | 89.09 | 4,360 mg |
| 3. L-Arginine | A-3909 | 210.7 | 1,760 mg |
| 4. L-Asparagine • 1 H2O | A-8381 | 150.1 | 960 mg |
| 5. L-Aspartic Acid | A-9256 | 133.1 | 440 mg |
| 6. L-Cysteine-HCl H2O | Fisher BP-376 | 175.63 | 320 mg |
| 7. L-Cystine | C-8767 | 240.3 | 120 mg |
| 8. L-Glutamic Acid | G-1251 | 147.1 | 1,000 mg |
| 9. L-Glutamine | G-1517 | 146.1 | 6,560 mg |
| 10. Glycine | Gibco 1106-038 | 75.07 | 480 mg |
| 11. L-Histidine | H-8000 | 155.2 | 640 mg |
| 12. DL-Isoleucine | I-2627 | 131.2 | 360 mg |
| 13. L-Leucine | L-8000 | 131.2 | 360 mg |
| 14. L-Lysine | L-5626 | 182.7 | 600 mg |
| 15. DL-Methionine | M-2768 | 149.2 | 800 mg |
| 16. L-Phenylalanine | P-2126 | 165.2 | 800 mg |
| 17. L-Proline | P-0380 | 115.1 | 27,600 mg |
| 18. DL-Serine | S-4375 | 105.1 | 800 mg |
| 19. L-Taurine | T-0625 | 125.1 | 1,080 mg |
| 20. DL-Threonine | T-1520 | 119.1 | 400 mg |
| 21. L-Tryptophan | Gibco 2110-010 | 204.0 | 400 mg |
| 22. L-Tyrosine | T-3754 | 181.2 | 800 mg |
| 23. DL-Valine | V-0375 | 117.2 | 840 mg |
1. Mix 23 Amino Acids in ~2.6 L H2O and stir at RT overnight to dissolve.
2. Mix independently, inorganic salts I, II and III in 280 ml, 160 ml and 160 ml H2O, respectively.
3. Mix sugars plus organic acids in 560 ml.
4. Combine all ingredients, including vitamins.
5. Adjust pH to 7.4 with 10 N NaOH (approx. 5–8 ml/L) and bring to 4 L final volume.
6. Filter sterilize and store in 500 ml aliquots at 4 deg C.
7. Add HI-FCS to 10% before use.
Phenol red is optional and should be omitted if doing sedimentation assays.
2. SDM-79 medium for PCF trypanosomes
Reference [34].
3. HMI-9 media for cultivating BSF trypanosomes
Reference: [36]
| Reagent | Cat Number | FW | Final | For 500 ml | |
|---|---|---|---|---|---|
| 1 | Iscove’s MDM, liquid * | Gibco #12440-053 | 1X Liquid | 1X | 500 ml bottle |
| 2 | Hypoxanthine | Sigma #H9377 | 136.1 g/mol | 1 mM | 68 mg |
| 3 | Bathocuproine Disulfonate | Sigma #B125 | 564.5 g/mol | 50 uM | 14.1 mg |
| 4 | L-Cysteine • H2O | Fisher BP376-100 | 175.6 g/mol | 1.5 mM | 91 mg |
| 5 | 2-mercaptoethanol | Sigma #M6250 | 14.3 M | 0.2 mM | 7.2 ul |
| 6 | Thymidine | Sigma #T9250 | 242.2 g/mol | 0.16 mM | 19.5 mg |
| 7 | Na-Pyruvate | Gibco #11360-070 | 100 mM | 1 mM | 5 ml |
| 8 | Pen/Strep (+ 29.2 mg/ml L-Gln) Stored at –20, thaw at 37 deg and invert several times to resuspend | Gibco #10378-016 | 10,000 u/ml | 100 u/ml | 5 ml |
1. Dissolve all reagents in 500 ml of pre-warmed (37 deg C) Iscove’s MDM by stirring at RT. This can be done right in the Iscove’s MDM bottle. Be sure all powders are dissolved.
