

Summary
Cell migration toward areas of higher extracellular matrix (ECM) rigidity via a process called “durotaxis” is thought to contribute to development, immune response, and cancer metastasis. To understand how cells sample ECM rigidity to guide durotaxis, we characterized cell-generated forces on the nanoscale within single mature integrin-based focal adhesions (FAs). We found that individual FAs act autonomously, exhibiting either stable or dynamically fluctuating (“tugging”) traction. We show that a FAK/phosphopaxillin/vinculin pathway is essential for high FA traction and to enable tugging FA traction over a broad range of ECM rigidities. We show that tugging FA traction is dispensable for FA maturation, chemotaxis, and haptotaxis but is critical to direct cell migration toward rigid ECM. We conclude that individual FAs dynamically sample rigidity by applying fluctuating pulling forces to the ECM to act as sensors to guide durotaxis, and that FAK/phosphopaxillin/vinculin signaling defines the rigidity range over which this dynamic sensing process operates.
Introduction
Directional control of cell migration is critical to developmental morphogenesis and tissue homeostasis, as well as disease progression in cancer. Cells sense gradients of environmental cues to guide directional movement. Such cues may be diffusible or substrate-bound biochemicals, as in chemotaxis and haptotaxis, or physical, including electric fields, topography, or extracellular matrix (ECM) rigidity (Petrie et al., 2009). Cell migration along an ECM-rigidity gradient is known as “durotaxis.” Durotaxis is thought to be critical to epithelial-to-mesenchymal transition (Guo et al., 2006; de Rooij et al., 2005), development of the nervous system (Flanagan et al., 2002; Koch et al., 2012), innate immunity (Mandeville et al., 1997), as well as cancer metastasis (Paszek et al., 2005; Wozniak et al., 2003; Ulrich et al., 2009).
ECM stiffness in tissues can vary locally or change over time during development or in disease states such as cancer or atherosclerosis. Thus, durotaxis requires cells to continuously sample and measure the spatial and temporal variability in the stiffness landscape of the ECM via a process known as “rigidity mechanosensing” (Janmey and McCulloch, 2007). Rigidity mechanosensing is critical to many integrin-dependent processes, including regulating proliferation and differentiation (Engler et al., 2006; Ingber and Folkman, 1989), growth of focal adhesions (FAs), contractility, spreading, and cell polarization (Pelham and Wang, 1997; Riveline et al., 2001; Jiang et al., 2006; Prager-Khoutorsky et al., 2011). There is extensive evidence that actomyosin cytoskeletal contractility and integrin engagement to ECM via FAs are required for rigidity mechanosensing (Hoffman et al., 2011). However, it is not known how cells dynamically sample local differences in a heterogeneous and changing ECM stiffness landscape to guide durotaxis, and the molecular mechanism controlling the range of rigidity cells feel remains elusive.
Here, we sought to understand how cells locally and dynamically sample a range of ECM rigidities to guide directed migration toward stiff ECMs. We utilized high-resolution time-lapse traction force microscopy (Sabass et al., 2008) to characterize the distribution and dynamics of traction forces within single mature FAs of migrating fibroblasts. This revealed that individual FAs act autonomously within a cell, exhibiting one of two distinct states of force transmission. Traction within FAs is either constant over time and positionally static or dynamically fluctuating in magnitude and position in a pattern reminiscent of repeated tugging on the ECM. We use pharmacological and genetic perturbations to show that a FAK/phosphopaxillin/vinculin pathway is essential for cells to exert high traction and to enable tugging force fluctuations by FAs over a broad range of ECM rigidities. We further demonstrate that FA tugging is dispensable for directional migration in response to biochemical gradients but is required for durotaxis. Together, our findings show that individual FAs repeatedly apply tugging forces to locally sense ECM stiffness to guide durotaxis, and that a specific pathway downstream of FAK broadens the range of rigidities over which this local dynamic rigidity-sensing process operates.
Results
Traction Stress Is Asymmetrically Distributed within Single Focal Adhesions
To analyze the distribution and dynamics of traction stress within individual FAs, we utilized high-resolution traction force microscopy (TFM, Gardel et al., 2008; Sabass et al., 2008). Mouse embryonic fibroblasts (MEFs) expressing enhanced green fluorescent protein (eGFP)-paxillin as FA marker were plated on ECMs of known rigidity consisting of fibronectin-coupled elastic polyacrylamide (PAA) substrates embedded with a mixture of red and far-red fluorescent beads. Cell-induced ECM deformation was visualized by spinning disk confocal microscopy, and traction fields were reconstructed at 0.7 μm resolution with Fourier transform traction cytometry (Sabass et al., 2008). To obtain multiple traction measurements within each FA, we limited our analysis to FAs ≥ 1.5 μm, which constituted at least 30% of all cellular FAs under all experimental conditions (Figure S5B available online). Thus, our study is focused on the role of mature FAs in mechanosensation.
High-resolution TFM of cells plated on 8.6 kPa ECMs revealed that traction stress magnitude and eGFP-paxillin intensity were distributed similarly across individual FAs, with a single peak value toward the FA center and low values toward the FA tips (Figure 1). Like previous reports (Stricker et al., 2011), individual FAs exhibited a mean peak traction stress of 0.8 ± 0.3 kPa and a mean traction stress of 0.16 ± 0.08 kPa per μm2 of FA area. However, line-scans across FAs showed that although the position of peak paxillin intensity was always in the FA center, the position of peak traction was often skewed toward the distal FA tip (toward leading edge) and rarely appeared skewed toward the proximal FA tip (toward cell center) in the region where stress fibers presumably attach (Figures 1A–1C). On average, the position of peak traction was skewed from the FA center by 1.1 ± 0.58 μm toward the distal FA tip (n = 1,269; Figure 1D). In the majority of FAs (73.6%), the skew of peak traction toward the distal tip of the FA was significant (>0.7 μm). In 26.1% of FAs, the peak traction location was not significantly different from the FA center, and in 0.3% of FAs, the traction peak was skewed toward the proximal FA tip (Figure 1D). The position of peak traction within FAs was not correlated with FA length or traction magnitude (Figures S1A and S1B) and was insensitive to image acquisition order or use of mApple-paxillin as an FA marker (not shown). Use of stiffer ECM (32 kPa) where cells induced smaller deformations (Figures S1C and S1D) did not change the distribution of traction stress across FAs, indicating that our results were not due to nonlinear deformation of PAA. Thus, traction stress can be asymmetrically distributed across individual FAs, indicating variability in force transmission from the actin cytoskeleton to the ECM on the nanoscale within a single FA.
