

Abstract
H. pylori infection is highly prevalent in Chile (73%). Usually a minority of infected patients develops complications such as ulcers and gastric cancer that have been associated with the presence of virulence factors (cagA, vacA) and host T helper response (Th1/Th2). Our aim was to evaluate the relationship between strain virulence and host immune response, using a multiple regression approach for the development of a model based on data collected from H. pylori infected patients in Chile. We analyzed levels of selected cytokines determined by ELISA (IL-12, IL-10, IFN-γ and IL-4) and the presence of cagA and vacA alleles polymorphisms determined by PCR in antral biopsies of 41 patients referred to endoscopy. By multiple regression analysis we established a correlation between bacterial and host factors using clinical outcome (gastritis and duodenal ulcer) as dependent variables. The selected model was described by: clinical outcome = 0.867491 (cagA) + 0.0131847 (IL-12/IL-10) + 0.0103503 (IFN-γ/IL-4) and it was able to explain over 90% of clinical outcomes observations (R2=96.4). This model considers that clinical outcomes are better explained by the interaction of host immune factors and strain virulence as a complex and interdependent mechanism.
Keywords: H. pylori, virulence factors, cytokines, gastroduodenal ulcer
1. Introduction
Helicobacter pylori, a Gram negative bacterium that colonizes gastric epithelium of the human stomach, has been associated to the development of chronic gastritis. In most patients (80%), H. pylori does not cause clinical symptoms and the infection can persist for lifetime without further problems. A minority of the infected patients will develop gastroduodenal disease such as peptic ulcer. Furthermore, a small percentage of infected patients will develop distal gastric cancer, depending on a variety of factors, including strain virulence and host immune responses [1], which is particularly important in Chile, a country with 73% of its population infected [2] and a GC mortality of 19/100.000 habitants [3].
Significant effort has been made to evaluate specific virulence factors present in a subset of strains which has been associated with more severe disease. Strains harboring a functional CagA-PAI and a VacA s1/m1 genotype have been detected at higher frequency in patients with duodenal ulcer, atrophic gastritis and gastric cancer compared with strains lacking this virulence factors [4]. VacA is a 95 kDa secreted cytotoxin which induces apoptosis and is also involved in immunomodulation and colonization [5]. CagA, a 128 kDa immunodominant antigen, encoded by a chromosomal pathogenicity island (Cag-PAI), which also containes genes coding for a type IV secretion system that is able to translocate CagA to the cytoplasm of the host cell, were it is phosphorylated at specific tyrosine residues by a Src Kinases family and activates SHP-2 tyrosine phosphatase which interferes with intracellular signaling leading to morphological as well as functional changes in epithelial cells [4].
H. pylori binds to the gastric epithelial cells through adhesins and persist in its niche for decades [6]. In strains carrying the Cag-PAI type IV secretion apparatus, translocation of CagA into epithelial cells are allowed eliciting the release of Interleukin (IL)-8 and others chemoquines by these cells. Chemoquines lead the recruitment of mononuclear and polymorphonuclear cells (PMNs), initiating the characteristic inflammation of H. pylori infected patients [7–8]. In addition, CagA induces disruption of tight junctions facilitating VacA-mediated disruption of epithelial barrier inducing apoptosis. TNF-α and other pro-inflammatory cytokines produced by local macrophages mediate apoptosis and may also lead to disruption of epithelial cells [9].
Although H. pylori itself can induce mucosal injury, the local immune response mechanisms have been involved in the pathogenesis of the disease. A large number of studies in mouse and human models have shown that a T lymphocyte helper (Th) type 1 response characterized by an up regulation of IFN-γ is associated with severe pathology outcomes such as ulcers [10]. A protective role has been assigned to the presence of Treg and Th2 type cells that balance some of the effects of Th1 bias. In athymic mice which have been transfuse with CD4+, the absence of CD4+CD25+ cells produces severe inflammation and elevated IFN-γ secretion [11]. In opposition, the presence of Th2 cytokine type IL-4 is associated with lower levels of gastric inflammation [12].
H. pylori is a relevant health problem among developing countries and few data concerning correlation between bacterial virulence factors and clinical outcomes is known in South America, particularly in Chile, which is a unique site for these studies, comparing outcomes and bacterial factors in host living in different regions because differences of vacA genotypes distribution in H. pylori along the country [13].
Because universal eradication therapy is not feasible and H. pylori vaccine it is not available at this point, it is of the utmost urgency to acquire a better understanding of the role of some virulence factors in the infection caused by this pathogen and the identification of mechanisms that regulate the immunological balance between host and H. pylori and their interactions to improve diagnostic and therapeutic modalities in our country. The aim of this study was to evaluate how selected strain virulence factors and selected host immune response factors relate using a preliminary model based on data collected from infected patients living in different Chilean cities.