2. If necessary, adjust pH to 7.4 with 1N NaOH
3. Filter Sterilize (0.22 um filter unit) and store at 2 – 8 deg C.
4. Add Heat-Inactivated Fetal Calf Serum to 15 % prior to use.
Alternatively, HMI-9 can be made from powdered IMDM (Gibco #12200-036; 17.7 g/pk) as a starting point.
| Reagent | Cat Number | FW | Final | For 1 LTR “1X” | |
|---|---|---|---|---|---|
| 1 | Iscove’s MDM, powder | Gibco #12200-036 | 17.7 g/pk | 1X = 1LTR | 1 pkg |
| 1* | NaHCO3 | EM Sciences #SX0320-1 | 84.01 g/mol | 36 mM | 3.024 g |
| 2 | Hypoxanthine | Sigma #H9377 | 136.1 g/mol | 1 mM | 136 mg |
| 3 | Bathocuproine Disulfonate | Sigma #B125 | 564.5 g/mol | 50 uM | 28.2 mg |
| 4 | L-Cysteine • H2O | Fisher BP376-100 | 175.6 g/mol | 1.5 mM | 182 mg |
| 5 | 2-mercaptoethanol | Sigma #M6250 | 14.3 M | 0.2 mM | 14.4 ul |
| 6 | Thymidine | Sigma #T9250 | 242.2 g/mol | 0.16 mM | 39 mg |
| 7 | Na-Pyruvate | Gibco #11360-070 | 100 mM | 1 mM | 10 ml |
| 8 | Pen/Strep (+ 29.2 mg/ml L-Gln) Stored at –20, thaw at 37 deg and invert several times to resuspend | Gibco #10378-016 | 10,000 u/ml | 100 u/ml | 10 ml |
1. Dissolve pkg of IMDM in 800 ml warm MiliQ H2O
2. Add 3.024 g NaHCO3 while stirring at RT.
3. Add remaining ingredients while stirring at RT. Be sure all powders are dissolved.
4. If necessary, adjust pH to 7.4 with 1N NaOH.
5. Bring to 1L with H2O
6. Filter Sterilize (0.22 um filter unit) and store at 2 – 8 deg C.
7. Add Heat-Inactivated Fetal Calf Serum to 15 % prior to use.
IV.B Transfection reagents
BioRad® Gene Pulser II gene pulser.
BioRad® electroporation cuvettes 0.4 cm.
-
Electroporation Medium (“EM”)
Reference: [37]
Cytomix for trypanosome transfections
120 mM KCl, 15 mM CaCl2, 10 mM KP04 pH 7.6, 25 mM HEPES, 2 mM EDTA, 5 mM MgCl2.
Phosphate-buffered sucrose
277 mM Sucrose, 16 mM KPO4 pH 7.4, 1 mM MgCl2
Combine cytomix and phosphate-buffered sucrose 3:1 for final EM.
IV.C Antibiotic concentrations for selection
See Table 2
IV.D Buffers for isolation of extracted flagellum skeletons
Reference: [45]
| PMN: | 10 mM NaPO4 pH 7.4, 1 mM MgCl, 150 mM NaCl |
| NP40: | add to PMN to 1% final, as needed |
IV.E Plasmids
See Table 3
Table 3.
Most commonly used vectors1
| Name | Description and application | Antibiotic resistance marker2 | Reference |
|---|---|---|---|
| pLEW100 (Fig. 5a) | All purpose inducible expression using divergent promoters | Phleo | Wirtz et al. 1999 |
| pLEW82 (Fig. 5a) | All purpose inducible expression using a single promoter | G418 | Wirtz et al. 1999 |
| pKR10 (Fig. 7) | pLEW100 modified to include HA tag and MCS for N-terminal or C-terminal tagging | Puro | Ralston and Hill, unpublished. |
| pZJM (Fig. 6b) | All purpose RNAi vector with opposing, Tet- inducible promoters | Phleo | Wang et al. 2000 |
| p2T7 (Fig. 6a) | All purpose RNAi vector with opposing, Tet- inducible promoters | Hygro | LaCount et al. 2002 |
| pMOTag series (Fig. 4, suppl. Fig. 1, suppl. Table 1) | Series of vectors for in situ tagging with epitope, fluorescent protein (See suppl. Table 1 for complete list of pMOTag variations and related vector systems) | G418, Puro, Hygro, Phleo, Blast | Oberholzer et al. MBP, 2006 http://www.izb.unibe.ch/res/seebeck/index.php |
| pC-PTP-Neo, pN-PTP-Puro | Plasmids for in situ tagging with PTP tandem affinity purification tag | G418, Puro | Schimanski et al. 2005 |
Many additional plasmids and variations exist. Plasmids listed are those commonly used and for which the authors’ laboratory has direct experience.
