

Abstract
Mitochondria are multifunctional organelles with diverse roles including energy production and distribution, apoptosis, eliciting host immune response, and causing diseases and aging. Mitochondria-mediated immune responses might be an evolutionary adaptation by which mitochondria might have prevented the entry of invading microorganisms thus establishing them as an integral part of the cell. This makes them a target for all the invading pathogens including viruses. Viruses either induce or inhibit various mitochondrial processes in a highly specific manner so that they can replicate and produce progeny. Some viruses encode the Bcl2 homologues to counter the proapoptotic functions of the cellular and mitochondrial proteins. Others modulate the permeability transition pore and either prevent or induce the release of the apoptotic proteins from the mitochondria. Viruses like Herpes simplex virus 1 deplete the host mitochondrial DNA and some, like human immunodeficiency virus, hijack the host mitochondrial proteins to function fully inside the host cell. All these processes involve the participation of cellular proteins, mitochondrial proteins, and virus specific proteins. This review will summarize the strategies employed by viruses to utilize cellular mitochondria for successful multiplication and production of progeny virus.
1. Introduction
1.1. Mitochondria
Mitochondria are cellular organelles found in the cytoplasm of almost all eukaryotic cells. One of their important functions is to produce and provide energy to the cell in the form of ATP, which help in proper maintenance of the cellular processes, thus making them indispensable for the cell. Besides acting as a powerhouse for the cell, they act as a common platform for the execution of a variety of cellular functions in normal or microorganism infected cells. Mitochondria have been implicated in aging [1, 2], apoptosis [3–7], the regulation of cell metabolism [4, 8], cell-cycle control [9–11], development of the cell [12–14], antiviral responses [15], signal transduction [16], and diseases [17–20].
Although all mitochondria have the same architecture, they vary greatly in shape and size. The mitochondria are composed of outer mitochondrial membrane, inner mitochondrial membrane, intermembrane space (space between outer and inner membrane), and matrix (space within inner mitochondrial membrane). The outer membrane is a smooth phospholipid bilayer, with different types of proteins imbedded in it [21]. The most important of them are the porins, which freely allow the transport (export and import) of the molecules (proteins, ions, nutrients, and ATP) less than 10 kDa across the membranes. The outer membrane surrounds the inner membrane creating an intermembrane space that contains molecules such as Cyt-C, SMAC/Diablo, and endonuclease G. It also acts as a buffer zone between the outer membrane and the inner membrane of mitochondria. The inner membrane is highly convoluted into structures called cristae, which increases the surface area of the membrane and are the seats of respiratory complexes. The inner membrane of mitochondria allows the free transport of oxygen and carbon dioxide. The movement of water through membranes is suggested to be controlled by aquaporins channel protein [22, 23] though a report suggested otherwise [24]. The matrix contains enzymes for the aerobic respiration, dissolved oxygen, water, carbon dioxide, and the recyclable intermediates that serve as energy shuttles and perform other functions.
Mitochondria contain a single 16 kb circular DNA genome, which codes for 13 proteins (mostly subunits of respiratory chains I, II, IV, and V), 22 mitochondrial tRNAs and 2 rRNAs [25, 26]. The mitochondrial genome is not enveloped (like nuclear envelop), contains few introns, and does not follow universal genetic code [27]. Although the majority of the mitochondrial proteins are encoded by nuclear DNA and imported into the mitochondria (reviewed by [21, 28–31]), mitochondria synthesize few proteins that are essential for their respiratory function [1, 27].
Proteins destined to mitochondria have either internally localized [28] or amino terminal localized [21] presequences known as mitochondria/matrix localization signals (MLS), which can be 10–80 amino acid long with predominantly positively charged amino acids. The combination of these presequences with adjacent regions determines the localization of a protein in respective mitochondrial compartments. The outer mitochondrial membrane contains two major translocators, namely, (a) the translocase of outer membrane (TOM) 40, which functions as an entry gate for most mitochondrial proteins with MLS and (b) sorting and assembly machinery (SAM) or translocase of β-barrel (TOB) protein, which is a specialized insertion machinery for beta-barrel membrane proteins [32]. Once proteins pass through the outer membrane, they are recruited by presequence translocase-associated motor (PAM) to the translocase of the inner mitochondrial membrane (TIM) 23 complexes, which mediates the import of proteins to the matrix. Finally, the presequences are cleaved in matrix and proteins are modified to their tertiary structure, and rendered functional [30].
1.2. Viruses
Viruses are acellular obligate intracellular microorganisms that infect the living cells/organisms and are the only exception to cell theory proposed by Schleiden and Schwann in 1838/1839 [33]. The viruses have an outer protein capsid and a nucleic acid core. Usually, the viral nucleic acids can be either DNA (double or single stranded) or RNA (+ or − sense single stranded or double stranded RNA). Some of the viruses are covered with an envelope embedded with glycoproteins. The viruses have long been associated with the living organisms, and it was in the later part of the century that their relationship with various cellular organelles was studied in detail. In order to survive and replicate in the cell, viruses need to take control of the various cellular organelles involved in defense and immune processes. They also require energy to replicate and escape from the cell. Once inside the host cell, they modulate various cellular signal pathways and organelles, including mitochondria, and use them for their own survival and replication. This review summarizes the functions of mitochondria and how viruses modulate them (Figure 1).
Figure 1.
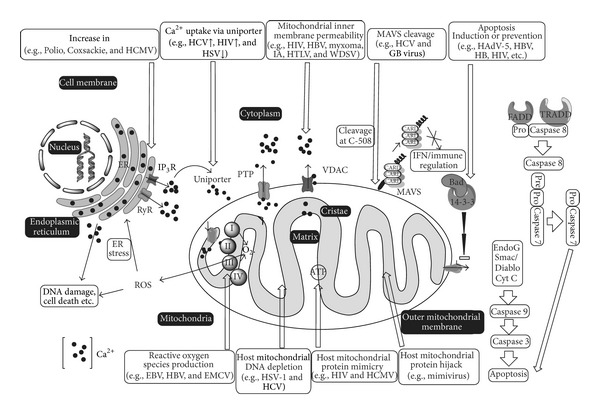
Schematic diagram of cell showing mitochondria, nucleus endoplasmic reticulum (ER) and cell membrane. iCa2+: intracellular calcium, FADD: Fas-associated protein with death domain, TRADD: tumor necrosis factor receptor type 1-associated death domain protein, PTP: permeability transition pore, VDAC: voltage-dependent anion channel, IP3R: inositol 1,4,5-trisphosphate receptor, RyR: ryanodine receptor, MAVS: mitochondrial antiviral signaling, I, II, III, and IV are complex I to IV of electron transport chain. O2 −: Superoxide radical, Bad, Bcl-2-associated death promoter, ROS: reactive oxygen species, IFN: interferon, HCMV: human cytomegalovirus, HIV: human immunodeficiency virus, HSV: herpes simplex virus, HBV: hepatitis B virus, HTLV: human T-lymphotropic virus, IA: influenza A virus, WDSV: Walleye dermal sarcoma virus, HCV: hepatitis C virus, HAdV: human adenovirus-5, EBV: Epstein-Barr virus, and EMCV: encephalomyocarditis virus.
2. Viruses Regulate Ca2+ Homeostasis in Host Cells
2.1. Ca2+ Homeostasis
Ca2+ is one of the most abundant and versatile elements in the cell and acts as a second messenger to regulate many cellular processes [34]. Earlier, outer membrane of mitochondria was thought to be permeable to Ca2+, but recent studies suggest that the outer membrane contains voltage-dependent anion channels (VDAC) having Ca2+ binding domains, which regulate the entry of Ca2+ into the mitochondrial intermembrane space [35–37]. The influx of Ca2+ through the inner membrane is regulated by the mitochondrial Ca2+ uniporter (MCU), which is a highly selective Ca2+ channel that regulates the Ca2+ uptake based on mitochondrial membrane potential (MMP). The net movement of charge due to Ca2+ uptake is directly proportional to the decrease of MMP [38]. A second mechanism that helps in Ca2+ movement across the mitochondria membrane is called “rapid mode” uptake mechanism (RaM) [39]. In this process, Ca2+ transports across the mitochondrial membrane by exchange with Na+, which in turn depends upon its exchange with H+ ion and thus MMP. This ion exchange across the mitochondrial membrane decreases MMP and is dependent on electron transport chain (ETC) for its maintenance. A third mechanism involves IP3R, a Ca2+ channel in endoplasmic reticulum. IP3R is connected to mitochondrial VDAC through a glucose regulating protein 75 (GRP75). This junction regulates/facilitates Ca2+ exchange from IP3R to VDAC [40].
Ca2+ efflux mechanism is regulated by the permeability transition pore (PTP). The PTP is assembled in the mitochondrial inner and outer membranes [41, 42], with Ca2+ binding sites on the matrix side of the inner membrane. The PTP regulates the mitochondrial Ca2+ release by a highly regulated “Flickering” mechanism that controls the opening and closing of the pore [43]. RaM works in sync with ryanodine receptor (RyR) isoform 1, which is another very important calcium release channel [44]. Both RyR and RaM regulate the phenomenon of excitation-metabolism coupling in which cytosolic Ca2+ induced contraction is matched by mitochondrial Ca2+ stimulation of ox-phos [45]. However, mitochondrial Ca2+ overload can result in prolonged opening of the pore leading to pathology [46]. Although Ca2+ is involved in the activation of many cellular processes including stimulation of the ATP synthase [47, 48], allosteric activation of Krebs cycle enzymes [49, 50], and the adenine nucleotide translocase (ANT) [51], the primary role of mitochondrial Ca2+ is in the stimulation of ox-phos [52–54]. Thus, the elevated mitochondrial Ca2+ results in up regulation of the entire ox-phos machinery, which then results in faster respiratory chain activity and higher ATP output, which can then meet the cellular ATP demand. Ca2+ also upregulates other mitochondrial functions including activation of N-acetylglutamine synthetase to generate N-acetylglutamine [55], potent allosteric activation of carbamoyl-phosphate synthetase, and the urea cycle [56]. Thus, any perturbation in mitochondrial or cytosolic Ca2+ homeostasis has profound implications for cell function. Moreover, mitochondrial Ca2+, particularly at high concentrations experienced in pathology appears to have several negative effects on mitochondrial functions [57].
2.2. Regulation by Viruses
A number of viruses alter the Ca2+regulatory activity of the cell for their survival. Herpes simplex type (HSV) 1 virus causes a gradual decline (65%) in mitochondrial Ca2+ uptake at 12 hrs lytic cycle [58], which helps in virus replication. Although mitochondrial Ca2+ uptake keeps fluctuating throughout the course of a measles virus infection of cells, the total amount of cellular Ca2+ remains the same [58] indicating the tight control that the virus exerts over the cellular processes during its life cycle.