2. Materials and methods
2.1 Collection of clinical specimens
Twenty patients from Iquique (IQ), five from Santiago (SA), five from Temuco (TE) and fifteen from Punta Arenas (PA) were enrolled in this study with an age (mean ± SD) of 27.7 ± 18.9 years. Criteria for patient inclusion included symptoms suggestive of peptic disease (nocturnal or burning abdominal pain, chronic vomiting or hematemesis) or a history of recurrent abdominal pain plus a first-degree relative with an endoscopically proven diagnosis of peptic ulcer disease. Exclusion criteria were hemodinamically unstable patients and recent therapy with antibiotics (1 month). Recluted patients were in the pediatric as well as in the adult age range. A detailed clinical history was obtained that included the characteristics of the abdominal pain as well as other clinical manifestations, family history of peptic ulcer disease, and recent therapy with non-steroidal anti-inflammatory drugs, bismuth compounds or acid suppressing drugs. H. pylori clinical strains were isolated from gastric biopsies obtained from patients who underwent upper gastrointestinal endoscopy for medical indication after informed consent. For culture purposes biopsies were transported to Santiago in BHI glycerol medium, for ELISA purposes biopsies were transported as frozen samples in dry ice and immediately suspended in normal saline (750 μl). Samples were manually grinded with a polished conical glass that fits in Eppendorf tubes.
A patient was considered infected if the rapid urease test (HE-PY test, Bios Chile, Santiago) was positive and histological analyses of at least 1 out of 2 biopsies revealed the presence of Gram-negative rods (Warthin-Starry silver stain or H&E stain). We confirmed this assignment by isolation of colonies. Four (8.9%) of the patients were considered H. pylori negative (control group), 31 (68.9%) had histological chronic gastritis and 10 (22.2%) had duodenal ulcer.
2.2 Growth conditions and identification of H. pylori strains
H. pylori clinical strains were isolated from biopsies and colonies were grown in a microaerophillic atmosphere (5% CO2) for 3–4 days on tryptic soy or Brucella agar plates containing 5% fresh horse blood, 5 mg vancomycin, 2.5 mg cefsulodin, 2.5 mg trimethoprime lactate, and 2.5 mg amphotericin B per 100 ml of medium. Single colonies were transferred to plates with the same medium but without horse blood. Morphologic characteristics were observed in both media. H. pylori were further identified based on biochemical tests for catalase, oxidase and urease [14].
2.3 Isolation of H. pylori chromosomal DNA
Cells coming from half of agar plate were collected and transferred to an Eppendorf tube using a sterile loop and processed as described by Owen and Bickley, with CTAB as detergent for cell lysis to obtain chromosomal DNA [15].
2.4 PCR reactions, primers, cloning and sequencing
PCR was used to amplify the cagA and vacA gene. The primers were designed according to the 26695 strain [16] and the Chilean strain CHCTX-1[17]. The primers used for cagA amplification are shown in table 1. The A17BN1 and Cag6 primers were used to amplify from the beginning of A17 central region of cagA fragment up the 3′ (C- terminus) of the glr gene (glutamate racemase gene). The A17BN1 and Cag7 primers were used to amplify from the beginning of the A17 fragment to 3′ end of cagA gene. Finally, to ensure the presence of cagA gene, the region A17 was amplified by PCR using the primers A17BN1 and A17BH2 [18]. The PCR mixture (20 ul) consisted of 15–20 ng of chromosomal DNA, 0.5uM each primer, 0.2mM each dNTP, 50mM MgCl2, and 0.5 Units of Taq DNA polymerase (Fermentas). The cycle conditions were 1 min at 94°C; 2 min at 50°C; and 3 min at 72°C for 30 cycles. A final elongation step of 5 min at 72°C was added. Amplifications were done in a PTC-100 MJ Research apparatus. As a PCR positive control, a 400 bp fragment of tsaA gene (alkyl hydroperoxide reductase) from H. pylori was amplified under the same conditions described for cagA amplification, using the AG261 and AG26M primers (Table 1) [19].
Table 1.