For most plasmids, several different antibiotic resistance markers are available.
Abbreviations: Phleo: phleomycin resistance, G418 : G418 resistance, Puro : Puromycin resistance, Hygro : Hygromycin resistance, Blast: Blasticidin resistance
IV.F Online Resources
Trypanosome Genome website at Sanger
”GeneDB” http://www.genedb.org/
Trypanosome Genome Resource, ”TriTryp DB”
http://beta.tritrypdb.org/tritrypdb1.1/
Trypanofan RNAi resource page and RNAit algorithm for selecting RNAi targets and primers, “Trypanofan”:
http://trypanofan.path.cam.ac.uk/trypanofan/main/
Useful lab web pages:
Prof. G. Cross
Prof. C. Clayton
http://www.zmbh.uni-heidelberg.de/Clayton/default.shtml
Prof. M. Carrington
http://web.me.com/mc115/mclab/resources.html
Prof. D. Horn
http://homepages.lshtm.ac.uk/~ipmbdhor/dhhome.htm
Prof. Thomas Seebeck
http://www.izb.unibe.ch/res/seebeck/index.php
Prof. L. Simpson
V. SUMMARY AND OUTLOOK
T. brucei offers a robust array of molecular genetic tools for functional analysis of flagellar proteins, as well as approaches for forward genetics. In particular, efficient homologous recombination and broad utility of heritable and inducible RNAi combine to provide tools complementary to those available in other systems. The ability to simultaneously ablate expression of an endogenous gene and replace it with a heterologous copy carrying directed point mutations will be useful for moving deeper into understanding how individual proteins work, particularly as the era of flagellum protein identification comes to a close and efforts shift to functional analysis.
As useful as the systems for functional analysis in T. brucei are, limitations, and therefore opportunities, remain. For example, in situ tagging requires replacement of the target gene’s 3′ UTR with a heterologous UTR,potentially resulting in altered expression. The recent development of a CRE recombinase-based system in T. brucei [58] should overcome this limitation in next-generation tagging cassettes. A second area of need is further development of forward genetic screening approaches. The RNAi library has circumvented many limitations and should be applied more broadly, while construction of additional libraries should continue to improve genome coverage. The mariner transposon mutagenesis approach [26] also warrants more attention. Regarding flagellum fractionation, lateral attachment of the flagellum to the cell body poses significant challenges for isolating memrane-enclosed flagella. Advances are being made in several laboratories by employing mutants that disrupt lateral flagellum attachment and this approach is likely to yield results in the near future. The erratic and three-dimensinal nature of T. brucei motility have so far limited quantitative descriptions of flagellar beating and cell movement. High-speed and three-dimensional video microscopy methods will need to be applied to overcome these limitations.
Supplementary Material
Supplemental Fig. 1. Illustration describing N-terminal in situ tagging of T. brucei genes via “knock-in” approach.
Supplementary Movie 1. Motility of cultured PCF trypanosomes. Video recorded using a 63× objective (recorded and played back at 30fps). Adapted from Ralston et al 2006.
Supplementary movie 2. Live fluorescence video microscopy of a PCF cell line expressing GFP-tagged PFR2 that has been stained with the viable dye CFSE to stain the nuclear and kinetoplast DNA (see also figure 5).
Supplemental Table 1. Commonly used vectors for C- and N-terminal in situ tagging of T. brucei genes. For complete list of all available plasmids see Oberholzer et al. MBP, 2006; Shen et al. MBP, 2001; Kelly et al. MBP, 2007 and homepage of Prof. G. Cross (http://tryps.rockefeller.edu/) and Prof. Thomas Seebeck (http://www.izb.unibe.ch/res/seebeck/index.php).
Fig. 8. Trypanosoma brucei as a model system for flagellum structure and function studies.
Cartoon of a T. brucei cell, emphasizing the evolutionarily-conserved character of the 9 + 2 flagellar axoneme, with various ciliated organisms representing the radial spokes that extend inward from the outer doublet microtubules toward the central pair. The paraflagellar rod, unique to trypanosomes, is not shown. Drawing design by Desiree Baron and Zachary Matheny. Adapted from Ralston et al. 2009, with permission.