The core protein of hepatitis C virus (HCV) targets mitochondria and increases Ca2+ [59, 60]. The NS5A protein of HCV causes alterations in Ca2+ homeostasis [61–63]. Both of these proteins may be responsible for the pathogenesis of liver disorders associated with HCV infection. Even in the cells coinfected with HCV and human immunodeficiency virus (HIV), these viruses enhance the MCU activity causing cellular stress and apoptosis [59, 64]. The p7 protein of HCV forms porin-like structures [65] and causes Ca2+ influx to cytoplasm from storage organelles [66]. These HCV proteins disturb the Ca2+ homeostasis at different stages of the infection and thus help to enhance the survival of the cell. Interestingly, interaction of protein X of hepatitis B virus (HBV) with VDAC causes the release of Ca2+ from storage organelles mitochondria/endoplasmic reticulum (ER)/golgi into the cytoplasmic compartment, which appears to help virus replication [67, 68].
The Nef protein of HIV interacts with IP3R [69] and induces an increase in cytosolic Ca2+ through promotion on T cell receptor-independent activation of the NFAT pathway [70]. Activated NFAT, in turn, causes the low-amplitude intracellular Ca2+ oscillation, promoting the viral gene transcription and replication [71].
Ca2+ is an important factor for different stages of rotavirus lifecycle and for stability to rotavirus virion [72]. The NSP4 protein of rotavirus increases the cytosolic Ca2+ concentration by activation of phospholipase C (PLC) and the resultant ER Ca2+ depletion through IP3R [73, 74]. This alteration in Ca2+ homeostasis has been attributed to an increase in the permeability of cell membrane [75]. A decrease in cellular Ca2+ concentrations toward the end of the life cycle has been reported to enable rotavirus release from the cell [76].
The 2BC protein of poliovirus increases the intracellular Ca2+ concentrations in the cells 4 hrs. After infection, which is necessary for viral gene expression [77, 78]. Toward the end of the virus life cycle, the release of Ca2+ from the lumen of ER through IP3R and RyR channels causes accumulation of Ca2+ in mitochondria through uniporter and VDAC resulting in mitochondrial dysfunction and apoptosis [79]. On the contrary, the 2B protein of Coxsackie virus decreases the membrane permeability by decreasing Ca2+ concentrations in infected cells [80, 81] due to its porin-like activity that results in Ca2+ efflux from the organelles. Reduced protein trafficking and low Ca2+ concentration in golgi and ER favor the formation of viral replication complexes, downregulate host antiviral immune response, and inhibit apoptosis [82, 83].
Enteroviruses orchestrate the apoptotic process during their life cycle to enhance its entry, survival, and release. The perturbation in cytoplasmic Ca2+ homeostasis at 2–4 hrs. postinfection coincides with the inhibition of the apoptotic response that can be attributed to decrease in cytotoxic levels of Ca2+ in the cell and the mitochondria. This also provides the virus with optimum conditions for the replication and protein synthesis. Finally, a decrease in mitochondrial and other storage organelles (ER and golgi) Ca2+ levels causes an increase in cytosolic Ca2+ concentration, leading to the formation of vesicles and cell death, thus assisting in virus release [81, 84, 85].
The pUL37 × 1 protein of human cytomegalovirus (HCMV) localizes to mitochondria [86] and causes the trafficking of Ca2+ from the ER to mitochondria at 4–6 hrs. After infection [87]. Active Ca2+ uptake by mitochondrion induces the production of ATP and other Ca2+ dependent enzymes accelerating virus replication, and a decrease in Ca2+ levels in the ER has antiapoptotic effects [88].
The 6.7K protein encoded by E3 region of HAdV-2 localizes to ER and helps maintain ER Ca2+ homeostasis in transfected cells, thus inhibiting apoptosis [89].
3. Viruses Cause Oxidative Stress in Host Cells
3.1. Electron Transport Chain
The mitochondrial respiratory chain is the main and most significant source of reactive oxygen species (RO) in the cell. Superoxide (O2 −•) is the primary ROS produced by mitochondria. In the normal state, there is little or no leakage of electrons between the complexes of the electron transport chain (ETC). However, during stress conditions, a small fraction of electrons leave complex III and reach complex IV [90]. This premature electron leakage to oxygen results in the formation of two types of superoxides, namely, O2 −, in its anionic form, and HO2 − in its protonated form.
Leakage of electrons takes place mainly from QO sites of complex III, which are situated immediately next to the intermembrane space resulting in the release of superoxides in either the matrix or the innermembrane space of the mitochondria [91–94]. About 25–75% of the total electron leak through Complex III could account for the net extramitochondrial superoxide release [95–97]. Thus, the main source of O2 −• in mitochondria is the ubisemiquinone radical intermediate (QH•) formed during the Q cycle at the QO site of complex III [98–100]. Complex I is also a source of ROS, but the mechanism of ROS generation is less clear. Recent reports suggest that glutathionylation [101] or PKA mediated phosphorylation [101–103] of complex I can elevate ROS generation. Backward flow of electron from complex I to complex II can also result in the production of ROS [99].
A variety of cellular defense mechanisms maintain the steady state concentration of these oxidants at nontoxic levels. This delicate balance between ROS generation and metabolism may be disrupted by various xenobiotics including viral proteins. The main reason for generation of ROS in virus-infected cells is to limit the virus multiplication. However, ROS also acts as a signal for various cellular pathways, and the virus utilizes the chaos generated inside the cell for its replication.
3.2. Viruses Induce Reactive Oxygen Species
A number of viruses cause oxidative stress to the host cells, which directly or indirectly helps them to survive. Human-Adenovirus- (HAdV-) 5 has been reported to induce the rupture of endosomal membrane upon infection resulting in the release of lysosomal cathepsins, which prompt the production of ROS. Cathepsins also induce the disruption of mitochondrial membrane leading to the release of ROS from mitochondria thus causing the oxidative stress [104].
The core protein of HCV causes oxidative stress in the cell and alters apoptotic pathways [64, 105–107]. The E1, E2, NS3, and core protein of HCV are potent ROS inducers and can cause host DNA damage independently [107, 108] or mediated by nitric oxide (NO) thus aiding in virus replication.
The ROS is generated during HIV infection [64, 109–111]. H2O2, an ROS generated during HIV infection strongly induces HIV long terminal repeat (LTR) via NF-kappa B activation. Impaired LTR activity ablates the LTR activation in response to ROS thus aiding in virus replication [112]. HIV also causes extensive cellular damage due to increased ROS production and decreased cytosolic antioxidant production [113]. Coinfection of HIV and HCV causes the hepatic fibrosis, the progression of which is regulated through the generation of ROS in an NF-κB dependent manner [113].
Epstein-Barr virus (EBV) causes increased oxidative stress in the host cells within 48 hrs. During the lytic cycle indicating the role of ROS in virus release [114]. Oxidative stress activates the EBV early gene BZLF-1, which causes the reactivation of EBV lytic cycle [114]. This has been proposed to play an important role in the pathogenesis of EBV-associated diseases including malignant transformations [115, 116].
Interestingly, HBV causes both an increase and a decrease in oxidative stress to enhance its survival in the host cells [117, 118]. HBV induces strong activation of Nrf2/ARE-regulated genes in vitro and in vivo through the activation of c-Raf and MEK by HBV protein X thus protecting the cells from HBV induced oxidative stress and promoting establishment of the infection [119]. The protein X of HBV also induces the ROS mediated upregulation of Forkhead box class O4 (Foxo4), enhancing resistance to oxidative stress-induced cell death [120]. However, reports also suggest that upon exposure to oxidative stress, HBV protein X accelerates the loss of Mcl-1 protein via caspase-3 cascade thus inducing pro apoptotic effects [118]. Coinfection of HCV also causes the genotoxic effects in peripheral blood lymphocytes due to increased oxidative damage and decreased MMP [121]. It is possible that contradictory functions of protein X of HBV cold occur at different stages of virus replication.
Encephalomyocarditis virus (EMCV) causes oxidative stress in the cells during infection damaging the neurons, which is an important process in the pathogenesis of EMCV infection [122].
4. Viruses Regulate Mitochondrial Membrane Potential in Host Cells
4.1. Mitochondrial Membrane Potential
Membrane potential (MP) is the difference in voltage or electrical potential between the interior and the exterior of a membrane. The membrane potential is generated either by electrical force (mutual attraction or repulsion between both positive or negative) and/or by diffusion of particles from high to low concentrations. The mitochondrial membrane potential (MMP) is an MP (≅ 180 mV) across the inner membrane of mitochondria, which provides energy for the synthesis of ATP. Movement of protons from complex I to V of electron transport chain (ETC) located in the inner mitochondrial membrane creates an electric potential across the inner membrane, which is important for proper maintenance of ETC and ATP production. Reported MMP values for mitochondria (in vivo) differ from species to species and from one organ to another depending upon the mitochondria function, protein composition, and the amount of oxidative phosphorylation activity required in that part of the body [43].
The voltage dependent anionic channels (VDACs) also known as mitochondrial porins form channels in the outer mitochondrial membranes and act as primary pathway for the movement of metabolites across the outer membrane [37, 96, 123–125]. In addition, a number of factors including oxidative stress, calcium overload, and ATP depletion induce the formation of nonspecific mitochondrial permeability transition pores (MPTP) in the inner mitochondrial membrane, which is also responsible for the maintenance of MMP [36, 37, 126]. The outer membrane VDACs, inner membrane adenine nucleotide translocase (ANT) [127], and cyclophilin D (CyP-D) in matrix are the structural elements of the mitochondrial permeability transition pore (MPTP).
When open, MPTP increases the permeability of the inner mitochondrial membrane to ions and solutes up to 1.5 kDa, which causes dissipation of the MMP and diffusion of solutes down their concentration gradients, by a process known as the permeability transition [128, 129]. The MPTP opening is followed by osmotic water flux, passive swelling, outer membrane rupture, and release of proapoptotic factors leading to the cell death [42, 130]. Because of the consequent depletion of ATP and Ca2+ deregulation, opening of the MPTP had been proposed to be a key element in determining the fate of the cell before a role for mitochondria in apoptosis was proposed [129].
The MMP can be altered by a variety of stimuli including sudden burst of ROS [43, 107], Ca2+ overload in the mitochondria or the cell [48, 57, 131], and/or by proteins of invading viruses [109, 132, 133]. In general, an increase or decrease in MMP is related to the induction or prevention of apoptosis, respectively. Prevention of apoptosis during early stages of virus infection is a usual strategy employed by viruses to prevent host immune response and promote their replication. On the contrary, induction of apoptosis during later stages of virus infection is a strategy used by viruses to release the progeny virions for dissemination to the surrounding cells.
4.2. Regulation by Viruses
Many viral proteins alter mitochondrial ion permeability and/or membrane potential for their survival in the cell. The p7, a hydrophobic integral membrane [134] viroprotein [135] of HCV, localizes to mitochondria [66] and controls membrane permeability to cations [66, 136] promoting cell survival for virus replication [135].
The R (Vpr) protein of HIV, a small accessory protein, localizes to the mitochondria, interacts with ANT, modulates MPTP, and induces loss of MMP promoting release of Cyto C [137] leading to cell death [138, 139]. The Tat protein of HIV also modulates MPTP leading to the accumulation of Tat in mitochondria and induction of loss of MMP resulting in caspase dependent apoptosis [140].
The M11L protein of myxoma poxvirus localizes to the mitochondria, interacts with the mitochondrial peripheral benzodiazepine receptor (PBR), and regulates MPTP [141] inhibiting MMP loss [142] and thus inhibiting induction of apoptosis during viral infection [143]. The FIL protein of vaccinia virus downregulates proapoptotic Bcl-2 family protein Bak and, inhibits the loss of the MMP and the release of Cyt-C [144, 145]. The crmA/Spi-2 protein of vaccinia virus, a caspase 8 inhibitor, modulates MPTP thus preventing apoptosis [146].