Figure 1. Traction Stresses Are Asymmetrically Distributed across Individual FAs.
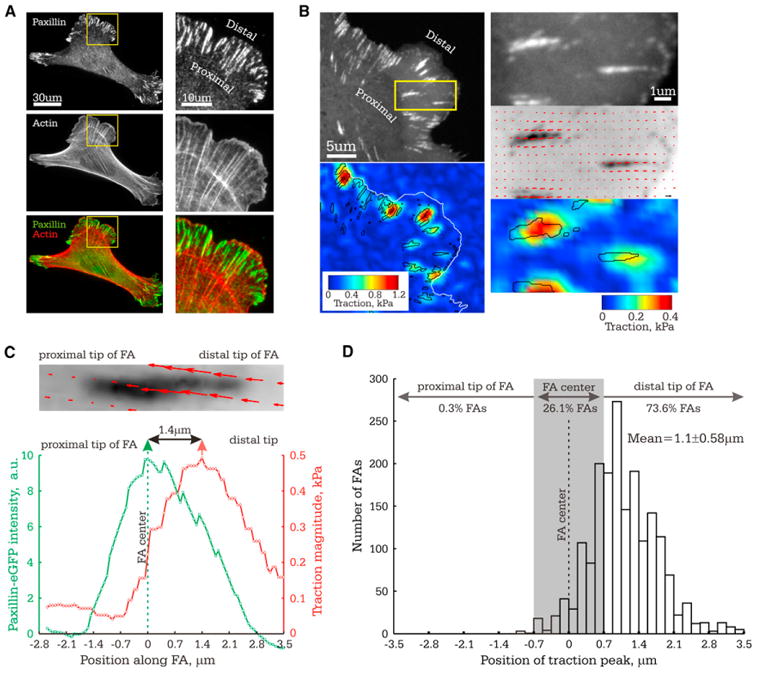
Analysis of traction stress distribution in FAs in MEF (8.6 kPa ECM). (A) Immunolocalization of paxillin and fluorescent phalloidin staining of actin. Right panel: Zoom of boxed region on left. Proximal and distal directions are marked.
(B) Images of eGFP-paxillin (top left, top right: zoomed image of the boxed region) and corresponding maps of reconstructed traction stresses on the ECM with positions of FA outlined in black (bottom left and bottom right: traction magnitude heatmaps; middle right: stress vector field overlaid on inverted contrast image of eGFP-paxillin).
(C and D) The center of the FA (position of peak eGFP-paxillin intensity) was set as the origin of the x axis, proximal and distal directions indicated. (C) Above: Stress vector field overlaid on inverted contrast image of eGFP-paxillin. Below: eGFP-paxillin intensity and traction stress as a function of distance along the FA shown above. Single-headed arrows: peak values; double-headed arrows: distance between peak values. (D) Histogram of the position of peak traction within single FAs, with number of FAs with peak traction stress located in each region shown (n = 1,269). Grey rectangle highlights the values that are not significantly different from the FA center.
See also Figure S1.
Time-Lapse TFM Reveals Two States of Traction in an FA: Stable and Tugging
To examine the dynamics of traction stress within FAs, we performed time-lapse TFM of cells plated on 8.6 kPa ECMs. We analyzed 2–3 min TFM movies (5 s image intervals) of steady-state FAs that were neither assembling, disassembling, nor moving. In some FAs, the position of peak traction was centered and stable within the FA, exhibiting insignificant changes in magnitude and location (Movie S1; Figures 2A, 2D, and 2G). However, more often, there were sudden, large changes in both the location and magnitude of the traction peak within the FA (Movie S2; Figures 2B, 2E, and 2H). In such FAs, the position of peak traction was usually near the distal FA tip but frequently shifted back toward the FA center and immediately returned to the distal tip. Accompanying the positional fluctuations were 1–3 kPa changes in peak traction magnitude. FAs exhibiting fluctuating peak traction and FAs with stable, centered peak traction coexisted in close proximity (n = 8, Figures 2C, 2F, and 2I), and fluctuations of peak traction magnitude and position were nonsynchronous in neighboring FAs (Figure S2). This indicates a lack of mechanical coupling among FAs and suggests that traction fluctuations are not due to global cell contractions. Thus, FAs can exhibit stable or fluctuating states of cytoskeleton-ECM force transmission within a single cell, and each FA acts autonomously.
Figure 2. Time-Lapse TFM Reveals Two States of Traction in Individual FA: Stable and Fluctuating.
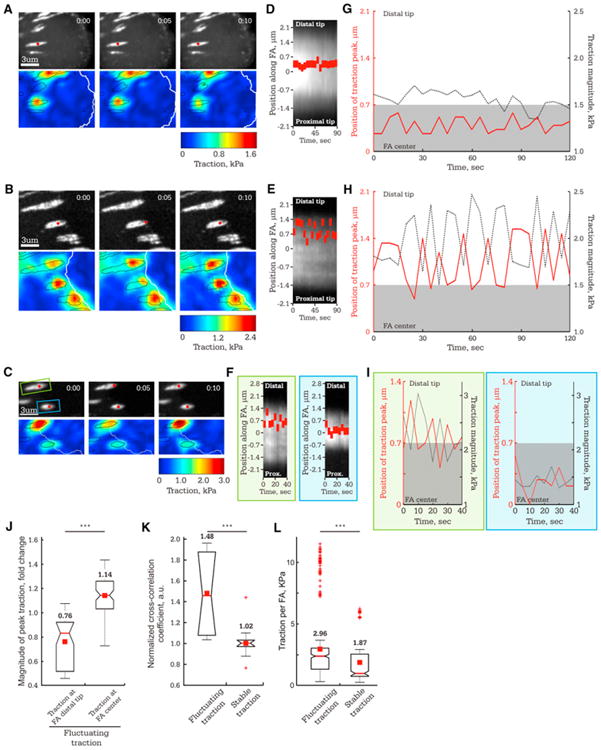
Images of FAs in MEF (8.6 kPa ECM) were captured at 5 s intervals.