Acknowledgments
We are greatful to all of our colleagues who have contributed to the development of systems and tools for analysis of gene function in T. brucei and apologize to those whose work was not cited. Work in the authors’ laboratory is supported by grants from the NIH-NIAID (AI052348), Burroughs Wellcome Fund, the Beckman Young Investigator program, and the Ellison Medical Foundation. ML is the recipient of an NIH-NRSA (GM07185). KR is the recipient of an NIH-NRSA (GM07104) and a Dissertation Year Fellowship from the UCLA graduate division. MO is the recipient of a Swiss National Sciences Foundation Fellowship.
Footnotes
Flagellum and cilium are interchangeable terms for the same eukaryotic organelle and distinct from the bacterial flagellum.
We use the term trypanosome to refer to Trypanosoma brucei, the topic of the present review, although related Trypanosoma species, most notably T. congelense are also responsible for diesease in wild and domestic animals.
VII. REFERENCES
- 1.Ibanez-Tallon I, Heintz N, Omran H. To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet. 2003;12(Spec1):R27–35. doi: 10.1093/hmg/ddg061. [DOI] [PubMed] [Google Scholar]
- 2.Badano JL, et al. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 3.Satir P, Christensen ST. Structure and function of mammalian cilia. Histochem Cell Biol. 2008;129(6):687–93. doi: 10.1007/s00418-008-0416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vlachou D, et al. The developmental migration of Plasmodium in mosquitoes. Curr Opin Genet Dev. 2006;16(4):384–91. doi: 10.1016/j.gde.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson DJ. Toxoplasma gondii and sex: essential or optional extra? Trends Parasitol. 2002;18(8):355–9. [PubMed] [Google Scholar]
- 6.Kohl L, Bastin P. The flagellum of trypanosomes. Int Rev Cytol. 2005;244:227–85. doi: 10.1016/S0074-7696(05)44006-1. [DOI] [PubMed] [Google Scholar]
- 7.Ralston KS, Hill KL. The flagellum of Trypanosoma brucei: new tricks from an old dog. Int J Parasitol. 2008;38(8–9):869–84. doi: 10.1016/j.ijpara.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ralston KS, et al. The Trypanosoma brucei Flagellum: Moving Parasites in New Directions. Annu Rev Microbiol. 2009 doi: 10.1146/annurev.micro.091208.073353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker PJ. Organization of function in trypanosome flagella. Nature. 1961;189:1017–8. doi: 10.1038/1891017a0. [DOI] [PubMed] [Google Scholar]
- 10.Bastin P, Sherwin T, Gull K. Paraflagellar rod is vital for trypanosome motility. Nature. 1998;391(6667):548. doi: 10.1038/35300. [DOI] [PubMed] [Google Scholar]
- 11.Maga JA, LeBowitz JH. Unravelling the kinetoplastid paraflagellar rod. Trends Cell Biol. 1999;9(10):409–13. doi: 10.1016/s0962-8924(99)01635-9. [DOI] [PubMed] [Google Scholar]
- 12.Oberholzer M, et al. Trypanosomes and mammalian sperm: one of a kind? Trends Parasitol. 2007;23(2):71–7. doi: 10.1016/j.pt.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Oberholzer M, et al. The Trypanosoma brucei cAMP phosphodiesterases TbrPDEB1 and TbrPDEB2: flagellar enzymes that are essential for parasite virulence. Faseb J. 2007;21(3):720–31. doi: 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- 14.Pullen TJ, et al. Protein targeting of an unusual, evolutionarily conserved adenylate kinase to a eukaryotic flagellum. Mol Biol Cell. 2004;15(7):3257–65. doi: 10.1091/mbc.E04-03-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Portman N, et al. Combining RNA interference mutants and comparative proteomics to identify protein components and dependences in a eukaryotic flagellum. J Biol Chem. 2009;284(9):5610–9. doi: 10.1074/jbc.M808859200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tetley L, Vickerman K. Differentiation in Trypanosoma brucei: host-parasite cell junctions and their persistence during acquisition of the variable antigen coat. J Cell Sci. 1985;74:1–19. doi: 10.1242/jcs.74.1.1. [DOI] [PubMed] [Google Scholar]