Primers used in PCR reactions for cagA, vacA and tsaΔ amplifications
| Name | Sequence 5′➝3′ |
|---|---|
| A17BN1 | agatctcatatgaaaaatggcaaaaataaggatttcagcaag |
| A17 BH2 | ggatccaagctctattattctgataccgcttgattgagattgtc |
| CagA5 | taaggagaaacaatgactaacgaa |
| CagA6 | atgaaaataggcgtttttgatagcggt |
| CagA7 | ttaagatttttggaaaccaccttttgt |
| A6261* | tgcggatccatatgttagttacaaaacttgcccccgat |
| A62M* | taaataagcttctctcaaagcgatcgcttcttcaaacagcacatc |
| VA3F** | ggtcaaaatgcggtcatgg |
| VA3R | ccattggtacctgtagaaac |
| VA4F** | ggagccccaggaaacattg |
| VA4R | cataactagcgccttgcac |
| VA1F** | atggaaatacaacaaacacac |
| VA1R | ctgcttgaatgcgccaaac |
| VA1F** | atggaaatacaacaaacacac |
| VA1R | ctgcttgaatgcgccaaac |
| SS1F** | gtcagcatcacaccgcaac |
| VA1R | ctgcttgaatgcgccaaac |
| SS3F** | agcgccataccgcaagag |
| VA1R | ctgcttgaatgcgccaaac |
| SS2F** | gctaacacgccaaatgatcc |
| VA1R | ctgcttgaatgcgccaaac |
Primers used for amplification of the H. pylori tsaA gene.
Primers pairs used for amplification of different regions of H. pylori vacA gene
Primers and reactions for the vacA amplifications were done according to the conditions established by Atherton et al [20]. Cloning of s1a, s1b, s2, m1 and m2 PCR fragments from the isolates were done by ligation into pGEM-T easy vector (Promega) and electroporation in E. coli strain DH5α plated on Luria agar containing 50μg/ml of Ampicilin. The sequences were done in ABI Prims-3100 genetic analyzer (Apllied Biosyztems). The sequences of the fragments were published in the service of the National Library of Medicine and the National Institute of Health (pubmed central) in a previous work done by our group [13]. Campylobacter yeyuni ATCC 29428 was used as DNA source in negative control samples and the strains ATCC 26695, ATCC 43504, J99, CHCTX-1 [17], 8823 and 8822 were used as positive control of vacA PCR amplification.
2.5 Cytokine determinations assays
For cytokine determination, the remaining antral biopsy specimens were homogenized with a tissue tearor (OMNI Th international) separately in 750 μl of normal saline. Supernatants obtained by centrifugation in a mini Eppendorf centrifuge (12,000 g for 5 minutes at 4°C) were frozen at −20°C in sterile vials (Sardstest) until used for an enzyme-linked immunosorbent assay (ELISA). Protein was measured using a modified bicinchoninic acid method (Pierce, Rockford, IL) and the total protein concentration in biopsy homogenates was expressed as mg/ml. The range of detection was between 0.02 and 2 mg/ml. IL-12, IL-10, IFN-γ and IL-4, were measured by ELISA (BD Biosciences Pharmingen, San Diego, CA) on supernatants of homogenates, as recommended by the manufacturer using recombinant human cytokines as positive controls for the development of standard curves. These assays demonstrated no measurable cross-reactivity to other cytokines and the limits of detection were 4.0 pg/ml for IL-12, 2.0 pg/ml for IL-10, 1.0 pg/ml for IFN-γ, and 2.0 pg/ml for IL-4. The final cytokine concentrations in biopsy homogenates were expressed as pg/mg of protein.
2.6 Statistical analysis
Chi square, Mann-Whitney, Kruscall Wallis and Dunn post test were used for comparison between groups. A multiple regression analysis was performed using H. pylori genotypes of cagA and vacA, levels of cytokines, age, gender and city of origin of the patients as explicatory variables of clinical outcome. The variables or interactions that did not contribute significantly to the model were excluded to reduce the model to a minimum number of interaction terms. A p value <0.05 was considered statistically significant.
3. Results
3.1 Bacterial virulence factor
Eighty four percent of H. pylori infected patients included in this study were positive for cagA gene (Table 2). The size of the fragments amplified from strains used in this study were according to those expected for the strain 26695 (3500 bp for A17BN1-Cag7 primers and 3000 bp for A17BN1-Cag6 primers and 1400 bp for A17 fragment, Figure 1). 91.2% were positive for vacA gene. The alleles s1bm1, s1am1 and s2m2 alleles were detected in 78%, 11% and 2.2% of the cases, respectively (Table 2). Fig. 2 shows PCR reactions for the different vacA alleles, according to the conditions described in method section. Virulence factors were present in infected patients regardless of severity of clinical outcome and there was no significant difference in cagA and vacA status in patients with chronic gastritis and duodenal ulcer (Table 2).
Table 2.