(A–C) Images of eGFP-paxillin (top panels, time in min:s shown) and corresponding heatmaps of reconstructed traction stresses with FAs outlined in black (bottom panels). Red dot: position of peak traction for the FAs analyzed in (D)–(I).
(A, D, and G) FA in which the position of peak traction remains stable near the FA center.
(B, E, and H) FA in which the position of peak traction fluctuates in the distal half of the FA.
(C, F, and I) Neighboring FAs in which position of peak traction is stable (blue box) or fluctuating (green box).
(D–F) Kymographs along the FAs marked by red dots in (A), (B), and (C), respectively. Red rectangle: Position of peak traction along the FA (y axis) over time(x axis).
(G–I) Plot of the position of peak traction stress along the FA (left axis, red, with the FA center set to zero) and the peak traction magnitude (right axis, black) over time for the FAs marked in (A), (B), and (C), respectively. Grey rectangle highlights values that are not significantly different from the FA center.
For (J)–(L), TFM time series of individual FAs were classified according to whether the FA exhibited fluctuating or stable traction, with means shown above each plot.
(J) Box plot of peak traction stress from the frames of TFM image series when the position of the traction peak was either in the distal tip of the FA (>0.7 μm from the FA center, n = 274 TFM frames) or at the FA center (<0.7 μm from the FA center, n = 28 TFM frames) for FAs that exhibited fluctuating traction (n = 9 FAs). Values are normalized to the mean of peak traction stress for the entire TFM series of each FA. (K) Box plots of cross-correlation coefficient between the magnitude and location of peak traction for FAs exhibiting fluctuating (n = 9 FAs) or stable traction (n = 9 FAs).
(L) Box plot of mean integrated traction stress per FA for each TFM frame in time-lapse series of FAs that exhibited either fluctuating (n = 302 frames, 9 FAs) or stable (n = 285 frames, 9 FAs) traction.
See also Figure S2 and Movies S1 and S2.
Plotting the magnitude and location of the traction peak within an FA together over time in FAs with fluctuating traction suggested intermittent “tugging” on the ECM. This was characterized by centripetal movement of the traction peak that was accompanied by an increase in its magnitude and immediate return of the traction peak to the distal FA tip corresponding to traction decrease (Figure 2H). During fluctuations, traction magnitude was 1.5-fold greater (p < 0.001) when the peak was in the FA center compared to when it was at the distal FA tip (Figure 2J). Temporal cross-correlation analysis confirmed that changes in peak traction location were accompanied by changes in traction magnitude for fluctuating but not stable traction FAs (Figure 2K). Although traction magnitude often dipped low during fluctuations, the mean total traction was significantly (1.5-fold, p < 0.001) higher in FAs exhibiting tugging traction than for those with stable traction (Figure 2L). Thus, FAs exhibit two distinct traction states: a weak state where traction is stable and centered in the FA and a strong, dynamic state with a specific pattern reminiscent of repeated tugging on the ECM. In tugging FAs, peak traction is low when positioned at the distal FA tip (engagement), builds strength as the peak moves rearward to the FA center (tug), and returns to a low level as the peak position snaps back to the FA distal tip (release), and this cycle is repeated intermittently.
The Position of Peak Traction within FAs Indicates the Dominant Dynamic Traction State in an FA Population
To dissect the molecular mechanism controlling FA traction dynamics, we required a less computationally intensive and laborious method than time-lapse TFM to determine dynamic traction states of FAs. Because the traction peak was generally central in stable traction FAs and was often skewed toward the distal tips of tugging FAs (Figures 2G and 2H), we hypothesized that the mean position of the traction peak measured for a large number of FAs would reflect the dominant FA dynamic state in the population. We classified TFM movies of FAs as “tugging” or “stable” and determined the mean position of peak traction within the FA at all time points in the movies. This showed that although stable and tugging FAs exhibited similar spatial distributions of GFP-paxillin intensity (Figure 3C), the mean position of peak traction in stable FAs was not significantly different from the FA center (skewed distally by 0.27 ± 0.33 μm), whereas for tugging FAs, the mean position of peak traction was skewed by 1.24 ± 0.46 μm toward the distal FA tip (Figure 3A). We also measured the fraction of FAs in which the position of peak traction was significantly skewed (>0.7 μm) toward the distal FA tip in 150 randomly selected snapshots from movies of stable or tugging FAs. This showed that the traction peak was distally skewed in 4% ± 1.1% and 91% ± 1.7% of TFM snapshots of stable and tugging FAs, respectively (Figure 3B). Thus, for a population of FAs, the more the mean position of peak traction is shifted toward the distal FA tip, the greater the proportion of tugging FAs. Conversely, the closer the mean position of peak traction to the FA center, the greater the proportion of FAs with stable traction.
Figure 3. FA Traction Dynamics Are Modulated by ROCK-Dependent ECM-Rigidity Mechanosensing.