- 17.Vassella E, et al. Major surface glycoproteins of insect forms of Trypanosoma brucei are not essential for cyclical transmission by tsetse. PLoS One. 2009;4(2):e4493. doi: 10.1371/journal.pone.0004493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohl L, Robinson D, Bastin P. Novel roles for the flagellum in cell morphogenesis and cytokinesis of trypanosomes. Embo J. 2003;22(20):5336–46. doi: 10.1093/emboj/cdg518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ralston KS, et al. Flagellar motility contributes to cytokinesis in Trypanosoma brucei and is modulated by an evolutionarily conserved dynein regulatory system. Eukaryot Cell. 2006;5(4):696–711. doi: 10.1128/EC.5.4.696-711.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broadhead R, et al. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440(7081):224–7. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 21.Pazour GJ. Comparative genomics: prediction of the ciliary and basal body proteome. Curr Biol. 2004;14(14):R575–7. doi: 10.1016/j.cub.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Li JB, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117(4):541–52. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 23.Pazour GJ, et al. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170(1):103–13. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith JC, et al. Robust method for proteome analysis by MS/MS using an entire translated genome: demonstration on the ciliome of Tetrahymena thermophila. J Proteome Res. 2005;4(3):909–19. doi: 10.1021/pr050013h. [DOI] [PubMed] [Google Scholar]
- 25.Ostrowski LE, et al. A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics. 2002;1(6):451–65. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 26.Leal S, et al. Transposon mutagenesis of Trypanosoma brucei identifies glycosylation mutants resistant to concanavalin A. J Biol Chem. 2004;279(28):28979–88. doi: 10.1074/jbc.M403479200. [DOI] [PubMed] [Google Scholar]
- 27.Morris JC, et al. Glycolysis modulates trypanosome glycoprotein expression as revealed by an RNAi library. Embo J. 2002;21(17):4429–38. doi: 10.1093/emboj/cdf474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen S, et al. In vivo epitope tagging of Trypanosoma brucei genes using a one step PCR-based strategy. Mol Biochem Parasitol. 2001;113(1):171–3. doi: 10.1016/s0166-6851(00)00383-2. [DOI] [PubMed] [Google Scholar]
- 29.Oberholzer M, et al. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol. 2006;145(1):117–20. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Schimanski B, Nguyen TN, Gunzl A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell. 2005;4(11):1942–50. doi: 10.1128/EC.4.11.1942-1950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berriman M, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309(5733):416–22. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 32.Carruthers VB, Cross GA. High-efficiency clonal growth of bloodstream- and insect-form Trypanosoma brucei on agarose plates. Proc Natl Acad Sci U S A. 1992;89(18):8818–21. doi: 10.1073/pnas.89.18.8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunningham I. New culture medium for maintenance of tsetse tissues and growth of trypanosomatids. J Protozool. 1977;24(2):325–9. doi: 10.1111/j.1550-7408.1977.tb00987.x. [DOI] [PubMed] [Google Scholar]
- 34.Brun R, Schonenberger Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop. 1979;36(3):289–92. [PubMed] [Google Scholar]
- 35.Wirtz E, et al. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99(1):89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 36.Hesse F, et al. A novel cultivation technique for long-term maintenance of bloodstream form trypanosomes in vitro. Mol Biochem Parasitol. 1995;70(1–2):157–66. doi: 10.1016/0166-6851(95)00027-x. [DOI] [PubMed] [Google Scholar]
- 37.Hill KL, et al. A novel protein targeting domain directs proteins to the anterior cytoplasmic face of the flagellar pocket in African trypanosomes. J Cell Sci. 1999;112(Pt 18):3091–101. doi: 10.1242/jcs.112.18.3091. [DOI] [PubMed] [Google Scholar]
- 38.Lee MG, Van der Ploeg LH. Homologous recombination and stable transfection in the parasitic protozoan Trypanosoma brucei. Science. 1990;250(4987):1583–7. doi: 10.1126/science.2177225. [DOI] [PubMed] [Google Scholar]