Characterisitics of the study groups
| N [%] | Chronic Gastritis 31 (68.9) |
Duodenal Ulcer 10 (22.2) |
|---|---|---|
| Age, X ± SD | 27.1 ± 21.3 | 34.4 ± 15.1 |
| Male, N° [%] | 9 (29) | 6 (60) |
| H. pylori infected, N° [%] | 31 (100) | 10 (100) |
| Presence of CagA, N° [%] | 28 (90.3) | 10 (100) |
| Vac Alleles, N° [%] | ||
| -s1bm1 | 27 (87.1) | 8 (80) |
| -s2m2 | 1 (3.2) | 0 (0) |
| -s1am1 | 3 (9.7) | 2 (20) |
| Cytokines, median (interquartile range) | ||
| -IL-10 | 24.5 (32.5) | 13.8 (24.9) |
| -IL-12 | 21.7 (53.4) | 23.5 (65) |
| -IL-4 | 14.2 (24.7) | 36.9 (41) |
| -IFN-γ | 12.6 (27.6) | 25.4 (40.5) |
p=ns
Figure 1.
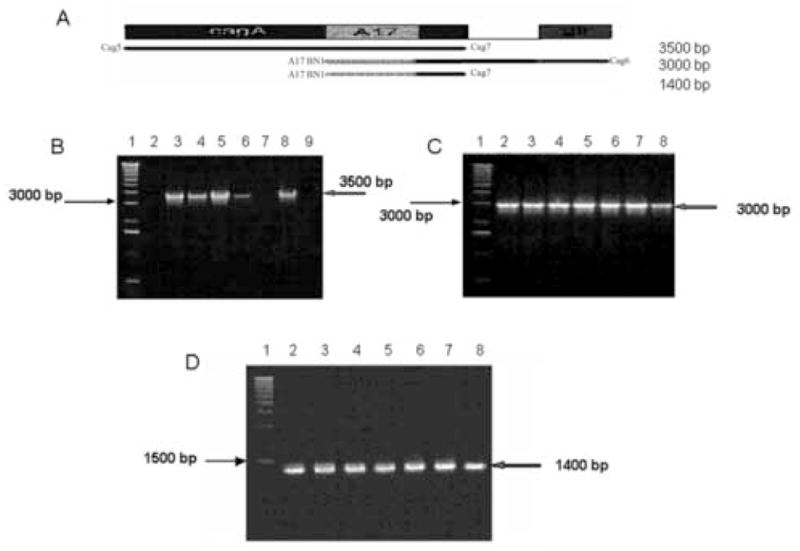
cagA detection. A. Scheme showing the amplified region of cagA gene. The primers were designed according to the 26695 strain as described in the Materials and Methods section. B. Amplification using A17BN1-Cag7 primers. Lane 1: 1kb DNA ladder Gibco. Lane 2: strain cagA -. Lanes 3–6 positive samples for this reaction. Lane 7: negative control with template DNA isolated from Campylobacter jejuni. Lane 8: positive control with chromosomal DNA isolated from strain 26695. C. Amplification using A17BN1-Cag6 primers. Lane 1: 1kb DNA ladder Gibco. Lanes 2–7: positive samples for this reaction. Lane 8: positive control with chromosomal DNA isolated from strain 26695. D. Amplification using A17BN1 and A17BH2 primers. Lane 1: 1kb DNA ladder Gibco. Lanes 2–7: positive strains for this reaction. Lane 8 positive control with chromosomal DNA isolated from strain 26695. Arrows indicate the expected size for each fragment and the size of the nearest band in DNA ladder.
Figure 2.
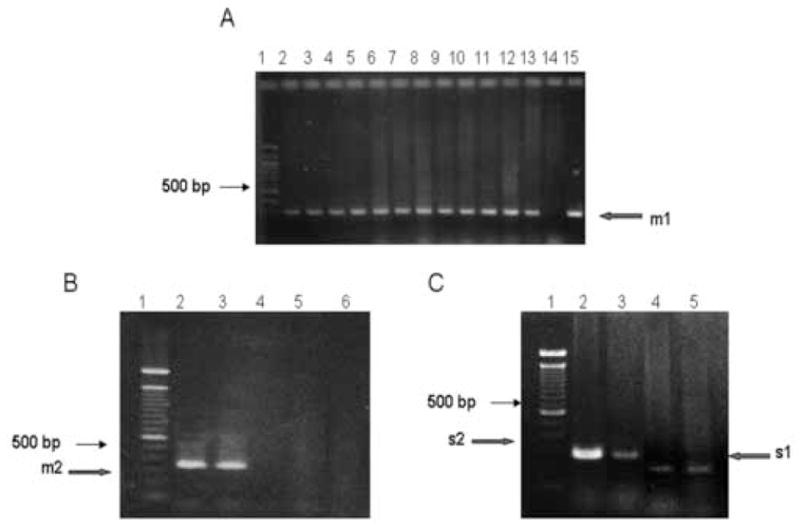
vacA amplification. A. PCR amplification of m1 allele. Lane 1: 100 bp DNA ladder standard, Lanes 2 through 13 samples obtained from patients living in IQ. Lane 14: negative control with template DNA isolated from Campylobacter jejuni. Lanes 15: positive control with chromosomal DNA isolated from strain J99. The arrow indicates the 290 bp expected PCR product. All PCR assays were repeated at least 3 times. B. PCR amplifications for m2 VacA allele. PCR fragments were separated in a 3% agarose gel. Lane 1: 100 bp ladder DNA standard, Lane 2: positive sample from a patient living in SA. Lane 3: positive control with chromosomal DNA isolated from strain 8822. Lanes 4 and 5: negative samples. Lane 6: strain 26695 as a negative control. The arrow indicates electrophoretic migration in a 3% agarose gel of the 352 bp expected fragment. C. Size differences in s1 and s2 PCR fragments derived from vacA gene from Chilean H. pylori clinical isolates. Fragments were separated in a 3% agarose gel. Lane 1: 100 bp ladder DNA standard. Lanes 2 and 3: s2 positive strains isolated from SA. Lane 4: a s1 positive strain from TE. Lane 5: a S1 positive control corresponding to H. pylori strain 26695. Arrows indicate the expected size for each fragment.