The PB1-F2 protein of influenza A viruses localizes to the mitochondria [147–150] and interacts with VDAC1 and ANT3 [151] resulting in decreased MMP, which induces the release of proapoptotic proteins causing cell death. Recent evidence shows that PB1-F2 is also able to form nonselective protein channel pores resulting in the alteration of mitochondrial morphology, dissipation of MMP, and cell death [150]. The M2 protein of influenza virus, a viroprotein, causes the alteration of mitochondrial morphology, dissipation of MMP, and cell death (reviewed by [135]).
The p13II, an accessory protein encoded by x-II ORF of human T-lymphotropic virus (HTLV), a new member of the viroprotein family [152], localizes to the mitochondria of infected cells and increases the MMP leading to apoptosis [153] and mitochondrial swelling [153–155].
The Orf C protein of Walleye dermal sarcoma virus (WDSV) localizes to the mitochondria [156] and induces perinuclear clustering of mitochondria and loss of MMP [156] leading to the release of proapoptotic factors thus causing apoptosis.
The 2B protein of Coxsackie virus decreases MMP by decreasing the Ca2+ concentrations in infected cells [80, 81].
5. Viruses Regulate Apoptosis
5.1. Apoptosis
During the coevolution of viruses with their hosts, viruses have developed several strategies to manipulate the host cell machinery for their survival, replication, and release from the cell. Viruses target the cellular apoptotic machinery at critical stages of viral replication to meet their ends [157, 158]. Depending upon the need, a virus may inhibit [159] or induce [160] apoptosis for the obvious purpose of replication and spread, respectively [158, 159]. Interference in mitochondrial function can cause either cell death due to deregulation of the Ca2+ signaling pathways and ATP depletion or apoptosis due to regulation of Bcl-2 family proteins. Apoptosis is a programmed cell death [161] characterized by membrane blebbing, condensation of the nucleus and cytoplasm, and endonucleosomal DNA cleavage. The process starts as soon as the cell senses physiological or stress stimuli, which disturbs the homeostasis of the cell [162, 163]. Apoptotic cell death can be considered as an innate response to limit the growth of microorganisms including viruses attacking the cell.
Two major pathways, namely, the extrinsic and the intrinsic are involved in triggering apoptosis [163, 164]. The extrinsic pathway is mediated by signaling through death receptors like tumor necrosis factor or Fas ligand receptor causing the assembly of death inducing signaling complex (DISC) with the recruitment of proteins like caspases leading to the mitochondrial membrane permeabilization. In the intrinsic pathway, the signals act directly on the mitochondria leading to mitochondrial membrane permeabilization before caspases are activated causing the release of Cyt-C [165, 166], which recruits APAF1 [167, 168] resulting in direct activation of caspase 9 [35, 169]. Both the extrinsic and the intrinsic processes congregate at the activation of downstream effector caspases, (i.e., caspase-3) [170] which is responsible for inducing the morphological changes observed in an apoptotic cell. In addition to Cyt-C, Smac/DIABLO as well as caspase independent death effectors inducing factor (AIF) and endonuclease G [171–173] acts as an activator of the caspase.
The B cell lymphoma- (Bcl-) 2 family of proteins tightly regulate the apoptotic events involving the mitochondria [174, 175]. More than 20 mammalian Bcl-2 family proteins have been described to date [176, 177]. They have been classified by the presence of Bcl-2 homology (BH) domains arranged in the order BH4-BH3-BH2-BH1 and the C-terminal hydrophobic transmembrane (TM) domain, which anchors them to the outer mitochondrial membrane [178]. The highly conserved BH1 and BH2 domains are responsible for antiapoptotic activity and multimerization of Bcl-2 family proteins. The BH3 domain is mainly responsible for proapoptotic activity and the less conserved BH4 domain is required for the antiapoptotic activities of Bcl-2 and Bcl-XL proteins [174, 178]. Most of the antiapoptotic proteins are multidomain proteins, which contain all four BH domains (BH1 to BH4) and a TM domain. In contrast, proapoptotic proteins are either multidomain proteins, which contain three BH domains (BH1 to BH3) or single domain proteins, which contain one domain (BH3) [158]. The Bcl-2 proteins regulate the MMP depending upon whether they belong to the pro- or antiapoptotic branch of the family, respectively. The MMP marks the dead end of apoptosis beyond which cells are destined to die [125, 166, 179–183].
5.2. Regulation by Viruses
Viruses encode homologs of Bcl-2 (vBcl-2) proteins, which can induce (pro-apototic) or prevent (antiapoptotic) apoptosis thus helping viruses to complete their life cycle in the host cells [117, 163, 175]. While the vBcl-2s and the cellular Bcl-2s share limited sequence homology, their secondary structures are predicted to be quite similar [158, 174, 184]. During primary infection, interplay between vBcl-2 and other proteins enhances the lifespan of the host cells resulting in efficient production of viral progeny and ultimately spread of infection to the new cells. It also favors viral persistence in the cells by enabling the latently infecting viruses to make the transition to productive infection. The pathways and strategies used by viruses to induce/inhibit apoptosis have been reviewed earlier [185].
Many viruses encode for the homologs of antiapoptotic Bcl-2 proteins, which preferentially localize to the mitochondria and may interact with the other proapoptotic Bax homologues. The E1B19K encoded by human-adenovirus- (HAdV-) 5 contains BH1 and BH3-like domains and blocks TNF-alpha-mediated death signaling by inhibiting a form of Bax that interrupts the caspase activation downstream of caspase-8 and upstream of caspase-9 [186, 187]. Like HAdV-5 E1B19K [186], some viruses encode Bcl-2 homologues lacking BH4 domain, which are thought to act by inhibiting proapoptotic members of Bcl-2 family proteins. The FPV309 protein encoded by fowl pox virus contains highly conserved BH1 and BH2-like domains, and a cryptic BH3 domain, interacts with Bax protein and inhibits apoptosis [188]. The A179L protein encoded by African swine fever virus (ASFV) contains BH1 and BH2 domains and, interacts with Bax-Bak proteins and inhibits apoptosis [189, 190]. The Bcl-2 homolog (vBcl-2) encoded by Herpesvirus saimiri (HVS) contains BH3 and BH4-like domains and interacts with Bax, thus stabilizing mitochondria against a variety of apoptotic stimuli preventing the cell death [191]. The E4 ORF encoded by equine Herpesvirus-3 contains BH1 and BH2 domains [192], which may interact with Bax and be essential for antiapoptotic activity [193].
Viruses also encode homologs of proapoptotic Bcl-2 proteins. The HBV encodes protein X, a vBcl-2 protein containing BH3, which localizes to the mitochondria and interacts with VDACs inducing the loss of the MMP leading to apoptosis [117, 121, 194, 195] or interacts with Hsp60 and induces apoptosis [196]. In contrast, another study revealed the protective effects of HB-X in response to proapoptotic stimuli (Fas, TNF, and serum withdrawal) but not from chemical apoptotic stimuli [197]. The protein X of HBV is known to stimulate NFκB [198, 199], SAPK [200, 201], and PI3K/PKB [202] to prevent apoptosis. It is possible that the diverse functions of HBV protein X occur at different times of virus replication cycle in the infected cells. The BALF1 protein encoded by EBV contains BH1 and BH4 domains [203], which interacts with the Bax-Bak proteins [192] and inhibits the antiapoptotic activity of the EBV BHRF1 and the Kaposi Sarcoma virus (KSV) Bcl-2 protein, both of which contain BH1 and BH2 domains [204] and interact with BH3 only proteins [205].
The effects of viral Bcl-2 homologues are thus apparently centered around mitochondria and include prevention or induction of MMP loss. The induction of MMP loss leads to the release of Cyto C and other proapoptotic signals into the cytosol and activation of downstream caspases leading to the cell death and dissemination of viruses to neighbouring cells for further infection.
Viruses encode pro/anti apoptotic proteins, which show no homology to Bcl-2 proteins [158]. The E6 protein of human papilloma virus (HPV) downregulates Bax signal upstream of mitochondria [206, 207] and prevents the release of Cyto C, AIF, and Omi, thus preventing apoptosis [208]. This E6 activity towards another Bcl2 family proapoptotic protein Bak is a key factor promoting the survival of HPV-infected cells, which in turn facilitates the completion of viral life cycle [207]. Enterovirus (EV) 71 induces conformational changes in Bax and increases its expression in cells following infection and induces the activation of caspases 3, 8, and PARP causing caspase dependent apoptosis [209]. On the contrary, Rubella viral capsid binds to Bax, forms oligoheteromers, and prevents the formation of pores on mitochondrial membrane thus preventing Bax induced apoptosis [210].
Viruses also encode proteins, which act as viral mitochondrial inhibitors of apoptosis (vMIA) thus protecting the cells. A splice variant of UL37 of HCMV acts as vMIA and protects the cells from apoptosis [211] thereby helping viruses to complete their replication cycle. It localizes to mitochondria and interacts with ANT [211] and Bax [212, 213]. HCMV vMIA has an N-terminal mitochondrial localization domain and a C-terminal antiapoptotic domain [211], which recruits Bax to mitochondria and prevents loss of MMP. It protects the cells against CD95 ligation [211] and oxidative stress-induced cell death [214, 215] and prevents mitochondrial fusion [216] thus promoting cell survival.
vMIA does not inhibit the apoptotic events upstream of mitochondria but can influence events like preservation of ATP generation, inhibition of Cyto C release, and caspase 9 activation, following induction of apoptosis. However, the exact mechanisms of the events around vMIA still remain a question.
6. Viruses Modulate Mitochondrial Antiviral Immunity
6.1. Mitochondrial Antiviral Immunity
Cells respond to virus attack by activating a variety of signal transduction pathways leading to the production of interferons [217], which limit or eliminate the invading virus. The presence of viruses inside the cell is first sensed by pattern recognition receptors (PRRs) that recognize the pathogen associated molecular patterns (PAMPs). PRRs include toll-like receptors (TLRs), nucleotide oligomerization domain (NOD) like receptors (NLRs), and retinoic acid-inducible gene I (RIG-I) like receptors (RLRs). Mitochondria have been associated with RLRs, which include retinoic acid-inducible gene I (RIG-I) [218] and melanoma differentiation-associated gene 5 (Mda-5) [219]. Both are cytoplasm-located RNA helicases that recognize dsRNA. The N-terminus of RIG-1 has caspase activation and recruitment domains (CARDs) whereas C-terminus has RNA helicase activity [218], which recognizes and binds to uncapped and unmodified RNA generated by viral polymerases in ATPase dependent manner. This causes conformational changes and exposes its CARD domains to bind and activate downstream effectors leading to the formation of enhanceosome [220] triggering NFκB production. RLRs have recently been reviewed in detail [221–223].