- 39.Zomerdijk JC, et al. The promoter for a variant surface glycoprotein gene expression site in Trypanosoma brucei. Embo J. 1990;9(9):2791–801. doi: 10.1002/j.1460-2075.1990.tb07467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arhin GK, et al. A PCR-based method for gene deletion and protein tagging in Trypanosoma brucei. Methods Mol Biol. 2004;270:277–86. doi: 10.1385/1-59259-793-9:277. [DOI] [PubMed] [Google Scholar]
- 41.Burkard G, Fragoso CM, Roditi I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol. 2007;153(2):220–3. doi: 10.1016/j.molbiopara.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 42.Glover L, Horn D. Site-specific DNA double-strand breaks greatly increase stable transformation efficiency in Trypanosoma brucei. Mol Biochem Parasitol. 2009;166(2):194–7. doi: 10.1016/j.molbiopara.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klingbeil MM, Motyka SA, Englund PT. Multiple mitochondrial DNA polymerases in Trypanosoma brucei. Mol Cell. 2002;10(1):175–86. doi: 10.1016/s1097-2765(02)00571-3. [DOI] [PubMed] [Google Scholar]
- 44.LaCount DJ, Barrett B, Donelson JE. Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J Biol Chem. 2002;277(20):17580–8. doi: 10.1074/jbc.M200873200. [DOI] [PubMed] [Google Scholar]
- 45.Robinson D, et al. Microtubules, tubulin, and microtubule-associated proteins of trypanosomes. Methods Enzymol. 1991;196:285–99. doi: 10.1016/0076-6879(91)96027-o. [DOI] [PubMed] [Google Scholar]
- 46.Schneider A, et al. Subpellicular and flagellar microtubules of Trypanosoma brucei brucei contain the same alpha-tubulin isoforms. J Cell Biol. 1987;104(3):431–8. doi: 10.1083/jcb.104.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hill KL, et al. T lymphocyte-triggering factor of african trypanosomes is associated with the flagellar fraction of the cytoskeleton and represents a new family of proteins that are present in several divergent eukaryotes. J Biol Chem. 2000;275(50):39369–78. doi: 10.1074/jbc.M006907200. [DOI] [PubMed] [Google Scholar]
- 48.Hart SR, et al. Analysis of the trypanosome flagellar proteome using a combined electron transfer/collisionally activated dissociation strategy. J Am Soc Mass Spectrom. 2009;20(2):167–75. doi: 10.1016/j.jasms.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 49.Dutcher SK, Huang B, Luck DJ. Genetic dissection of the central pair microtubules of the flagella of Chlamydomonas reinhardtii. J Cell Biol. 1984;98(1):229–36. doi: 10.1083/jcb.98.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bastin P, et al. Protein transport and flagellum assembly dynamics revealed by analysis of the paralysed trypanosome mutant snl-1. J Cell Sci. 1999;112(Pt 21):3769–77. doi: 10.1242/jcs.112.21.3769. [DOI] [PubMed] [Google Scholar]
- 51.Branche C, et al. Conserved and specific functions of axoneme components in trypanosome motility. J Cell Sci. 2006;119(Pt 16):3443–55. doi: 10.1242/jcs.03078. [DOI] [PubMed] [Google Scholar]
- 52.Hutchings NR, Donelson JE, Hill KL. Trypanin is a cytoskeletal linker protein and is required for cell motility in African trypanosomes. J Cell Biol. 2002;156(5):867–77. doi: 10.1083/jcb.200201036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gadelha C, Wickstead B, Gull K. Flagellar and ciliary beating in trypanosome motility. Cell Motil Cytoskeleton. 2007;64(8):629–43. doi: 10.1002/cm.20210. [DOI] [PubMed] [Google Scholar]
- 54.Baron DM, Kabututu ZP, Hill KL. Stuck in reverse: loss of LC1 in Trypanosoma brucei disrupts outer dynein arms and leads to reverse flagellar beat and backward movement. J Cell Sci. 2007;120(Pt 9):1513–20. doi: 10.1242/jcs.004846. [DOI] [PubMed] [Google Scholar]
- 55.Sugrue P, et al. Flagellar wave reversal in the kinetoplastid flagellate Crithidia oncopelti. Biol Cell. 1988;63(2):127–31. doi: 10.1016/0248-4900(88)90051-2. [DOI] [PubMed] [Google Scholar]
- 56.Clayton CE. Genetic manipulation of kinetoplastida. Parasitol Today. 1999;15(9):372–8. doi: 10.1016/s0169-4758(99)01498-2. [DOI] [PubMed] [Google Scholar]
- 57.Li F, et al. Procyclic Trypanosoma brucei cell lines deficient in ornithine decarboxylase activity. Mol Biochem Parasitol. 1996;78(1–2):227–36. doi: 10.1016/s0166-6851(96)02630-8. [DOI] [PubMed] [Google Scholar]