3.2 Levels of cytokines
Individual cytokines such as IL-10, IL-12, IFN-γ and IL-4 showed no statistical difference among patients with different clinical outcomes (Table 2). The ratio of cytokines was used for assessment of T helper profile. IFN-γ/IL-4 ratio showed a non significant increase in patients with chronic gastritis or duodenal ulcer in comparison to patients diagnosed as H. pylori negative (Fig 3A). IL-12/IL-10 ratio increased in parallel with the severity of clinical outcomes showing significant differences between non infected patients and patients with duodenal ulcer disease (p=0.04, Fig. 3B).
Figure 3.
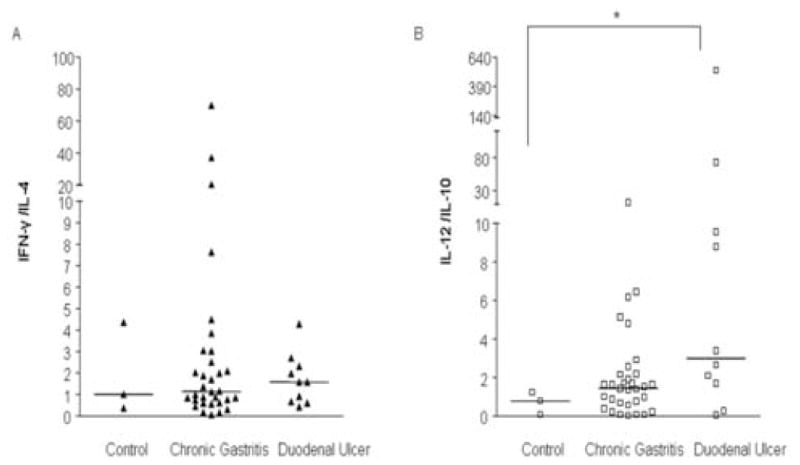
A. IFN-γ/IL-4 ratio showed no statistical difference among patients with different clinical outcomes. B. IL-12/IL-10 ratio was higher in patients with duodenal ulcer compared to non-infected patients. Data are presented as individual points plus median. p<0.05, Kruscall-Wallis, Dunn post test.
3.3 Correlation between host and bacterial factors
By multiple regression analysis we obtained a model that considered that clinical outcome to H. pylori infection, such as chronic gastritis or duodenal ulcer, was determined by the interaction of host immune response measured by Th cytokine balance and bacterial virulence factors, such as CagA but not VacA. This interaction was defined by the equation where Clinical outcome = 0.867491 (cagA) + 0.0131847 (IL-12/IL-10) + 0.0103503 (IFN-γ/IL-4). Variables included in this equation had a p value of less than 0.01 showing a statistically significant relationship between them. The model had a R-squared of 0.964 explaining 96.4% of the variability in clinical outcome.
To determine the accuracy of our equation in relation to the endoscopic diagnosis of duodenal ulcer we obtained theoretical titers by fitting cytokine profile and cagA status for each patient. Duodenal ulcer showed a significant increase of equation titers from chronic gastritis to duodenal ulcer (p=0.01, Fig 4).
Figure 4.