A CARD domain containing protein named mitochondrial antiviral signaling (MAVS) [15, 224], virus-induced signaling adaptor (VISA) [225], IFN-β promoter stimulator 1 (IPS-1) [226], or CARD adaptor inducing IFN-β (CARDIF) protein [227] acts downstream of the RIG-I. Besides the presence of N-terminal CARD domain, MAVS contains a proline-rich region and a C-terminal hydrophobic transmembrane (TM) region, which targets the protein to the mitochondrial outer membrane and is critical for its activity [15]. The TM region of the MAVS resembles the TM domains of many C-terminal tail-anchored proteins on the outer membrane of the mitochondria including Bcl-2 and Bcl-xL [15]. Recent reports indicate that MAVS has an important role in inducing the antiviral defenses in the cell. Overexpression of MAVS leads to the activation of NFκB and IRF-3, leading to the induction of type I interferon response, which is abrogated in the absence of MAVS [15] thus indicating the specific role of MAVS in inducing antiviral response. MAVS has also been shown to prevent apoptosis by its interaction with VDAC [228] and preventing the opening of MPTP.
6.2. Regulation by Viruses
Some viruses induce cleavage of MAVs from outer membranes of mitochondria [227, 229] thus greatly reducing their ability to induce interferon response. HCV persists in the host by lowering the host cell immune response including inhibiting the production of IFN-β by RIG-I pathway [230–232]. The NS3/4A protein of HCV colocalizes with mitochondrial MAVS [227, 229] leading to the cleavage of MAVS at amino acid 508. Since free form of the MAVS is not functional, the dislodging of MAV from the mitochondria inactivates MAVS [227] thus helping in paralyzing the host defense against HCV. Interestingly, another member of family Flaviviridae GB virus B shares 28% amino acid homology with HCV over the lengths of their open-reading frames [233]. The NS3/4A protein of GB virus also cleaves MAVS in a manner similar to HCV, thus effectively compromising the host immune response by preventing the production of interferons [234]. Other viruses like influenza A translocate RIG-I/MAVS components to the mitochondria of infected human primary macrophages and regulate the antiviral/apoptotic signals increasing the viral survivability [235].
7. Viruses Hijack Host Mitochondrial Proteins
Over the years, viruses have perfected different strategies to establish complex relationships with their host with the sole purpose of preserving their existence. One such strategy involves the hijacking of the host cell mitochondrial proteins. The p32, a mitochondria-associated cellular protein, is a member of a complex involved in the import of cytosolic proteins to the nucleus. Upon entry into the cell, adenovirus hijacks this protein and piggybacks it to transport its genome to the nucleus [236], thereby increasing its chances of survival and establishment in the host cell. During HIV-1 assembly, tRNALys iso-acceptors are selectively incorporated into virions, and tRNA3 Lys binds to HIV genome and is used as the primer for reverse transcription [237]. In humans, a single gene produces both cytoplasmic and mitochondrial Lys tRNA synthetases (LysRSs) by alternative splicing [238]. The mitochondrial LysRS is produced as a preprotein, which is transported into the mitochondria. The premitochondrial or mitochondrial LysRS is specifically packaged into HIV [239] and acts as a primer to initiate the replication of HIV-I RNA genome, which then binds to a site complementary to the 3′-end 18 nucleotides of tRNA3 Lys. It is proposed that HIV viral protein R (Vpr) alters the permeability of the mitochondria [138] leading to the release of premito- or mito-LysRS, which then interacts with Vpr [240] and gets packed into the progeny virions.
Viperin, an interferon inducible protein, is induced in the cells in response to viral infection [241]. This protein has been shown to prevent the release of influenza virus particles from the cells by trapping them in lipid rafts inside the cells thereby preventing its dissemination [242]. During infection, HCMV induces IFN independent expression of viperin, which interacts with HCMV encoded vMIA protein resulting in relocation of viperin from ER to mitochondria. In mitochondria, viperin interacts with mitochondrial tri-functional protein and decreases ATP generation by disrupting oxidation of fatty acids, which results in disrupting actin cytoskeleton of the cells and enhancing the viral infectivity [243].
8. Viruses Alter Intracellular Distribution of Mitochondria
Viruses alter the intracellular distribution of mitochondria either by concentrating the mitochondria near the viral factories to meet energy requirements during viral replication or by cordoning off the mitochondria within cytoplasm to prevent the release of mediators of apoptosis. The protein X of HBV causes microtubule mediated perinuclear clustering of the mitochondria by p38 mitogen-activated protein kinase (MAPK) mediated dynein activity [244]. HCV nonstructural protein 4A (NS4A) either alone or together with NS3, (in the form of the NS3/4A polyprotein) accumulates on mitochondria and changes their intracellular distribution [245]. HIV-1 infection causes clustering of the mitochondria in the infected cells [246]. Interestingly, ASFV causes the microtubule-mediated clustering of the mitochondria around virus factories in the cell providing energy for virus release [247]. Similar changes were observed in the chick embryo fibroblasts infected with frog virus 3, where degenerate mitochondria surrounding virus factories were found [248].
9. Viruses Mimic the Host Mitochondrial Proteins
Molecular mimicry is “the theoretical possibility that sequence similarities between foreign and self-peptides are sufficient to result in the cross-activation of autoreactive T or B cells by pathogen-derived peptides” [249, 250]. Since structure follows the function, viruses, during their coevolution with hosts have evolved to mimic the host proteins to meet their ends during progression of their life cycle inside the cell. Mimicking aids the viruses to gain access to host cellular machinery and greatly helps in their survival in the hostile host environment.
Mimivirus, a member of the newly created virus family Mimiviridae, encodes a eukaryotic mitochondria carrier protein (VMC-I) [251], which mimics the host cell's mitochondrial carrier protein and thus controls the mitochondrial transport machinery in infected cells. It helps to transport ADP, dADP, TTP, dTTP, and UTP in exchange for dATP, thus exploiting the host for energy requirements during replication of its A+T rich genome [251]. Besides VMC-I, mimivirus encodes several other proteins (L359, L572, R776, R596, R740, R824 L81, R151, R900, and L908) with putative mitochondria localization signals, which suggest that mimivirus has evolved a strategy to take over the host mitochondria and exploited its physiology to compensate for its energy requirements and biogenesis [251]. Viral Bcl-2 homologues (vBcl-2) are other groups of viral proteins that mimic the host cell Bcl-2s and have been described elsewhere in this review.
10. Viruses Cause Host Mitochondrial DNA Depletion
Mammalian mitochondria contain a small circular genome, which synthesizes enzymes for oxidative phosphorylation and mitochondrial RNAs (mtRNAs) [27]. To increase the chance of survival, some viruses appear to have adopted the strategy of damaging the host cell mitochondrial DNA. Since mitochondria act as a source of energy and play an important role in antiviral immunity as well, it is possible that damage to mitochondrial DNA may help in evading mitochondrial antiviral immune responses [252].
During productive infection of mammalian cells in vitro, HSV-1 induces the rapid and complete degradation of host mitochondrial DNA [252]. The UL12.5 protein of HSV-1 localizes to the mitochondria and induces DNA depletion in the absence of other viral gene products [252, 253]. The immediate early Zta protein of EBV interacts with mitochondrial single stranded DNA binding protein resulting in reduced mitochondrial DNA (mtDNA) replication and enhanced viral DNA replication [254]. HCV causes the reactive oxygen species and nitrous oxide mediated DNA damage in host mtDNA [107, 255]. Interestingly, depletion of mtDNA has also been observed in HIV/HCV coinfected humans [256].
11. Conclusions
Though progress has been made in understanding the interaction of viruses with mitochondria-mediated pathways, the pathways linking the detection of viral infection by PRRs (or exact mechanism by which PRRs recognize the PAMPs) and their link to mitochondria-mediated cell death remain poorly understood. Role of the mitochondria in immunity and viral mechanisms to evade them highlights the fact that even after billions of years of coevolution, the fight for the survival is still going on. Both the host and the viruses are evolving, finding new ways to survive. It may be interesting to note that mitochondria mediated apoptosis might be an evolutionary adaptation by which they might have effectively prevented the entry of other microorganisms trying to gain entry into the host cell and thus effectively establishing themselves as an integral part of the cell.
Acknowledgments
The authors thank Dr. Vikram Misra, Veterinary Microbiology, University of Saskatchewan, for his vision and advice. They thank Sherry Hueser for carefully proofreading the paper. The paper is published with the permission of Director VIDO as VIDO article no. 617. Suresh K. Tikoo is funded by grants from Natural Sciences and Engineering Research Council of Canada.