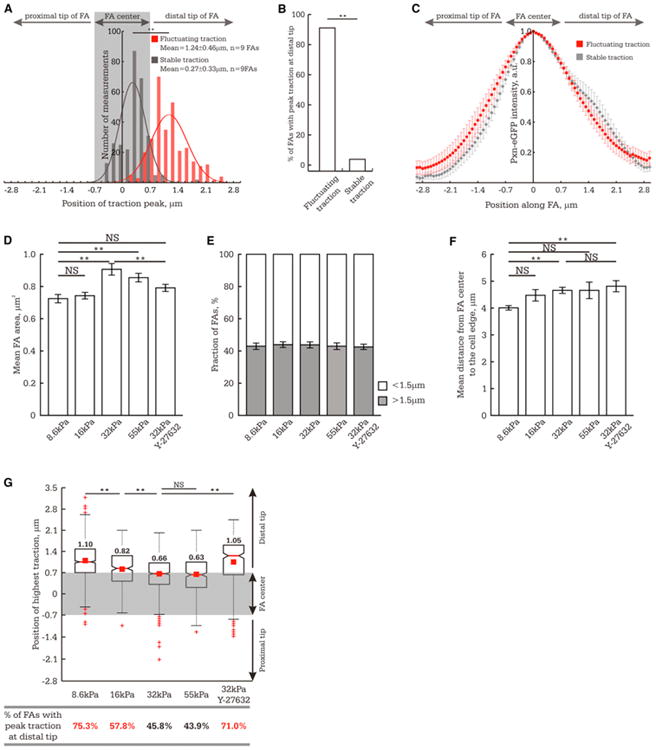
In (A) and (G), the FA center (position of peak eGFP-paxillin intensity) was set as the origin; gray rectangle highlights the values that are not significantly different from the FA center, and distal and proximal directions are indicated. (A–C) TFM movies of FA in cells plated on 8.6 kPa ECM were classified according to whether the FA exhibited tugging or stable traction.
(A) Histogram of the position of peak traction in each frame of time-lapse TFM series for FAs exhibiting tugging (red, n = 302 measurements, n = 9 FAs) or stable traction (gray, n = 285 measurements, n = 9 FAs).
(B) One hundred and fifty snapshots were randomly selected from sets of TFM movies of FAs that exhibited either tugging (n = 9 FAs) or stable (n = 9 FAs) traction.
This panel shows the percent of TFM snapshots of FAs in which the position of peak traction was located at the distal tip of the FA (>0.7 μm from the FA center). (C) Profile of eGFP-paxillin intensity across FAs exhibiting tugging or stable traction dynamics. n = 9 FAs for each type of traction behavior.
(D–F) Effect of ECM stiffness and treatment with 1 μM Y-27632 on FA morphometry. (D) Mean FA size (n > 600 FAs from 7 cells).
(E) Fraction of FAs ≥ 1.5 μm (n > 600 FAs from 7 cells).
(F) FA subcellular localization quantified as the mean distance between the FA center and the cell edge (n > 200 FAs per experimental condition).
(G) Analysis of peak traction position within single FAs (n > 200 FAs per experimental condition).Top: Box plot of peak traction position within FAs. Bottom: Fraction of TFM snapshots of FAs in which the position of peak traction was significantly skewed (>0.7 μm) toward the distal FA tip. Values greater than 50% are marked in red.
**p < 0.01, NS p > 0.05 as detected by Mann-Whitney test. (D–F) Data shown as mean ± standard error of the mean (SEM). See also Figure S3.
FA Traction Dynamics Are Modulated by ROCK-Dependent ECM-Rigidity Mechanosensing
We next sought to determine whether FA traction dynamics are modulated by ECM rigidity. Varying ECM rigidity (8.6 to 55 kPa) showed that FAs on average were slightly but significantly smaller on softer ECMs, as expected (Figure 3D) (Prager-Khoutorsky et al., 2011), however more than 40% of FAs under each condition were ≥ 1.5 μm and thus suitable for TFM analysis (Figure 3E). Furthermore, time-lapse TFM movies showed FAs exhibiting both tugging and stable traction on all ECM rigidities (Figure S3A). Thus, mature FAs exhibit stable and tugging traction regimes on a range of ECM rigidities.
To determine whether ECM rigidity affected the proportion of FAs exhibiting stable or tugging traction, we located the position of peak traction within mature FAs of similar subcellular localization (Figure 3F) in TFM snapshots and determined the mean position of peak traction within the FA and the fraction of FAs with the traction peak significantly skewed toward the distal FA tip. This showed that the mean position of peak traction within FAs varied as an inverse function of ECM rigidity between 8.6 and 32 kPa: the stiffer the ECM, the more central (less skewed) the traction peak within the FA, and conversely, the softer the ECM, the further the mean traction peak was skewed toward the distal FA tip (Figure 3G). Similarly, increasing ECM rigidity from 8.6 to 16 to 32 kPa was associated with a progressively decreased fraction of FAs with the traction peak skewed toward the distal FA tip. Further increasing ECM rigidity from 32 to 55 kPa did not change either the mean position of peak traction within the FA or the fraction of FAs with a distally located traction peak (Figure 3G). ECM rigidities greater than 55 kPa were not evaluated because their deformation was not sufficient for accurate TFM measurements. Thus, soft ECMs promote tugging traction dynamics in FAs, and rigid ECMs promote stable traction in FAs.
To determine whether the rigidity response of FA traction dynamics is regulated by p160 Rho kinase (ROCK)-dependent contractility, we analyzed the position of peak traction within FAs in cells plated on rigid ECM (32 kPa) and treated with a low concentration (1 μM) of Y-27632 (“ROCK inhibitor”). This level of ROCK inhibitor only slightly reduced FA size but had no effect FA subcellular location (Figures 3D–3F). Treatment with ROCK inhibitor shifted the mean position of peak traction toward the distal FA tip and increased the fraction of FAs with a distally located traction peak compared to cells plated on 32 kPa ECM in the absence of ROCK inhibitor (Figure 3G). FAs in cells plated on 32 kPa ECM with ROCK inhibitor exhibited a similar value for mean peak traction position within FA as for cells plated on softer, 8.6 kPa ECMs in the absence of ROCK inhibitor (Figure 3G). We were unable to perform these experiments with blebbistatin due to its phototoxic effects (Kolega, 2004). Thus, reducing ECM rigidity or downregulation of Rho kinase-mediated contractility switches FAs from stable to tugging traction states. Furthermore, these experiments define an ∼20 kPa range of ECM rigidities (8.6 to 32 kPa) over which FA traction dynamics are rigidity sensitive in MEFs.