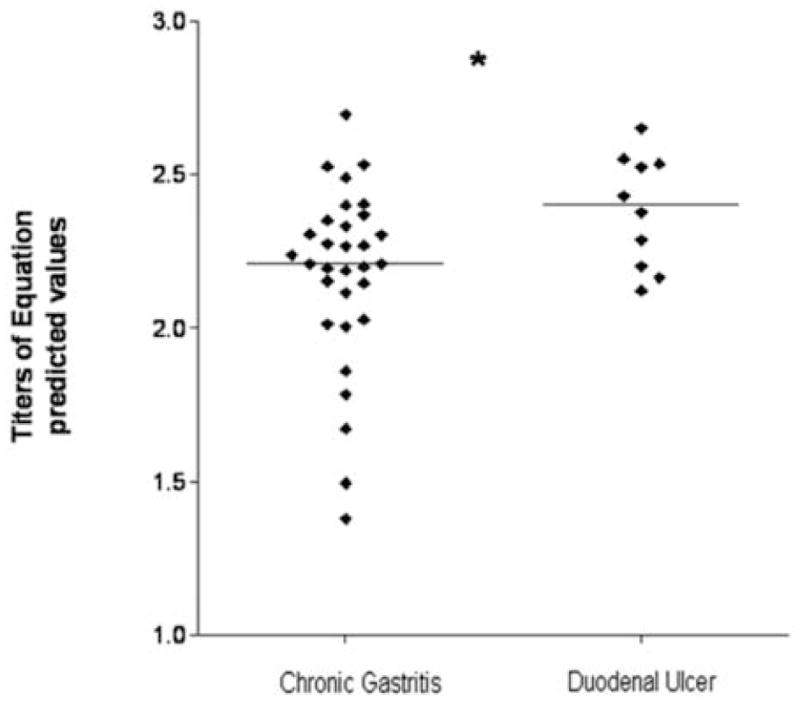
Observed and predicted values of clinical outcomes explained by the equation Clinical outcome: 0.867491 (cagA) + 0.0131847 (IL-12/IL-10) + 0.0103503 (IFN-γ/IL-4) that relates virulence factors such as CagA and host immune response defined as Th1/Th2 cytokine ratios. Titers of equation predicted values, obtained by fitting to the equation measured parameters, increase with severity of endoscopy observed outcome. Data are presented as individual points plus median. p<0.05, Mann-Whitney test.
4. Discussion
The present study investigates the relationship between selected bacterial virulence and host immune response on the clinical outcome of H. pylori-associated disease. Substantial morbidity is associated with H. pylori-induced diseases; approximately 90–95% of duodenal ulcers and 70–75% of gastric ulcers are attributable to this infection [21]. Therefore it has been considerable efforts focusing on delineating the complexes mechanisms by which this pathogen induces gastric inflammation and disease.
The relationship between the cag Pathogenicity Island and peptic ulcer are inconclusive. There are a number of studies showing that patients with duodenal ulcers have increased frequency of infection with cag+ strains [22–24]. However, in different populations the frequency of cag+ strains is highly variable, and it is not surprising that in populations with a high frequency of cag+ strains, the association with ulceration has not been observed and the associated risk for peptic ulcer is the same for H. pylori infection regardless the cagA status [25–27]. Previous results of our group showed prevalence of 84% of CagA positive patients measured by antibody titers [30]. In the current study infected patients showed 84 % positivity for cagA measured by PCR reaction with an almost identical extent of the cagA gene in patients with higher levels of inflammation or mucosal damage, such as duodenal ulcer patients than in patients with gastritis, showing no capability for discrimination between clinical outcomes by itself.
VacA gene, which encodes a secreted bacterial cytotoxin, has also been involved in the pathogenesis of peptic ulcer disease. Although vacA gene is present in virtually all H. pylori strains they vary considerably in their cytotoxic activity due to variations in vacA gene structure [5]. Strains that possess an s1/m1 vacA genotype are considered as most virulent with an increased risk of peptic ulcer disease and gastric cancer and enhanced gastric epithelial cell injury compared to those that possess s2/m2 alleles [24]. Prior work from our group showed that the most prevalent VacA genotype in Chilean patients was s1bm1 with 76%, followed by s1a m1 with 21% and s2 m2 that occurred in only 3% [13]. In the current study 91.2% of H. pylori-infected patients were carrying VacA positive strains also with higher proportion of s1bm1 genotype. Because the presence of cagA and vacA, particularly virulent alleles like s1m1, is so extended in our population, and ulcer percentages is not particularly high, we were not able to use these virulence markers as sole predictors of duodenal ulcer disease. Similar results were observed in a Japanese population which had a 79% of positivity for type I strain (CagA and VacA positive) showing that in high prevalence countries the presence of virulence factors can not be used as predictors of different outcomes on their own [28].