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Antignani A, Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Current Opinion in Cell Biology. 2006;18(6):685–689. doi: 10.1016/j.ceb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Human Molecular Genetics. 2005;14(2):R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 5.Gradzka I. Mechanisms and regulation of the programmed cell death. Postepy Biochemii. 2006;52(2):157–165. [PubMed] [Google Scholar]
- 6.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Current Biology. 2006;16(14):R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 7.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological Reviews. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 8.Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochimica et Biophysica Acta. 2006;1763(5-6):542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. The FEBS Letters. 2003;546(1):113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 10.Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular Cell. 2005;18(3):283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 11.Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Developmental Cell. 2005;9(6):843–854. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Bakeeva LE, Chentsov YS, Skulachev VP. Mitochondrial framework (reticulum mitochondriale) in rat diaphragm muscle. Biochimica et Biophysica Acta. 1978;501(3):349–369. doi: 10.1016/0005-2728(78)90104-4. [DOI] [PubMed] [Google Scholar]
- 13.Bakeeva LE, Chentsov YS, Shulachev VP. Intermitochondrial contacts in myocardiocytes. Journal of Molecular and Cellular Cardiology. 1983;15(7):413–420. doi: 10.1016/0022-2828(83)90261-4. [DOI] [PubMed] [Google Scholar]
- 14.Honda S, Hirose S. Stage-specific enhanced expression of mitochondrial fusion and fission factors during spermatogenesis in rat testis. Biochemical and Biophysical Research Communications. 2003;311(2):424–432. doi: 10.1016/j.bbrc.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Current Opinion in Cell Biology. 2003;15(6):706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annual Review of Neuroscience. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 18.van den Eeden SK, Tanner CM, Bernstein AL, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. The American Journal of Epidemiology. 2003;157(11):1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- 19.Martin LJ. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. Journal of Neuropathology and Experimental Neurology. 2006;65(12):1103–1110. doi: 10.1097/01.jnen.0000248541.05552.c4. [DOI] [PubMed] [Google Scholar]
- 20.McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease—its impact, etiology, and pathology. In: St John JC, editor. Current Topics in Developmental Biology. New York, NY, USA: Academic Press; 2007. pp. 113–155. [DOI] [PubMed] [Google Scholar]
- 21.Rapaport D. Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins. EMBO Reports. 2003;4(10):948–952. doi: 10.1038/sj.embor.embor937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amiry-Moghaddam M, Lindland H, Zelenin S, et al. Brain mitochondria contain aquaporin water channels: evidence for the expression of a short AQP9 isoform in the inner mitochondrial membrane. FASEB Journal. 2005;19(11):1459–1467. doi: 10.1096/fj.04-3515com. [DOI] [PubMed] [Google Scholar]
- 23.Calamita G, Ferri D, Gena P, et al. The inner mitochondrial membrane has aquaporin-8 water channels and is highly permeable to water. The Journal of Biological Chemistry. 2005;280(17):17149–17153. doi: 10.1074/jbc.C400595200. [DOI] [PubMed] [Google Scholar]
- 24.Yang B, Zhao D, Verkman AS. Evidence against functionally significant aquaporin expression in mitochondria. The Journal of Biological Chemistry. 2006;281(24):16202–16206. doi: 10.1074/jbc.M601864200. [DOI] [PubMed] [Google Scholar]
- 25.Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annual Review of Biochemistry. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 26.Shoubridge EA. The ABcs of mitochondrial transcription. Nature Genetics. 2002;31(3):227–228. doi: 10.1038/ng0702-227. [DOI] [PubMed] [Google Scholar]
- 27.Burger G, Gray MW, Lang BF. Mitochondrial genomes: anything goes. Trends in Genetics. 2003;19(12):709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annual Review of Biochemistry. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 29.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138(4):628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nature Reviews Molecular Cell Biology. 2010;11(9):655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 31.van der Laan M, Hutu DP, Rehling P. On the mechanism of preprotein import by the mitochondrial presequence translocase. Biochimica et Biophysica Acta. 2010;1803(6):732–739. doi: 10.1016/j.bbamcr.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Habib SJ, Waizenegger T, Lech M, Neupert W, Rapaport D. Assembly of the TOB complex of mitochondria. The Journal of Biological Chemistry. 2005;280(8):6434–6440. doi: 10.1074/jbc.M411510200. [DOI] [PubMed] [Google Scholar]
- 33.Schwann T. Microscopical researches into the accordance in the structure and growth of animals and plants. In: Schleiden MJ, editor. Contributions to Phytogenesis. London, UK: Sydenham Society; 1847. [Google Scholar]
- 34.Berridge MJ, Bootman MD, Lipp P. Calcium—a life and death signal. Nature. 1998;395(6703):645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 35.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 36.Chorna SV, Dosenko VI, Strutyns’ka NA, Vavilova HL, Sahach VF. Increased expression of voltage-dependent anion channel and adenine nucleotide translocase and the sensitivity of calcium-induced mitochondrial permeability transition opening pore in the old rat heart. Fiziolohichnyǐ Zhurnal. 2010;56(4):19–25. [PubMed] [Google Scholar]
- 37.Liu Y, Gao L, Xue Q, et al. Voltage-dependent anion channel involved in the mitochondrial calcium cycle of cell lines carrying the mitochondrial DNA A4263G mutation. Biochemical and Biophysical Research Communications. 2011;404(1):364–369. doi: 10.1016/j.bbrc.2010.11.124. [DOI] [PubMed] [Google Scholar]
- 38.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 39.Gunter TE, Gunter KK. Uptake of calcium by mitochondria: transport and possible function. IUBMB Life. 2002;52(3–5):197–204. doi: 10.1080/15216540152846000. [DOI] [PubMed] [Google Scholar]
- 40.Szabadkai G, Bianchi K, Várnai P, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. Journal of Cell Biology. 2006;175(6):901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halestrap AP. What is the mitochondrial permeability transition pore? Journal of Molecular and Cellular Cardiology. 2009;46(6):821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 42.Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochemical Society Transactions. 2010;38(4):841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 43.Hüttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, Doan JW. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. Journal of Bioenergetics and Biomembranes. 2008;40(5):445–456. doi: 10.1007/s10863-008-9169-3. [DOI] [PubMed] [Google Scholar]
- 44.Petronilli V, Persson B, Zoratti M, Rydstrom J, Azzone GF. Flow-force relationships during energy transfer between mitochondrial proton pumps. Biochimica et Biophysica Acta. 1991;1058(2):297–303. doi: 10.1016/s0005-2728(05)80250-6. [DOI] [PubMed] [Google Scholar]
- 45.Xia W, Shen Y, Xie H, Zheng S. Involvement of endoplasmic reticulum in hepatitis B virus replication. Virus Research. 2006;121(2):116–121. doi: 10.1016/j.virusres.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 46.Koopman WJH, Nijtmans LGJ, Dieteren CEJ, et al. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxidants and Redox Signaling. 2010;12(12):1431–1470. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 47.Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochodrial apoptosis-inducing factor. Nature. 1999;397(6718):441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 48.Balaban RS. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochimica et Biophysica Acta. 2009;1787(11):1334–1341. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wernette ME, Ochs RS, Lardy HA. Ca2+ stimulation of rat liver mitochondrial glycerophosphate dehydrogenase. The Journal of Biological Chemistry. 1981;256(24):12767–12771. [PubMed] [Google Scholar]
- 50.McCormack JG, Denton RM. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Developmental Neuroscience. 1993;15(3–5):165–173. doi: 10.1159/000111332. [DOI] [PubMed] [Google Scholar]
- 51.Mildaziene V, Baniene R, Nauciene Z, et al. Calcium indirectly increases the control exerted by the adenine nucleotide translocator over 2-oxoglutarate oxidation in rat heart mitochondria. Archives of Biochemistry and Biophysics. 1995;324(1):130–134. doi: 10.1006/abbi.1995.9918. [DOI] [PubMed] [Google Scholar]
- 52.Haworth RA, Hunter DR, Berkoff HA. Contracture in isolated adult rat heart cells. Role of Ca2+, ATP, and compartmentation. Circulation Research. 1981;49(5):1119–1128. doi: 10.1161/01.res.49.5.1119. [DOI] [PubMed] [Google Scholar]
- 53.Copello JA, Barg S, Sonnleitner A, et al. Differential activation by Ca2+, ATP and caffeine of cardiac and skeletal muscle ryanodine receptors after block by Mg2+ . Journal of Membrane Biology. 2002;187(1):51–64. doi: 10.1007/s00232-001-0150-x. [DOI] [PubMed] [Google Scholar]
- 54.Nasr P, Gursahani HI, Pang Z, et al. Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochemistry International. 2003;43(2):89–99. doi: 10.1016/s0197-0186(02)00229-2. [DOI] [PubMed] [Google Scholar]
- 55.Johnston JD, Brand MD. The mechanism of Ca2+ stimulation of citrulline and N-acetylglutamate synthesis by mitochondria. Biochimica et Biophysica Acta. 1990;1033(1):85–90. doi: 10.1016/0304-4165(90)90198-6. [DOI] [PubMed] [Google Scholar]
- 56.McGivan JD, Bradford NM, Mendes-Mourão J. The regulation of carbamoyl phosphate synthase activity in rat liver mitochondria. Biochemical Journal. 1976;154(2):415–421. doi: 10.1042/bj1540415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Annals of the New York Academy of Sciences. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 58.Lund K, Ziola B. Cell sonicates used in the analysis of how measles and herpes simplex type 1 virus infections influence Vero cell mitochondrial calcium uptake. Canadian Journal of Biochemistry and Cell Biology. 1985;63(11):1194–1197. doi: 10.1139/o85-149. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB Journal. 2007;21(10):2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- 60.Campbell RV, Yang Y, Wang T, et al. Effects of hepatitis C core protein on mitochondrial electron transport and production of reactive oxygen species. Methods in Enzymology. 2009;456:363–380. doi: 10.1016/S0076-6879(08)04420-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-κB. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(17):9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalamvoki M, Mavromara P. Calcium-dependent calpain proteases are implicated in processing of the hepatitis C virus NS5A protein. Journal of Virology. 2004;78(21):11865–11878. doi: 10.1128/JVI.78.21.11865-11878.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, et al. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. Journal of Hepatology. 2009;50(5):872–882. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 64.Baum MK, Sales S, Jayaweera DT, et al. Coinfection with hepatitis C virus, oxidative stress and antioxidant status in HIV-positive drug users in Miami. HIV Medicine. 2011;12(2):78–86. doi: 10.1111/j.1468-1293.2010.00849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook GA, Opella SJ. NMR studies of p7 protein from hepatitis C virus. European Biophysics Journal. 2010;39(7):1097–1104. doi: 10.1007/s00249-009-0533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Griffin SDC, Harvey R, Clarke DS, Barclay WS, Harris M, Rowlands DJ. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. Journal of General Virology. 2004;85(2):451–461. doi: 10.1099/vir.0.19634-0. [DOI] [PubMed] [Google Scholar]
- 67.Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294(5550):2376–2378. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- 68.Choi Y, Park SG, Yoo JH, Jung G. Calcium ions affect the hepatitis B virus core assembly. Virology. 2005;332(1):454–463. doi: 10.1016/j.virol.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 69.Foti M, Cartier L, Piguet V, et al. The HIV Nef protein alters Ca2+ signaling in myelomonocytic cells through SH3-mediated protein-protein interactions. The Journal of Biological Chemistry. 1999;274(49):34765–34772. doi: 10.1074/jbc.274.49.34765. [DOI] [PubMed] [Google Scholar]
- 70.Manninen A, Saksela K. HIV-1 Nef interacts with inositol trisphosphate receptor to activate calcium signaling in T cells. Journal of Experimental Medicine. 2002;195(8):1023–1032. doi: 10.1084/jem.20012039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity. 1997;6(3):235–244. doi: 10.1016/s1074-7613(00)80326-x. [DOI] [PubMed] [Google Scholar]