A FAK/Phosphopaxillin/Vinculin Signaling Pathway Promotes FA Force Transmission and Traction Dynamics
We next sought to determine whether FA traction dynamics are controlled by a signaling pathway composed of FAK, paxillin, and vinculin, which is thought to regulate rigidity-dependent maturation and strengthening of FAs (Dumbauld et al., 2010b, 2010a; Mierke et al., 2010; Pasapera et al., 2010). It is thought that FAK activity, induced by integrin engagement of rigid ECM (Provenzano et al., 2009), promotes phosphorylation of paxillin on tyrosines 31 and 118 (Schneider et al., 2009) to induce a phosphopaxillin-vinculin interaction and vinculin recruitment and strengthening of the FA (Pasapera et al., 2010).
We first examined the role of FAK activity and paxillin phosphorylation in regulation of FA traction dynamics. We inhibited FAK activity with PF-228 (10 μM for 30 min, Slack-Davis et al., 2007) or altered paxillin phosphorylation by suppressing endogenous paxillin with small interfering RNA (siRNA) and re-expressing either phosphomimetic (paxillinY31/118E) or non-phosphorylatable (paxillinY31/118F) mutants tagged with eGFP (Pasapera et al., 2010). FAK inhibition induced larger FAs, whereas expression of paxillin mutants had no effect on FA size (Figures 4A and 4B) (Zaidel-Bar et al., 2007). None of the treatments significantly affected the fraction of FAs amenable to TFM analysis (Figure 4C) or cell contractility as assayed by western blot for serine 19 phosphorylated myosin II regulatory light chain (pMLC, Figure 4D). Furthermore, distribution of GFP-tagged paxillin mutants across single FAs was indistinguishable from that with wild-type GFP-paxillin (Figure S4). TFM analysis of FAs in cells plated on 8.6 kPa ECMs showed that all three treatments reduced total traction stress (Figure 4E), and TFM movies showed that FAs exhibiting both tugging and stable traction in cells were present (Figure S3B). However, quantifying the position of peak traction within the FA in a large number of TFM snapshots revealed that compared to controls, all three treatments significantly decreased the mean position of peak traction within the FA and the fraction of FAs with their traction peaks skewed toward the distal FA tip (Figure 4F). Thus, FAK activity and a paxillin Y31/118 phosphoregulation cycle promote FA force transmission and tugging FA traction dynamics in cells plated on 8.6 kPa ECMs.
Figure 4. Inhibiting FAK or Altering Paxillin Y31/118 Phosphorylation Reduces FA Force Transmission and Depletes Tugging FA Traction Dynamics.
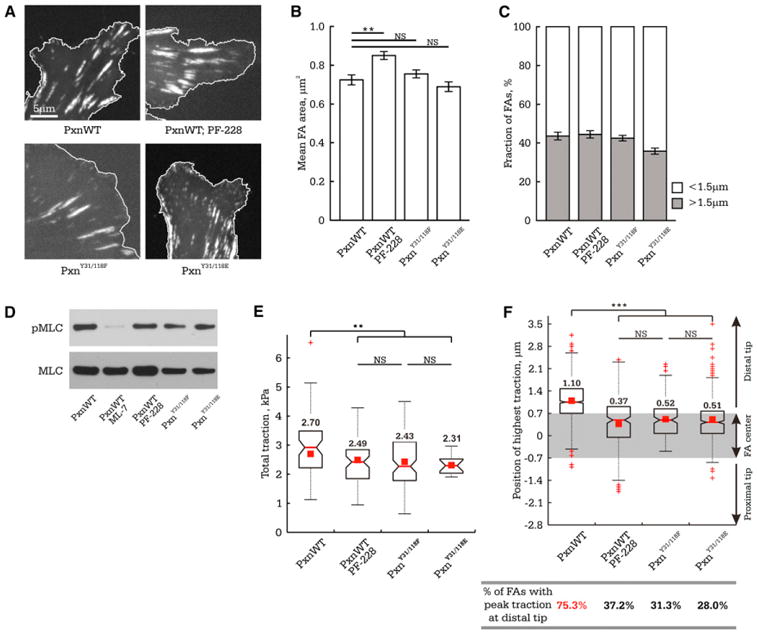
MEFs were cotransfected with nontargeting siRNAs and eGFP-tagged wild-type paxillin (PxnWT) or with paxillin-targeting siRNAs and eGFP-tagged siRNA-resistant paxillin mutants (phosphomimetic [PxnY31/118E] or nonphosphorylatable [PxnY31/118F]), or they were treated with 10 μM FAK inhibitor (PF-228). Cells were plated on 8.6 kPa ECMs.
(A) Localization of eGFP-tagged paxillins in FAs (cell edge is outlined in white).
(B) Mean FA size (n > 850 FAs from 7 cells).
(C) Fraction of FAs ≥ 1.5 μm (n > 850 FAs from 7 cells).
(D) Western blot of cell lysates for myosin II regulatory light chain (MLC) and serine 19 phosphorylated MLC (pMLC). MEFs treated with 20 μM ML-7 and 10 μM Y-27632 for 2 hr (PxnWT ML-7) were used as a control.
(E) Box plot of total cellular traction normalized to total FA area upon inhibiting FAK (n = 6 cells) or altering paxillin phosphorylation (PxnY31/118E, n = 14 cells or PxnY31/118F, n = 12 cells).