The host immune response is an important determinant of H. pylori-associated disease. H. pylori colonization induces a systemic and mucosal response directed to multiple antigens [29]. Among the local immune mediators T helper cytokines may play a pivotal role in pathogenesis in patients with different H. pylori-associated outcomes. Individual cytokine levels are not related with clinical outcomes in this study. However, there is an increase of the Th1 response, as measured by IL-12/IL-10 ratio, as mucosal damage progresses from chronic gastritis to duodenal ulcer. This predominant Th1 response observed during infection is unlikely to be effective in eradication of the pathogen and may contribute to gastroduodenal pathogenesis [12]. However, the Th1-predominant response is not able to explain all the variability of our subjects in terms of H. pylori-associated morbidity.
Although the presence of cagA and vacA genes in Chilean population are equally extended and the deviation towards a Th1 type response in peptic ulcer patients is not consistent for all measured cytokines, the development of duodenal ulcer disease is associated mainly to the presence of CagA and to the Th1/Th2 cytokine ratios. Our model accounts for 96% of our sample variability and is able to discriminate between patients with mild inflammation (gastritis) from those who develop severe outcomes such as duodenal ulcer.
The present study demonstrates that the relationship between bacterial virulence and host immune factors is determinant in the development of clinical outcomes of H. pylori- associated diseases. Our results show that in countries with high prevalence of H. pylori infection with virulent strains, we can not consider virulence factors and host immune response to be unconnected processes, so for analytical approximations to predict or describe the appearance of diverse outcomes in H. pylori infected patients, a multivariate course of action must be considered.
Acknowledgments
This work was supported in part by grants from FONDECYT #1030401, #1030894, FONDEF DO2I-1067 and NIH #DK-54495.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sipponen P, Hyvarinen H, Seppala K, Blazer MJ. Pathogenesis of the transformation from gastritis to malignacy. Aliment Pharmacol Ther. 1999;12:61–71. doi: 10.1111/j.1365-2036.1998.00005.x. [DOI] [PubMed] [Google Scholar]
- 2.Rollan A, Ferreccio C, Harris P, Serrano C, Jara A, Venegas A. Early Helicobacter pylori infection is related to gastric cancer risk in Chile, a high-risk area: A population-based study. Can J Gastroenterol. 2005;4B [Google Scholar]
- 3.República de Chile. Ministerio de Salud. Departamento de Estadísticas. [Accession: 25/08/2006];Bases de datos de Mortalidad 1990–2004. Available from http://deis.minsal.cl/
- 4.Monack DM, Mueller A, Falkow S. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nature Rev Microbiol. 2004;2:747–765. doi: 10.1038/nrmicro955. [DOI] [PubMed] [Google Scholar]
- 5.Atherton JC, Peek RM, Tham K, Cover T, Blaser MJ. Clinical and pathological importance of heterogeneity in VacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology. 1997;112:92–99. doi: 10.1016/s0016-5085(97)70223-3. [DOI] [PubMed] [Google Scholar]
- 6.Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood group antigen-binding adhesin. Proc Natl Acad Sci. 1999;96:1278–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crabtree J, Covacci A, Farmery SM, Xiang Z, Tompkins DS, Perry S, Lindley I, Rappuoli R. Helicobacter pylori induced interleukin-8 expression in gastric epithelial cells is associated with CagA positive phenotype. J Clin Pathol. 1995;48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson W, Falkow S. Disrruption of epithelial apical-juntional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner Regulation of gastric epithelial cell growth by Helicobacter pylori: evidence for a major role of apoptosis. Gastroenterology. 1997;113:1836–1847. doi: 10.1016/s0016-5085(97)70003-9. [DOI] [PubMed] [Google Scholar]
- 10.Sommer F, Faller G, Konturek P, et al. Antrum and corpus mucosa-infiltrating CD4+ lymphocytes in Helicobacter pylori gastritis display a Th1 phenotype. Infect Immun. 1998;6:5543–5546. doi: 10.1128/iai.66.11.5543-5546.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghavan S, Fredriksson M, Svennerholm AM, Holmgren J, Suri-Payer E. Absence of CD4+CD25+ regulatory T cells is associated with a loss of regulation leading to increased pathology in Helicobacter pylori-infected mice. Clin Exp Immunol. 2003;132:393–400. doi: 10.1046/j.1365-2249.2003.02177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smythies L, Waites K, Lindsey J, Harris P, Ghiara P, Smith P. Helicobacter pylori-induced mucosal inflamation is Th1 mediated and exacerbated in IL-4, but not IFN-γ, gene-deficient mice. J Immunol. 2000;165:1022–1029. doi: 10.4049/jimmunol.165.2.1022. [DOI] [PubMed] [Google Scholar]