- 72.Ruiz MC, Cohen J, Michelangeli F. Role of Ca2+ in the replication and pathogenesis of rotavirus and other viral infections. Cell Calcium. 2000;28(3):137–149. doi: 10.1054/ceca.2000.0142. [DOI] [PubMed] [Google Scholar]
- 73.Tian P, Estes MK, Hu Y, Ball JM, Zeng CQ, Schilling WP. The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. Journal of Virology. 1995;69(9):5763–5772. doi: 10.1128/jvi.69.9.5763-5772.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Díaz Y, Chemello ME, Peña F, et al. Expression of nonstructural rotavirus protein NSP4 mimics Ca2+ homeostasis changes induced by rotavirus infection in cultured cells. Journal of Virology. 2008;82(22):11331–11343. doi: 10.1128/JVI.00577-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zambrano JL, Díaz Y, Peña F, et al. Silencing of rotavirus NSP4 or VP7 expression reduces alterations in Ca2+ homeostasis induced by infection of cultured cells. Journal of Virology. 2008;82(12):5815–5824. doi: 10.1128/JVI.02719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruiz MC, Aristimuño OC, Díaz Y, et al. Intracellular disassembly of infectious rotavirus particles by depletion of Ca2+ sequestered in the endoplasmic reticulum at the end of virus cycle. Virus Research. 2007;130(1-2):140–150. doi: 10.1016/j.virusres.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 77.Irurzun A, Arroyo J, Alvarez A, Carrasco L. Enhanced intracellular calcium concentration during poliovirus infection. Journal of Virology. 1995;69(8):5142–5146. doi: 10.1128/jvi.69.8.5142-5146.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aldabe R, Irurzun A, Carrasco L. Poliovirus protein 2BC increases cytosolic free calcium concentrations. Journal of Virology. 1997;71(8):6214–6217. doi: 10.1128/jvi.71.8.6214-6217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brisac C, Téoulé F, Autret A, et al. Calcium flux between the endoplasmic reticulum and mitochondrion contributes to poliovirus-induced apoptosis. Journal of Virology. 2010;84(23):12226–12235. doi: 10.1128/JVI.00994-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nieva JL, Agirre A, Nir S, Carrasco L. Mechanisms of membrane permeabilization by picornavirus 2B viroporin. The FEBS Letters. 2003;552(1):68–73. doi: 10.1016/s0014-5793(03)00852-4. [DOI] [PubMed] [Google Scholar]
- 81.van Kuppeveld FJM, de Jong AS, Melchers WJG, Willems PHGM. Enterovirus protein 2B po(u)res out the calcium: a viral strategy to survive? Trends in Microbiology. 2005;13(2):41–44. doi: 10.1016/j.tim.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 82.de Jong AS, Visch HJ, de Mattia F, et al. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. The Journal of Biological Chemistry. 2006;281(20):14144–14150. doi: 10.1074/jbc.M511766200. [DOI] [PubMed] [Google Scholar]
- 83.de Jong AS, de Mattia F, van Dommelen MM, et al. Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. Journal of Virology. 2008;82(7):3782–3790. doi: 10.1128/JVI.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Kuppeveld FJM, Hoenderop JGJ, Smeets RLL, et al. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO Journal. 1997;16(12):3519–3532. doi: 10.1093/emboj/16.12.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campanella M, de Jong AS, Lanke KWH, et al. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. The Journal of Biological Chemistry. 2004;279(18):18440–18450. doi: 10.1074/jbc.M309494200. [DOI] [PubMed] [Google Scholar]
- 86.Bozidis P, Williamson CD, Wong DS, Colberg-Poley AM. Trafficking of UL37 proteins into mitochondrion-associated membranes during permissive human cytomegalovirus infection. Journal of Virology. 2010;84(15):7898–7903. doi: 10.1128/JVI.00885-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharon-Friling R, Goodhouse J, Colberg-Poley AM, Shenk T. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(50):19117–19122. doi: 10.1073/pnas.0609353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO Journal. 2001;20(11):2690–2701. doi: 10.1093/emboj/20.11.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moise AR, Grant JR, Vitalis TZ, Jefferies WA. Adenovirus E3-6.7K maintains calcium homeostasis and prevents apoptosis and arachidonic acid release. Journal of Virology. 2002;76(4):1578–1587. doi: 10.1128/JVI.76.4.1578-1587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chan PH, Niizuma K, Endo H. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. Journal of Neurochemistry. 2009;109(1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry. 2002;41(25):7866–7874. doi: 10.1021/bi025581e. [DOI] [PubMed] [Google Scholar]
- 92.Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Q-o site of the cytochrome bc1 complex probed by superoxide production. Biochemistry. 2003;42(21):6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 93.Muller FL, Liu Y, van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. The Journal of Biological Chemistry. 2004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 94.Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11(4):473–485. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- 95.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. The Journal of Biological Chemistry. 2002;277(47):44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 96.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. The Journal of Biological Chemistry. 2003;278(8):5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 97.Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radical Biology and Medicine. 2003;35(8):938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 98.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxidants and Redox Signaling. 2006;8(9-10):1737–1744. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 99.Stowe DF, Camara AKS. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxidants and Redox Signaling. 2009;11(6):1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovascular Research. 2009;81(3):449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 101.Taylor JM, Quilty D, Banadyga L, Barry M. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. The Journal of Biological Chemistry. 2006;281(51):39728–39739. doi: 10.1074/jbc.M607465200. [DOI] [PubMed] [Google Scholar]
- 102.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raha S, Myint AT, Johnstone L, Robinson BH. Control of oxygen free radical formation from mitochondrial complex I: roles for protein kinase A and pyruvate dehydrogenase kinase. Free Radical Biology and Medicine. 2002;32(5):421–430. doi: 10.1016/s0891-5849(01)00816-4. [DOI] [PubMed] [Google Scholar]
- 104.McGuire KA, Barlan AU, Griffin TM, Wiethoff CM. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. Journal of Virology. 2011;85(20):10806–10813. doi: 10.1128/JVI.00675-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nishina S, Hino K, Korenaga M, et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008;134(1):226–238. doi: 10.1053/j.gastro.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 106.de Mochel NSR, Seronello S, Wang SH, et al. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology. 2010;52(1):47–59. doi: 10.1002/hep.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hsieh MJ, Hsieh YS, Chen TY, Chiou HL. Hepatitis C virus E2 protein induce reactive oxygen species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate cell line. Journal of Cellular Biochemistry. 2010;112(1):233–243. doi: 10.1002/jcb.22926. [DOI] [PubMed] [Google Scholar]
- 108.Machida K, Mcnamara G, Cheng KT, et al. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. Journal of Immunology. 2010;185(11):6985–6998. doi: 10.4049/jimmunol.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kruman II, Nath A, Mattson MP. HIV-1 protein tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Experimental Neurology. 1998;154(2):276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- 110.Baugh MA. HIV: reactive oxygen species, enveloped viruses and hyperbaric oxygen. Medical Hypotheses. 2000;55(3):232–238. doi: 10.1054/mehy.2000.1048. [DOI] [PubMed] [Google Scholar]
- 111.Gil L, Tarinas A, Hernandez D, et al. Altered oxidative stress indexes related to disease progression marker in human immunodeficiency virus infected patients with antiretroviral therapy. Biomedicine and Aging Pathology. 2011;1(1):8–15. doi: 10.1016/j.biopha.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 112.Pyo CW, Yang YL, Yoo NK, Choi SY. Reactive oxygen species activate HIV long terminal repeat via post-translational control of NF-κB. Biochemical and Biophysical Research Communications. 2008;376(1):180–185. doi: 10.1016/j.bbrc.2008.08.114. [DOI] [PubMed] [Google Scholar]
- 113.Lin W, Wu G, Li S, et al. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NF κB. The Journal of Biological Chemistry. 2011;286(4):2665–2674. doi: 10.1074/jbc.M110.168286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lassoued S, Gargouri B, El Feki AEF, Attia H, van Pelt J. Transcription of the epstein-barr virus lytic cycle activator BZLF-1 during oxidative stress induction. Biological Trace Element Research. 2010;137(1):13–22. doi: 10.1007/s12011-009-8555-y. [DOI] [PubMed] [Google Scholar]
- 115.Lassoued S, Ameur RB, Ayadi W, Gargouri B, Mansour RB, Attia H. Epstein-Barr virus induces an oxidative stress during the early stages of infection in B lymphocytes, epithelial, and lymphoblastoid cell lines. Molecular and Cellular Biochemistry. 2008;313(1-2):179–186. doi: 10.1007/s11010-008-9755-z. [DOI] [PubMed] [Google Scholar]
- 116.Gargouri B, van Pelt J, El Feki AEF, Attia H, Lassoued S. Induction of Epstein-Barr virus (EBV) lytic cycle in vitro causes oxidative stress in lymphoblastoid B cell lines. Molecular and Cellular Biochemistry. 2009;324(1-2):55–63. doi: 10.1007/s11010-008-9984-1. [DOI] [PubMed] [Google Scholar]
- 117.Kim YJ, Jung JK, Lee SY, Jang KL. Hepatitis B virus X protein overcomes stress-induced premature senescence by repressing p16INK4a expression via DNA methylation. Cancer Letters. 2010;288(2):226–235. doi: 10.1016/j.canlet.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 118.Hu L, Chen L, Yang G, et al. HBx sensitizes cells to oxidative stress-induced apoptosis by accelerating the loss of Mcl-1 protein via caspase-3 cascade. Molecular Cancer. 2011;10(article 43) doi: 10.1186/1476-4598-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schaedler S, Krause J, Himmelsbach K, et al. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. The Journal of Biological Chemistry. 2010;285(52):41074–41086. doi: 10.1074/jbc.M110.145862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Srisuttee R, Koh SS, Park EH, et al. Up-regulation of Foxo4 mediated by hepatitis B virus X protein confers resistance to oxidative stress-induced cell death. International Journal of Molecular Medicine. 2011;28(2):255–260. doi: 10.3892/ijmm.2011.699. [DOI] [PubMed] [Google Scholar]
- 121.Bhargava A, Khan S, Panwar H, et al. Occult hepatitis B virus infection with low viremia induces DNA damage, apoptosis and oxidative stress in peripheral blood lymphocytes. Virus Research. 2010;153(1):143–150. doi: 10.1016/j.virusres.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 122.Ano Y, Sakudo A, Kimata T, Uraki R, Sugiura K, Onodera T. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neuroscience Letters. 2010;469(1):39–43. doi: 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 123.Colombini M, Blachly-Dyson E, Forte M. VDAC, a channel in the outer mitochondrial membrane. Ion channels. 1996;4:169–202. doi: 10.1007/978-1-4899-1775-1_5. [DOI] [PubMed] [Google Scholar]
- 124.Forte M, Blachly-Dyson E, Colombini M. Structure and function of the yeast outer mitochondrial membrane channel, VDAC. Society of General Physiologists Series. 1996;51:145–154. [PubMed] [Google Scholar]
- 125.Villinger S, Briones R, Giller K, et al. Functional dynamics in the voltage-dependent anion channel. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(52):22546–22551. doi: 10.1073/pnas.1012310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426(6962):39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 127.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. The protective mechanisms. Archives of Biochemistry and Biophysics. 1979;195(2):453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 128.Garlid KD, Sun X, Paucek P, Woldegiorgis G. Mitochondrial cation transport systems. Methods in Enzymology. 1995;260:331–348. doi: 10.1016/0076-6879(95)60149-x. [DOI] [PubMed] [Google Scholar]
- 129.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiological Reviews. 1999;79(4):1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 130.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochemical Society Transactions. 2006;34(2):232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 131.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47(2):122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 132.Piccoli C, Scrima R, Quarato G, et al. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology. 2007;46(1):58–65. doi: 10.1002/hep.21679. [DOI] [PubMed] [Google Scholar]