(F) Above: Box plot of peak traction position relative to the FA center within single FAs (PxnWT, n = 150 FAs; PF-228, n = 223 FAs; PxnY31/118E, n = 443 FAs; PxnY31/118F, n = 153 FAs). Grey rectangle highlights values that are not significantly different from the FA center, with distal and proximal directions indicated. Bottom: The fraction of TFM snapshots of FAs in which the position of peak traction was significantly skewed (>0.7 μm) toward the distal FA tip.
Values greater than 50% are marked in red. **p < 0.01, ***p < 0.001, NS p > 0.05 as detected by Mann-Whitney test. (B and C) Data shown as mean ± SEM. See also Figure S4.
We next examined the role of vinculin and the vinculin-paxillin interaction in regulation of FA traction dynamics. We suppressed vinculin expression with siRNA or altered the paxillin-vinculin interaction by depleting endogenous paxillin and re-expressing a GFP-tagged paxillin point mutant defective in vinculin binding (paxillinE151Q, Brown et al., 1996) (Figures 5A and 5B). Neither treatment affected FA size, the fraction of TFM-suitable FAs, or cell contractility (Figures 5C–5F). Furthermore, as with GFP-paxillin, GFP-paxillinE151Q localized within FAs (Figure S4). TFM analysis of FAs in cells plated on 8.6 kPa ECMs showed that although both tugging and stable FA traction states were observed (Figure S3B), both perturbations reduced traction stress, decreased the mean position of peak traction within the FA, and reduced the fraction of FAs with the traction peak skewed toward the distal FA tip (Figures 5G and 5H). Together, these data suggest that the signaling pathway composed of FAK, phospho-/dephospho-paxillinY31/118, and a paxillin-vinculin interaction promotes FA force transmission and tugging FA traction dynamics in cells adhered to 8.6 kPa ECMs.
Figure 5. Depleting Vinculin or Inhibiting Paxillin-Vinculin Interaction Reduces FA Force Transmission and Depletes Tugging FA Traction Dynamics.
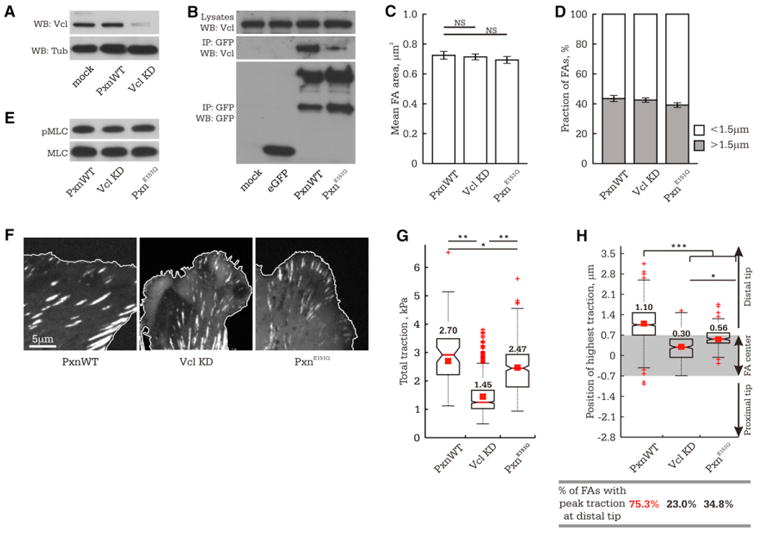
MEFs were cotransfected with nontargeting or vinculin-targeting (VclKD) siRNAs and eGFP-tagged wild-type paxillin (PxnWT) or with paxillin-targeting siRNAs and eGFP-tagged siRNA-resistant paxillin mutant (PxnE151Q).
(A) Western blot (WB) of siRNA-mediated depletion of vinculin (VclKD) in MEFs (72 hr after transfection), with tubulin (Tub) as loading control.
(B) Anti-GFP immunoprecipitations (IP) of mock-, eGFP-, eGFP-paxillin, or eGFP-paxillinE151Q-transfected MEFs, followed by analysis by WB with antibodies to vinculin and GFP. The top band in the anti-GFP immunoblot is eGFP-paxillin, the lower band a commonly observed degradation product.
In (C)–(H), cells were plated on 8.6 kPa ECMs.
(C) Mean FA size (n > 850 FAs from 7 cells).
(D) Fraction of FAs ≥ 1.5 μm (n > 850 FAs from 7 cells).
(E) WB of cell lysates for MLC and pMLC. (F) eGFP-tagged paxillins in FAs (cell edge, white outline).
(G) Box plot of total cellular traction normalized to total FA area (n = 6 cells per treatment).
(H) Above: Box plot of peak traction position relative to the FA center within single FAs (PxnWT, n = 150 FAs; VclKD, n = 283 FAs; PxnE151Q, n = 204 FAs). Grey rectangle highlights values that are not significantly different from the FA center, with distal and proximal directions indicated. Bottom: The fraction of TFM snapshots of FAs in which the position of peak traction was significantly skewed (>0.7 μm) toward the distal FA tip.
Values greater than 50% are marked in red. *p < 0.05, **p < 0.01, ***p < 0.001, NS p > 0.05 as detected by Mann-Whitney test. (C and D) Data shown as mean ± SEM. See also Figure S4.