- 58.Scahill MD, Pastar I, Cross GA. CRE recombinase-based positive-negative selection systems for genetic manipulation in Trypanosoma brucei. Mol Biochem Parasitol. 2008;157(1):73–82. doi: 10.1016/j.molbiopara.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haenni S, et al. The procyclin-associated genes of Trypanosoma brucei are not essential for cyclical transmission by tsetse. Mol Biochem Parasitol. 2006;150(2):144–56. doi: 10.1016/j.molbiopara.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 60.Kelly S, et al. Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol Biochem Parasitol. 2007;154(1):103–9. doi: 10.1016/j.molbiopara.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Z, et al. p166, a link between the trypanosome mitochondrial DNA and flagellum, mediates genome segregation. Embo J. 2008;27(1):143–54. doi: 10.1038/sj.emboj.7601956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Umeyama T, Wang CC. Polo-like kinase is expressed in S/G2/M phase and associated with the flagellum attachment zone in both procyclic and bloodstream forms of Trypanosoma brucei. Eukaryot Cell. 2008;7(9):1582–90. doi: 10.1128/EC.00150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Z, Umeyama T, Wang CC. The chromosomal passenger complex and a mitotic kinesin interact with the Tousled-like kinase in trypanosomes to regulate mitosis and cytokinesis. PLoS One. 2008;3(11):e3814. doi: 10.1371/journal.pone.0003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nguyen TN, et al. Purification of an eight subunit RNA polymerase I complex in Trypanosoma brucei. Mol Biochem Parasitol. 2006;149(1):27–37. doi: 10.1016/j.molbiopara.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 65.Urwyler S, et al. A family of stage-specific alanine-rich proteins on the surface of epimastigote forms of Trypanosoma brucei. Mol Microbiol. 2007;63(1):218–28. doi: 10.1111/j.1365-2958.2006.05492.x. [DOI] [PubMed] [Google Scholar]
- 66.Barrett B, LaCount DJ, Donelson JE. Trypanosoma brucei: a first-generation CRE-loxP site-specific recombination system. Exp Parasitol. 2004;106(1–2):37–44. doi: 10.1016/j.exppara.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 67.Zamudio JR, et al. Trypanosoma brucei spliced leader RNA maturation by the cap 1 2′-O-ribose methyltransferase and SLA1 H/ACA snoRNA pseudouridine synthase complex. Mol Cell Biol. 2009;29(5):1202–11. doi: 10.1128/MCB.01496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirtz E, Clayton C. Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science. 1995;268(5214):1179–83. doi: 10.1126/science.7761835. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z, et al. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J Biol Chem. 2000;275(51):40174–9. doi: 10.1074/jbc.M008405200. [DOI] [PubMed] [Google Scholar]
- 70.Wickstead B, Ersfeld K, Gull K. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol. 2002;125(1–2):211–6. doi: 10.1016/s0166-6851(02)00238-4. [DOI] [PubMed] [Google Scholar]
- 71.Schlaeppi K, Deflorin J, Seebeck T. The major component of the paraflagellar rod of Trypanosoma brucei is a helical protein that is encoded by two identical, tandemly linked genes. J Cell Biol. 1989;109(4 Pt 1):1695–709. doi: 10.1083/jcb.109.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baron DM, et al. Functional genomics in Trypanosoma brucei identifies evolutionarily conserved components of motile flagella. J Cell Sci. 2007;120(Pt 3):478–91. doi: 10.1242/jcs.03352. [DOI] [PubMed] [Google Scholar]
- 73.Boothroyd CE, et al. A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature. 2009;459(7244):278–81. doi: 10.1038/nature07982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selvapandiyan A, et al. Centrin1 is required for organelle segregation and cytokinesis in Trypanosoma brucei. Mol Biol Cell. 2007;18(9):3290–301. doi: 10.1091/mbc.E07-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grandgenett PM, et al. A function for a specific zinc metalloprotease of African trypanosomes. PLoS Pathog. 2007;3(10):1432–45. doi: 10.1371/journal.ppat.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ngo H, et al. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc Natl Acad Sci U S A. 1998;95(25):14687–92. doi: 10.1073/pnas.95.25.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Motyka SA, Englund PT. RNA interference for analysis of gene function in trypanosomatids. Curr Opin Microbiol. 2004;7(4):362–8. doi: 10.1016/j.mib.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 78.Ullu E, Tschudi C, Chakraborty T. RNA interference in protozoan parasites. Cell Microbiol. 2004;6(6):509–19. doi: 10.1111/j.1462-5822.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- 79.LaCount DJ, Donelson JE. RNA interference in African trypanosomes. Protist. 2001;152(2):103–11. doi: 10.1078/1434-4610-00047. [DOI] [PubMed] [Google Scholar]