- 133.Gac M, Bigda J, Vahlenkamp TW. Increased mitochondrial superoxide dismutase expression and lowered production of reactive oxygen species during rotavirus infection. Virology. 2010;404(2):293–303. doi: 10.1016/j.virol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 134.Carrère-Kremer S, Montpellier-Pala C, Cocquerel L, Wychowski C, Penin F, Dubuisson J. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. Journal of Virology. 2002;76(8):3720–3730. doi: 10.1128/JVI.76.8.3720-3730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gonzalez ME, Carrasco L. Viroporins. The FEBS Letters. 2003;552(1):28–34. doi: 10.1016/s0014-5793(03)00780-4. [DOI] [PubMed] [Google Scholar]
- 136.Pavlovic D, Neville DCA, Argaud O, et al. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):6104–6108. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Azuma A, Matsuo A, Suzuki T, Kurosawa T, Zhang X, Aida Y. Human immunodeficiency virus type 1 Vpr induces cell cycle arrest at the G1 phase and apoptosis via disruption of mitochondrial function in rodent cells. Microbes and Infection. 2006;8(3):670–679. doi: 10.1016/j.micinf.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 138.Jacotot E, Ravagnan L, Loeffler M, et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Journal of Experimental Medicine. 2000;191(1):33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deniaud A, Brenner C, Kroemer G. Mitochondrial membrane permeabilization by HIV-1 Vpr. Mitochondrion. 2004;4(2-3):223–233. doi: 10.1016/j.mito.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 140.Macho A, Calzado MA, Jiménez-Reina L, Ceballos E, León J, Muñoz E. Susceptibility of HIV-1-TAT transfected cells to undergo apoptosis. Biochemical mechanisms. Oncogene. 1999;18(52):7543–7551. doi: 10.1038/sj.onc.1203095. [DOI] [PubMed] [Google Scholar]
- 141.Everett H, Barry M, Sun X, et al. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. Journal of Experimental Medicine. 2002;196(9):1127–1139. doi: 10.1084/jem.20011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Everett H, Barry M, Lee SF, et al. M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. Journal of Experimental Medicine. 2000;191(9):1487–1498. doi: 10.1084/jem.191.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Macen JL, Graham KA, Lee SF, Schreiber M, Boshkov LK, McFadden G. Expression of the myxoma virus tumor necrosis factor receptor homologue and M11L genes is required to prevent virus-induced apoptosis in infected rabbit T lymphocytes. Virology. 1996;218(1):232–237. doi: 10.1006/viro.1996.0183. [DOI] [PubMed] [Google Scholar]
- 144.Wasilenko ST, Stewart TL, Meyers AFA, Barry M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(2):14345–14350. doi: 10.1073/pnas.2235583100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wasilenko ST, Banadyga L, Bond D, Barry M. The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. Journal of Virology. 2005;79(22):14031–14043. doi: 10.1128/JVI.79.22.14031-14043.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wasilenko ST, Meyers AFA, Helm KV, Barry M. Vaccinia virus infection disarms the mitochondrion-mediated pathway of the apoptotic cascade by modulating the permeability transition pore. Journal of Virology. 2001;75(23):11437–11448. doi: 10.1128/JVI.75.23.11437-11448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bruns K, Studtrucker N, Sharma A, et al. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. The Journal of Biological Chemistry. 2007;282(1):353–363. doi: 10.1074/jbc.M606494200. [DOI] [PubMed] [Google Scholar]
- 148.Chen W, Calvo PA, Malide D, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nature Medicine. 2001;7(12):1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 149.Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. Journal of Virology. 2003;77(13):7214–7224. doi: 10.1128/JVI.77.13.7214-7224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Henkel M, Mitzner D, Henklein P, et al. Proapoptotic influenza A virus protein PB1-F2 forms a nonselective ion channel. PLoS ONE. 2010;5(6) doi: 10.1371/journal.pone.0011112.e11112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Danishuddin M, Khan SN, Khan AU. Molecular interactions between mitochondrial membrane proteins and the C-terminal domain of PB1-F2: an in silico approach. Journal of Molecular Modeling. 2010;16(3):535–541. doi: 10.1007/s00894-009-0555-5. [DOI] [PubMed] [Google Scholar]
- 152.Silic-Benussi M, Marin O, Biasiotto R, D’Agostino DM, Ciminale V. Effects of human T-cell leukemia virus type 1 (HTLV-1) p13 on mitochondrial K+ permeability: a new member of the viroporin family? The FEBS Letters. 2010;584(10):2070–2075. doi: 10.1016/j.febslet.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ciminale V, Zotti L, D’Agostino DM, et al. Mitochondrial targeting of the p13(II) protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I) Oncogene. 1999;18(31):4505–4514. doi: 10.1038/sj.onc.1203047. [DOI] [PubMed] [Google Scholar]
- 154.Biasiotto R, Aguiari P, Rizzuto R, Pinton P, D’Agostino DM, Ciminale V. The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake. Biochimica et Biophysica Acta. 2010;1797(6-7):945–951. doi: 10.1016/j.bbabio.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 155.Silic-Benussi M, Cavallari I, Zorzan T, et al. Suppression of tumor growth and cell proliferation by p13II, a mitochondrial protein of human T cell leukemia virus type 1. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(17):6629–6634. doi: 10.1073/pnas.0305502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nudson WA, Rovnak J, Buechner M, Quackenbush SL. Walleye dermal sarcoma virus Orf C is targeted to the mitochondria. Journal of General Virology. 2003;84(2):375–381. doi: 10.1099/vir.0.18570-0. [DOI] [PubMed] [Google Scholar]
- 157.White E. Mechanisms of apoptosis regulation by viral oncogenes in infection and tumorigenesis. Cell Death and Differentiation. 2006;13(8):1371–1377. doi: 10.1038/sj.cdd.4401941. [DOI] [PubMed] [Google Scholar]
- 158.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathogens. 2008;4(5) doi: 10.1371/journal.ppat.1000018.e1000018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Benedict CA, Norris PS, Ware CF. To kill or be killed: viral evasion of apoptosis. Nature Immunology. 2002;3(11):1013–1018. doi: 10.1038/ni1102-1013. [DOI] [PubMed] [Google Scholar]
- 160.Hay S, Kannourakis G. A time to kill: viral manipulation of the cell death program. Journal of General Virology. 2002;83(7):1547–1564. doi: 10.1099/0022-1317-83-7-1547. [DOI] [PubMed] [Google Scholar]
- 161.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. The British Journal of Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gulbins E, Dreschers S, Bock J. Role of mitochondria in apoptosis. Experimental Physiology. 2003;88(1):85–90. doi: 10.1113/eph8802503. [DOI] [PubMed] [Google Scholar]
- 163.Borutaite V. Mitochondria as decision-makers in cell death. Environmental and Molecular Mutagenesis. 2010;51(5):406–416. doi: 10.1002/em.20564. [DOI] [PubMed] [Google Scholar]
- 164.Sanfilippo CM, Blaho JA. The facts of death. International Reviews of Immunology. 2003;22(5-6):327–340. doi: 10.1080/08830180305211. [DOI] [PubMed] [Google Scholar]
- 165.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 166.Castanier C, Arnoult D. Mitochondrial dynamics during apoptosis. Medecine/Sciences. 2010;26(10):830–835. doi: 10.1051/medsci/20102610830. [DOI] [PubMed] [Google Scholar]
- 167.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90(3):405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 168.Karbowski M. Mitochondria on guard: role of mitochondrial fusion and fission in the regulation of apoptosis. Advances in Experimental Medicine and Biology. 2010;687:131–142. doi: 10.1007/978-1-4419-6706-0_8. [DOI] [PubMed] [Google Scholar]
- 169.Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. The Journal of Biological Chemistry. 1999;274(8):5053–5060. doi: 10.1074/jbc.274.8.5053. [DOI] [PubMed] [Google Scholar]
- 170.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 171.Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nature Cell Biology. 2001;3(11):E255–E263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 172.Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. Journal of Cellular Physiology. 2002;192(2):131–137. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- 173.Ohta S. A multi-functional organelle mitochondrion is involved in cell death, proliferation and disease. Current Medicinal Chemistry. 2003;10(23):2485–2494. doi: 10.2174/0929867033456440. [DOI] [PubMed] [Google Scholar]
- 174.Danial NN, Gimenez-Cassina A, Tondera D. Homeostatic functions of BCL-2 proteins beyond apoptosis. Advances in Experimental Medicine and Biology. 2010;687:1–32. doi: 10.1007/978-1-4419-6706-0_1. [DOI] [PubMed] [Google Scholar]
- 175.Soriano ME, Scorrano L. The interplay between BCL-2 family proteins and mitochondrial morphology in the regulation of apoptosis. Advances in Experimental Medicine and Biology. 2010;687:97–114. doi: 10.1007/978-1-4419-6706-0_6. [DOI] [PubMed] [Google Scholar]
- 176.Krishna S, Low ICC, Pervaiz S. Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochemical Journal. 2011;435(3):545–551. doi: 10.1042/BJ20101996. [DOI] [PubMed] [Google Scholar]
- 177.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Current Opinion in Genetics and Development. 2011;21(1):12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochemical and Biophysical Research Communications. 2003;304(3):437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 179.Crompton M. Bax, Bid and the permeabilization of the mitochondrial outer membrane in apoptosis. Current Opinion in Cell Biology. 2000;12(4):414–419. doi: 10.1016/s0955-0674(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 180.Waterhouse NJ, Ricci JE, Green DR. And all of a sudden it’s over: mitochondrial outer-membrane permeabilization in apoptosis. Biochimie. 2002;84(2-3):113–121. doi: 10.1016/s0300-9084(02)01379-2. [DOI] [PubMed] [Google Scholar]
- 181.Belzacq AS, Vieira HLA, Verrier F, et al. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Research. 2003;63(2):541–546. [PubMed] [Google Scholar]
- 182.Zamzami N, Kroemer G. Apoptosis: mitochondrial membrane permeabilization—the (w)hole story? Current Biology. 2003;13(2):R71–R73. doi: 10.1016/s0960-9822(02)01433-1. [DOI] [PubMed] [Google Scholar]
- 183.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45(6):643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 184.Cuconati A, White E. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes and Development. 2002;16(19):2465–2478. doi: 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- 185.Thomson BJ. Viruses and apoptosis. International Journal of Experimental Pathology. 2001;82(2):65–76. doi: 10.1111/j.1365-2613.2001.iep0082-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Perez D, White E. TNF-α signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19K. Molecular Cell. 2000;6(1):53–63. [PubMed] [Google Scholar]
- 187.Pützer BM, Stiewe T, Parssanedjad K, Rega S, Esche H. E1A is sufficient by itself to induce apoptosis independent of p53 and other adenoviral gene products. Cell Death and Differentiation. 2000;7(2):177–188. doi: 10.1038/sj.cdd.4400618. [DOI] [PubMed] [Google Scholar]
- 188.Banadyga L, Gerig J, Stewart T, Barry M. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein bak. Journal of Virology. 2007;81(20):11032–11045. doi: 10.1128/JVI.00734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Brun A, Rivas C, Esteban M, Escribano JM, Alonso C. African swine fever virus gene A179L, a viral homologue of bcl-2, protects cells from programmed cell death. Virology. 1996;225(1):227–230. doi: 10.1006/viro.1996.0592. [DOI] [PubMed] [Google Scholar]
- 190.Revilla Y, Cebrián A, Baixerás E, Martínez C, Viñuela E, Salas ML. Inhibition of apoptosis by the African swine fever virus Bcl-2 homologue: role of the BH1 domain. Virology. 1997;228(2):400–404. doi: 10.1006/viro.1996.8395. [DOI] [PubMed] [Google Scholar]
- 191.Derfuss T, Fickenscher H, Kraft MS, et al. Antiapoptotic activity of the herpesvirus saimiri-encoded Bcl-2 homolog: stabilization of mitochondria and inhibition of caspase-3-like activity. Journal of Virology. 1998;72(7):5897–5904. doi: 10.1128/jvi.72.7.5897-5904.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Marshall WL, Yim C, Gustafson E, et al. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. Journal of Virology. 1999;73(6):5181–5185. doi: 10.1128/jvi.73.6.5181-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369(6478):321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 194.Rahmani Z, Huh KW, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. Journal of Virology. 2000;74(6):2840–2846. doi: 10.1128/jvi.74.6.2840-2846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lu YW, Chen WN. Human hepatitis B virus X protein induces apoptosis in HepG2 cells: role of BH3 domain. Biochemical and Biophysical Research Communications. 2005;338(3):1551–1556. doi: 10.1016/j.bbrc.2005.10.117. [DOI] [PubMed] [Google Scholar]