A FAK/Phosphopaxillin/Vinculin Signaling Pathway Regulates the Range of the ECM Rigidities over which FAs Exhibit Tugging Traction Dynamics
We next sought to determine whether the FAK/paxillin/vinculin signaling module regulates the response of FA traction dynamics to differing ECM rigidities. To test this, we performed TFM analysis of FAs in cells plated on a range of ECM rigidities and treated with FAK inhibitor, expressing phospho- or vinculin-binding mutants of paxillin, or with vinculin depleted, as described above. Although varying ECM stiffness under some perturbations affected mean FA size, under all conditions, at least 30% of FAs were large enough for TFM analysis (Figure S5B). In control cells, increasing ECM rigidity from 8.6 to 32 kPa decreased the mean position of peak traction within FAs and reduced the fraction of FAs with the traction peak skewed toward the distal tip, indicating that stiffer ECMs promote stable FA traction (Figures 3G and 6A). In contrast, increasing ECM rigidity from 8.6 to 32 kPa for any of the experimentally perturbed cells changed neither the mean position of peak traction within the FA nor the fraction of FAs with the traction peak skewed toward the distal FA tip (Figure 6A). Conversely, for control cells, decreasing ECM rigidity from 8.6 to 4.1 kPa did not significantly change the mean position of peak traction and fraction of FAs with peak traction located at the distal tip (n = 7 FAs, p > 0.1, Figure 6B). However, for cells with FAK/paxillin/vinculin signaling perturbed, decreasing ECM rigidity from 8.6 to 4.1 kPa increased the mean position of peak traction within the FA and increased the fraction of FAs with the traction peak skewed toward the distal tip (Figure 6B). Thus, in control cells, tugging FA traction dynamics are induced by an ECM stiffness of 8.6 kPa or less, whereas for cells with the FAK/paxillin/vinculin signaling perturbed, the rigidity-dependent induction of tugging FA traction dynamics is not prevented, but the threshold for promoting these dynamics is shifted to a softer ECM (4.1 kPa).
Figure 6. A FAK/Paxillin/Vinculin Signaling Module Regulates the Range of the ECM Rigidities over which FAs Exhibit Traction Dynamics.

MEFs were plated on 4.1 (blue), 8.6 (green), or 32 kPa (orange) ECMs. Cells were cotransfected with nontargeting siRNAs and eGFP-tagged wild-type paxillin (PxnWT) or with paxillin-targeting siRNAs and eGFP-tagged siRNA-resistant paxillin mutants (phosphomimetic [PxnY31/118E], non-phosphorylatable [PxnY31/118F], or vinculin-binding deficient [PxnE151Q]), or they were treated with vinculin-targeting siRNAs and eGFP-paxillin (VclKD). Cells were additionally treated with 10 μM PF-228 to inhibit FAK (PF-228) or 1 μM Y-27632 to inhibit ROCK (light green bars in C and D).
(A–C) Top panels: Box plots of the position of peak traction within FAs in cells plated on ECMs of various stiffness. Grey rectangle highlights the values that are not significantly different from the FA center, with distal and proximal directions indicated. Bottom panels: The fraction of TFM snapshots of FAs in which the position of peak traction was significantly skewed (>0.7 μm) toward the distal FA tip measured at the same experimental condition as in the panels directly above. n>200 FA for each experimental condition. Values greater than 50% are marked in red. *p < 0.05, **p < 0.01, ***p < 0.001, NS p > 0.05 as detected by Mann-Whitney test.
(D) Phosphorylation of MLC assayed by WB analysis (top panel). Loading controls: total MLC and GFP (middle and bottom panels, respectively).
See also Figure S5.
We next sought to determine whether ROCK activity mediates the rigidity-dependent regulation of FA traction dynamics independent of FAK/paxillin/vinculin signaling. TFM analysis of cells plated on 8.6 kPa ECMs with FAK/paxillin/vinculin signaling perturbed showed that low-level ROCK inhibition had no significant effect on FA size (Figure S5A) yet increased the mean position of peak traction within the FA and increased the fraction of FAs with the traction peak skewed toward the distal tip (Figure 6C). Thus, ROCK activity shifts FAs from tugging to stable traction states, independent of FAK activity, paxillin phosphoregulation cycle, vinculin, or a paxillin-vinculin interaction. Together, these results suggest that the FAK/paxillin/vinculin signaling pathway does not promote either stable or tugging traction behavior per se but is essential to strengthen FAs and sustain ROCK-mediated FA traction dynamics over a wider range of ECM rigidities.
Reducing Intracellular or Extracellular Tension Promotes Tugging FA Traction Dynamics
To determine whether the ECM stiffness-dependent change in FA traction dynamics is caused by downregulation of myosin II contractility, we examined whether myosin II activity correlated with changes in FA traction dynamics induced by either reducing ECM rigidity or inhibiting ROCK activity. We analyzed pMLC level in cells in which the predominant state of FA traction dynamics was controlled by a specific combination of ECM rigidity, treatment with ROCK inhibitor, and molecular perturbation. This showed that FA traction dynamics did not correlate with myosin II activity. Decreasing ECM rigidity from 8.6 to 4.1 kPa had little effect on pMLC for both controls and cells in which FAK/phosphopaxillin/vinculin signaling was perturbed (Figure 6D) but had opposing effects on FA traction dynamics. In controls, this change in ECM rigidity had no effect on FA traction dynamics, but in cells with perturbed signaling, reducing ECM rigidity promoted tugging FA traction (Figure 6B). In contrast, ROCK inhibition of cells adhered to 8.6 kPa ECMs substantially reduced pMLC under all conditions (Figure 6D) yet promoted traction fluctuations for cells with perturbed FAK/phosphopaxillin/vinculin signaling, but not for controls (Figure 6C). These results suggest that tugging FA traction dynamics are not caused directly by downregulation of myosin II contractility, but rather that release of tension on FAs either externally (by ECM rigidity) or internally (by myosin II) is sufficient to induce FA traction dynamics.
Tugging FA Traction Dynamics Are Dispensable for FA Maturation, Chemotaxis, and Haptotaxis but Are Critical to Migration Speed and Durotaxis
To determine the physiological significance of FA traction dynamics, we assessed rigidity-dependent FA maturation and random or directed migration in cells in which FA traction dynamics were controlled by specific combinations of ECM rigidity and molecular perturbation. We focused on paxillin mutants defective in either Y31/118 phosphoregulation (paxillinY31/118E) or vinculin binding (paxillinE151Q) plated on different ECM stiffnesses (4.1 or 8.6 kPa) in the presence or absence of ROCK inhibitor (1 μM). These treatments had no effect on the fraction of FAs amenable to TFM or the distributions of paxillin or vinculin within individual FAs (Figures S5 and S6). As shown above (Figure 6), expression of these mutants tagged with eGFP in a paxillin siRNA background inhibited tugging FA traction fluctuations on rigid ECMs (8.6 or 32 kPa), but decreasing ECM rigidity to 4.1 kPa or inhibiting ROCK activity induced tugging FA traction dynamics.