- 13.Díaz MI, Valdivia A, Palacios JL, Martínez P, Harris P, Novales J, Garrido E, Valderrama D, Shilling C, Kirberg A, Hebel E, Bravo R, Siegel F, Leon G, Venegas A. The H. pylori vacA s1a and s1b alleles from clinical isolates from different regions of Chile show a distinct distribution along the country. W J Gastroenterol. 2005;11:6366–6372. doi: 10.3748/wjg.v11.i40.6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goowin S. In: Methods in Molecular Medicine, Helicobacter pylori protocols. Clayton CL, Mobley LT, editors. Tolowa, NJ: Humana Press; 1999. pp. 7–13. [Google Scholar]
- 15.Owen R, Bickley J. In: Methods in Molecular Medicine, Helicobacter pylori protocols. Clayton CL, Mobley LT, editors. Tolowa, NJ: Humana Press; 1999. pp. 88–89. [Google Scholar]
- 16.Tomb JF, White O, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 17.Muller I, Medina-Selby A, Palacios JL, Martínez P, Opazo P, Bruce E, Mancilla M, Valenzuela P, Yudelevich A, Venegas A. Cloning and comparison of ten gene sequences of a Chilean H. pylori strain with other H. pylori strains revealed higher variability for VacA and CagA virulence factors. Biol Res. 2002;35:67–84. doi: 10.4067/s0716-97602002000100010. [DOI] [PubMed] [Google Scholar]
- 18.Valdivia A, Palacios JL, Harris P, et al. El gen cagA de Helicobacter pylori de cepas de pacientes de Los Angeles (Chile) presenta distinto tamaño en la región codificante del dominio hidrofilico A17. Acta Microbiol. 2002;8:11–16. [Google Scholar]
- 19.Yan J, Kumagai T, Ohnishi M, Ueno IHO. Immune Response to a 26-kDa Protein, Alkyl Hydroperoxide Reductase in Helicobacter pylori-Infected Mongolian Gerbil. Helicobacter. 2001;6:274–82. doi: 10.1046/j.1523-5378.2001.00038.x. [DOI] [PubMed] [Google Scholar]
- 20.Atherton JC, Cao P, Peek R, Tummuru M, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori: association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 21.Peek R, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 22.Crabtree J, Taylor J, Wyatt J, Heatley R, Shallcross T, Tompkins D, Rathbone B. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration and gastric pathology. Lancet. 1991;338:332–335. doi: 10.1016/0140-6736(91)90477-7. [DOI] [PubMed] [Google Scholar]
- 23.Cover T, Glupczynski Y, Lage A, Burette A, Tummuru M, Perez-Perez G, Blazer M. Serologic detection of infection with cagA + Helicobacter pylori strains. J Clin Microbiol. 1995;33:1496–500. doi: 10.1128/jcm.33.6.1496-1500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Doorn L, Figueiredo C, Sanna R, Plaisier A, Schneeberger P, De Boer W, Quint W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology. 1998;115:58–66. doi: 10.1016/s0016-5085(98)70365-8. [DOI] [PubMed] [Google Scholar]
- 25.Pan Z, et al. Equaly high prevalences of infection with cag positive Helicobacter pylori in Chinese patients with peptic ulcer disease and those with chronic gastritis-associated dyspepsia. J Clin Microbiol. 1997;35:1344–1347. doi: 10.1128/jcm.35.6.1344-1347.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham D, Genta R, Graham D, Crabtree J. Serum CagA antibodies in asymptomatic subjects and patients with peptic ulcer: lack of correlation of IgG antibody in patients with peptic ulcer or asymptomatic Helicobacter pylori gastritis. J Clin Pathol. 1996;49:829–32. doi: 10.1136/jcp.49.10.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaoka Y, Souchek J, Odenbreit S, Haas R, Arnqvist A, Boren T, et al. Discrimination between cases of duodenal ulcer and gastritis on the basis of putative virulence factors of Helicobacter pylori. J Clin Microbiol. 2002;40:2244–6. doi: 10.1128/JCM.40.6.2244-2246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeda S, Ogura K, Yoshida H, Kanai F, Ikenoue T, Kato N, et al. Major virulence factors, VacA and CagA, are commonly positive in Helicobacter pylori isolates in Japan. Gut. 1998;42:338–43. doi: 10.1136/gut.42.3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appelmelk B, Montiro M, Martin S, Moran A, Vandenbroucke-Grauls C. Why Helicobacter pylori has a Lewis antigens. Trends Microbiol. 2000;8:565–570. doi: 10.1016/s0966-842x(00)01875-8. [DOI] [PubMed] [Google Scholar]
- 30.Harris P, Godoy A, Arenillas S, et al. CagA Antibodies as a Marker of Virulence in Chilean Patients With Helicobacter pylori Infection. JPGN. 2003;37:596–602. doi: 10.1097/00005176-200311000-00018. [DOI] [PubMed] [Google Scholar]