- 80.Redmond S, Vadivelu J, Field MC. RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol Biochem Parasitol. 2003;128(1):115–8. doi: 10.1016/s0166-6851(03)00045-8. [DOI] [PubMed] [Google Scholar]
- 81.LaCount DJ, et al. Double-stranded RNA interference in Trypanosoma brucei using head-to-head promoters. Mol Biochem Parasitol. 2000;111(1):67–76. doi: 10.1016/s0166-6851(00)00300-5. [DOI] [PubMed] [Google Scholar]
- 82.Bastin P, et al. Flagellum ontogeny in trypanosomes studied via an inherited and regulated RNA interference system. J Cell Sci. 2000;113(Pt 18):3321–8. doi: 10.1242/jcs.113.18.3321. [DOI] [PubMed] [Google Scholar]
- 83.Shi H, et al. Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. Rna. 2000;6(7):1069–76. doi: 10.1017/s1355838200000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bosher JM, Labouesse M. RNA interference: genetic wand and genetic watchdog. Nat Cell Biol. 2000;2(2):E31–6. doi: 10.1038/35000102. [DOI] [PubMed] [Google Scholar]
- 85.Ullu E, et al. RNA interference: advances and questions. Philos Trans R Soc Lond B Biol Sci. 2002;357(1417):65–70. doi: 10.1098/rstb.2001.0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Absalon S, et al. Intraflagellar transport and functional analysis of genes required for flagellum formation in trypanosomes. Mol Biol Cell. 2008;19(3):929–44. doi: 10.1091/mbc.E07-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ralston KS, Hill KL. Trypanin, a component of the flagellar Dynein regulatory complex, is essential in bloodstream form African trypanosomes. PLoS Pathog. 2006;2(9):e101. doi: 10.1371/journal.ppat.0020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jetton N, et al. The cell cycle as a therapeutic target against Trypanosoma brucei: Hesperadin inhibits Aurora kinase-1 and blocks mitotic progression in bloodstream forms. Mol Microbiol. 2009;72(2):442–58. doi: 10.1111/j.1365-2958.2009.06657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Englund PT, et al. RNAi libraries and kinetoplast DNA. Biochem Soc Trans. 2005;33(Pt 6):1409–12. doi: 10.1042/BST0331409. [DOI] [PubMed] [Google Scholar]
- 90.Drew ME, et al. The adenosine analog tubercidin inhibits glycolysis in Trypanosoma brucei as revealed by an RNA interference library. J Biol Chem. 2003;278(47):46596–600. doi: 10.1074/jbc.M309320200. [DOI] [PubMed] [Google Scholar]
- 91.Monnerat S, et al. Searching for novel cell cycle regulators in Trypanosoma brucei with an RNA interference screen. BMC Res Notes. 2009;2:46. doi: 10.1186/1756-0500-2-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Motyka SA, et al. Integration of pZJM library plasmids into unexpected locations in the Trypanosoma brucei genome. Mol Biochem Parasitol. 2004;134(1):163–7. doi: 10.1016/j.molbiopara.2003.11.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Illustration describing N-terminal in situ tagging of T. brucei genes via “knock-in” approach.
Supplementary Movie 1. Motility of cultured PCF trypanosomes. Video recorded using a 63× objective (recorded and played back at 30fps). Adapted from Ralston et al 2006.
Supplementary movie 2. Live fluorescence video microscopy of a PCF cell line expressing GFP-tagged PFR2 that has been stained with the viable dye CFSE to stain the nuclear and kinetoplast DNA (see also figure 5).
Supplemental Table 1. Commonly used vectors for C- and N-terminal in situ tagging of T. brucei genes. For complete list of all available plasmids see Oberholzer et al. MBP, 2006; Shen et al. MBP, 2001; Kelly et al. MBP, 2007 and homepage of Prof. G. Cross (http://tryps.rockefeller.edu/) and Prof. Thomas Seebeck (http://www.izb.unibe.ch/res/seebeck/index.php).