- 196.Tanaka Y, Kanai F, Kawakami T, et al. Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochemical and Biophysical Research Communications. 2004;318(2):461–469. doi: 10.1016/j.bbrc.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 197.Diao J, Khine AA, Sarangi F, et al. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. The Journal of Biological Chemistry. 2001;276(11):8328–8340. doi: 10.1074/jbc.M006026200. [DOI] [PubMed] [Google Scholar]
- 198.Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature. 1993;361(6414):742–745. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- 199.Su F, Schneider RJ. Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of rel-related proteins. Journal of Virology. 1996;70(7):4558–4566. doi: 10.1128/jvi.70.7.4558-4566.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Benn J, Su F, Doria M, Schneider RJ. Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. Journal of Virology. 1996;70(8):4978–4985. doi: 10.1128/jvi.70.8.4978-4985.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Henkler F, Lopes AR, Jones M, Koshy R. Erk-independent partial activation of AP-1 sites by the hepatitis B virus HBx protein. Journal of General Virology. 1998;79(11):2737–2742. doi: 10.1099/0022-1317-79-11-2737. [DOI] [PubMed] [Google Scholar]
- 202.Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL. Hepatitis b virus x protein inhibits transforming growth factor-β-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. The Journal of Biological Chemistry. 2000;275(33):25858–25864. doi: 10.1074/jbc.M003578200. [DOI] [PubMed] [Google Scholar]
- 203.Komano J, Sugiura M, Takada K. Epstein-barr virus contributes to the malignant phenotype and to apoptosis resistance in Burkitt’s lymphoma cell line Akata. Journal of Virology. 1998;72(11):9150–9156. doi: 10.1128/jvi.72.11.9150-9156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. Journal of Virology. 2002;76(5):2469–2479. doi: 10.1128/jvi.76.5.2469-2479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Flanagan AM, Letai A. BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death and Differentiation. 2008;15(3):580–588. doi: 10.1038/sj.cdd.4402292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Thomas M, Banks L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. Journal of General Virology. 1999;80(6):1513–1517. doi: 10.1099/0022-1317-80-6-1513. [DOI] [PubMed] [Google Scholar]
- 207.Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes and Development. 2000;14(23):3065–3073. doi: 10.1101/gad.182100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Leverrier S, Bergamaschi D, Ghali L, et al. Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis. 2007;12(3):549–560. doi: 10.1007/s10495-006-0004-1. [DOI] [PubMed] [Google Scholar]
- 209.Sun ZM, Xiao Y, Ren LL, Lei XB, Wang JW. Enterovirus 71 induces apoptosis in a Bax dependent manner. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2011;25(1):49–52. [PubMed] [Google Scholar]
- 210.Ilkow CS, Goping IS, Hobman TC. The rubella virus capsid is an anti-apoptotic protein that attenuates the pore-forming ability of Bax. PLoS Pathogens. 2011;7(2) doi: 10.1371/journal.ppat.1001291.e1001291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Goldmacher VS, Bartle LM, Skaletskaya A, et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(22):12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Arnoult D, Bartle LM, Skaletskaya A, et al. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(21):7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Poncet D, Larochette N, Pauleau A, et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. The Journal of Biological Chemistry. 2004;279(21):22605–22614. doi: 10.1074/jbc.M308408200. [DOI] [PubMed] [Google Scholar]
- 214.Vieira HLA, Belzacq AS, Haouzi D, et al. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene. 2001;20(32):4305–4316. doi: 10.1038/sj.onc.1204575. [DOI] [PubMed] [Google Scholar]
- 215.Boya P, Morales MC, Gonzalez-Polo R, et al. The chemopreventive agent N-(4-hydroxyphenyl)retinamide induces apoptosis through a mitochondrial pathway regulated by proteins from the Bcl-2 family. Oncogene. 2003;22(40):6220–6230. doi: 10.1038/sj.onc.1206827. [DOI] [PubMed] [Google Scholar]
- 216.McCormick AL, Smith VL, Chow D, Mocarski ES. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. Journal of Virology. 2003;77(1):631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Katze MG, He Y, Gale M., Jr. Viruses and interferon: a fight for supremacy. Nature Reviews Immunology. 2002;2(9):675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 218.Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunology. 2004;5(7):730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 219.Andrejeva J, Childs KS, Young DF, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-β promoter. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(49):17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Maniatis T, Falvo JV, Kim TH, et al. Structure and function of the interferon-β enhanceosome. Cold Spring Harbor Symposia on Quantitative Biology. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- 221.Scott I. The role of mitochondria in the mammalian antiviral defense system. Mitochondrion. 2010;10(4):316–320. doi: 10.1016/j.mito.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Castanier C, Arnoult D. Mitochondrial localization of viral proteins as a means to subvert host defense. Biochimica et Biophysica Acta. 2011;1813(4):575–583. doi: 10.1016/j.bbamcr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 223.Wang C, Liu X, Wei B. Mitochondrion: an emerging platform critical for host antiviral signaling. Expert Opinion on Therapeutic Targets. 2011;15(5):647–665. doi: 10.1517/14728222.2011.561321. [DOI] [PubMed] [Google Scholar]
- 224.Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Research. 2006;16(2):141–147. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- 225.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-β signaling. Molecular Cell. 2005;19(6):727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 226.Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nature Immunology. 2005;6(10):981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 227.Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 228.Xu Y, Zhong H, Shi W. MAVS protects cells from apoptosis by negatively regulating VDAC1. Molecular and Cellular Biochemistry. 2010;375(1-2):p. 219. doi: 10.1007/s11010-010-0658-4. [DOI] [PubMed] [Google Scholar]
- 229.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(49):17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300(5622):1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 231.Breiman A, Grandvaux N, Lin R, et al. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKε . Journal of Virology. 2005;79(7):3969–3978. doi: 10.1128/JVI.79.7.3969-3978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Foy E, Li K, Sumpter R, Jr., et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(8):2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Beames B, Chavez D, Lanford RE. GB virus B as a model for hepatitis C virus. ILAR Journal. 2001;42(2):152–160. doi: 10.1093/ilar.42.2.152. [DOI] [PubMed] [Google Scholar]
- 234.Chen Z, Benureau Y, Rijnbrand R, et al. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. Journal of Virology. 2007;81(2):964–976. doi: 10.1128/JVI.02076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Öhman T, Rintahaka J, Kalkkinen N, Matikainen S, Nyman TA. Actin and RIG-I/MAVS signaling components translocate to mitochondria upon influenza a virus infection of human primary macrophages. Journal of Immunology. 2009;182(9):5682–5692. doi: 10.4049/jimmunol.0803093. [DOI] [PubMed] [Google Scholar]
- 236.Matthews DA, Russell WC. Adenovirus core protein V interacts with p32—a protein which is associated with both the mitochondria and the nucleus. Journal of General Virology. 1998;79(7):1677–1685. doi: 10.1099/0022-1317-79-7-1677. [DOI] [PubMed] [Google Scholar]
- 237.Cen S, Khorchid A, Javanbakht H, et al. Incorporation of lysyl-tRNA synthetase into human immunodeficiency virus type 1. Journal of Virology. 2001;75(11):5043–5048. doi: 10.1128/JVI.75.11.5043-5048.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Tolkunova E, Park H, Xia J, King MP, Davidson E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual: alternative splicing of the primary transcript. The Journal of Biological Chemistry. 2000;275(45):35063–35069. doi: 10.1074/jbc.M006265200. [DOI] [PubMed] [Google Scholar]
- 239.Kaminska M, Shalak V, Francin M, Mirande M. Viral hijacking of mitochondrial lysyl-tRNA synthetase. Journal of Virology. 2007;81(1):68–73. doi: 10.1128/JVI.01267-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Stark LA, Hay RT. Human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) interacts with Lys-tRNA synthetase: implications for priming of HIV-1 reverse transcription. Journal of Virology. 1998;72(4):3037–3044. doi: 10.1128/jvi.72.4.3037-3044.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Qiu LQ, Cresswell P, Chin KC. Viperin is required for optimal Th2 responses and T-cell receptor-mediated activation of NF-κB and AP-1. Blood. 2009;113(15):3520–3529. doi: 10.1182/blood-2008-07-171942. [DOI] [PubMed] [Google Scholar]
- 242.Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host and Microbe. 2007;2(2):96–105. doi: 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 243.Seo JY, Yaneva R, Hinson ER, Cresswell P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science. 2011;332(6033):1093–1097. doi: 10.1126/science.1202007. [DOI] [PubMed] [Google Scholar]
- 244.Kim S, Kim HY, Lee S, et al. Hepatitis B virus X protein induces perinuclear mitochondrial clustering in microtubule- and dynein-dependent manners. Journal of Virology. 2007;81(4):1714–1726. doi: 10.1128/JVI.01863-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Nomura-Takigawa Y, Nagano-Fujii M, Deng L, et al. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. Journal of General Virology. 2006;87(7):1935–1945. doi: 10.1099/vir.0.81701-0. [DOI] [PubMed] [Google Scholar]
- 246.Radovanović JS, Todorović V, Boričić I, Janković-Hladni M, Korać A. Comparative ultrastructural studies on mitochondrial pathology in the liver of AIDS patients: clusters of mitochondria, protuberances, “minimitochondria,” vacuoles, and virus-like particles. Ultrastructural Pathology. 1999;23(1):19–24. doi: 10.1080/019131299281798. [DOI] [PubMed] [Google Scholar]
- 247.Rojo G, Chamorro M, Salas ML, Vinuela E, Cuezva JM, Salas J. Migration of mitochondria to viral assembly sites in African swine fever virus-infected cells. Journal of Virology. 1998;72(9):7583–7588. doi: 10.1128/jvi.72.9.7583-7588.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kelly DC. Frog virus 3 replication: electron microscope observations on the sequence of infection in chick embryo fibroblasts. Journal of General Virology. 1975;26(1):71–86. doi: 10.1099/0022-1317-26-1-71. [DOI] [PubMed] [Google Scholar]
- 249.Fujinami RS, Oldstone MBA. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230(4729):1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 250.Kohm AP, Fuller KG, Miller SD. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. Trends in Microbiology. 2003;11(3):101–105. doi: 10.1016/s0966-842x(03)00006-4. [DOI] [PubMed] [Google Scholar]
- 251.Monné M, Robinson AJ, Boes C, Harbour ME, Fearnley IM, Kunji ERS. The mimivirus genome encodes a mitochondrial carrier that transports dATP and dTTP. Journal of Virology. 2007;81(7):3181–3186. doi: 10.1128/JVI.02386-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Reports. 2007;8(2):188–193. doi: 10.1038/sj.embor.7400878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Corcoran JA, Saffran HA, Duguay BA, Smiley JR. Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. Journal of Virology. 2009;83(6):2601–2610. doi: 10.1128/JVI.02087-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wiedmer A, Wang P, Zhou J, et al. Epstein-Barr virus immediate-early protein Zta co-opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. Journal of Virology. 2008;82(9):4647–4655. doi: 10.1128/JVI.02198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MMC. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STATS activation. Journal of Virology. 2006;80(14):7199–7207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.de Mendoza C, Martin-Carbonero L, Barreiro P, et al. Mitochondrial DNA depletion in HIV-infected patients with chronic hepatitis C and effect of pegylated interferon plus ribavirin therapy. AIDS. 2007;21(5):583–588. doi: 10.1097/QAD.0b013e32805e8742. [DOI] [PubMed] [Google Scholar]