To determine whether FA traction dynamics and FA growth are mechanistically linked, we analyzed FA size in control and mutant-expressing cells plated on a range of ECM rigidities. Increasing ECM stiffness increased FA area in control cells as expected; however, phosphomimetic or vinculin binding-deficient paxillin mutants abrogated this response (Figure 7A). Thus, paxillin phosphoregulation and a vinculin-paxillin interaction are required for ECM rigidity-dependent FA growth but only mediate traction fluctuations at specific ECM rigidities (Figure 6B).
Figure 7. Tugging FA Traction Dynamics Slow Random Cell Migration, Are Dispensable for FA Maturation, Chemotaxis, and Haptotaxis, but Are Critical to Durotaxis.
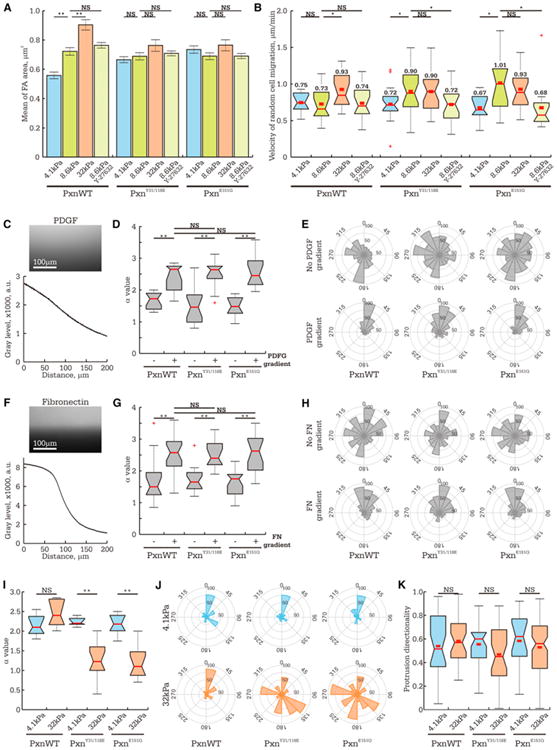
MEFs were treated with nontargeting siRNAs and expressed eGFP-tagged wild-type paxillin (PxnWT), or endogenous paxillin was suppressed by siRNAs, and eGFP-tagged siRNA-resistant paxillin mutants were expressed (phosphomimetic [PxnY31/118E] or vinculin binding-deficient [PxnE151Q]) and imaged by phase-contrast microscopy.
(A and B) Cells were plated on 4.1 (blue), 8.6 (green), or 32 kPa (orange) ECMs or treated with 1 μM Y-27632 to inhibit ROCK (light green bars). (A) Mean FA size (>600 FA per condition). Data shown as mean ± SEM (B) Box plots of instantaneous cell migration velocities (n > 7 cells for each condition).
(C–J) Analysis of cell migration in response to biochemical gradient as assayed with microfluidic chambers (C–H) or stiffness gradient (I and J).
(C and F) Fluorescent images (top panel) of dextran-Cy5 (C, molecular weight [MW] = 10 kDa) or substrate-bound rhodamine-FN (F), with line scans of intensity across the microfluidic chamber (bottom panels).
(D,G, and I) Box plots of directional persistence, α, quantified by fitting a plot of MSD overtime (Figures S7A-S7C) data with a power law model (MSD(τ) = 4D*τα, Suraneni et al., 2012).
(E, H, and J) Rose diagrams of cell migration (22.5° bins) showing the angle of each turn in the migration track relative to the gradient, with the radius indicating number of measurements.
(D, E, G, and H) Comparison of even distribution (−) or concentration gradient (+) of soluble PDGF and substrate-bound FN.
(I and J) Comparison of migration on strained ECMs of different stiffness.
(K) Box plots of asymmetry in membrane protrusion relative to direction of ECM stiffness gradient. The data represent the fraction of cell protrusion area (10 min interval), which extends toward stiffer ECMs (n > 10 cells per condition).
See also Figures S6 and S7 and Movies S3 and S4.
To determine the role of tugging FA traction dynamics in cell migration, we measured velocity of control and mutant-expressing cells undergoing random migration on a range of ECM rigidities. This showed that conditions that promoted traction fluctuations within FA corresponded to slower random migration. Cells expressing either wild-type or mutant paxillins migrated at similar speeds on 4.1 kPa ECM (Figure 7B), where all exhibit tugging FA traction dynamics (Figure 6B), whereas migration velocity was significantly faster on the most rigid ECM (32 kPa, Figure 7B), where stable FA traction predominates for all conditions (Figure 6A). On 8.6 kPa ECM, control cells exhibited tugging FA traction (Figures 3G and 6B) and migrated at a slow speed similar to that on softer 4.1 kPa ECMs (Figure 7B). In contrast, on 8.6 kPa ECM, cells bearing paxillin mutants exhibited stable FA traction (Figure 6B) and migrated faster than those on 4.1 kPa ECM and at speeds similar to those of both control and experimental cells on more rigid 32 kPa ECM (Figure 7B). Inhibition of ROCK activity in cells bearing paxillin mutants plated on 8.6 kPa ECM also decreased the velocity of cell migration. The drug-induced reduction in cell velocity was specific to induction of FA traction fluctuation, as treatment of control cells plated on 8.6 kPa ECMs with ROCK inhibitor did not reduce migration velocity (Figure 7B). This demonstrates that ROCK-dependent FA traction fluctuations slow random cell migration.
To determine the role of paxillin Y31/118 phosphoregulation or vinculin binding in directed cell migration toward biochemical cues, we assessed the effects of mutants on chemotaxis toward platelet-derived growth factor (PDGF) and haptotaxis toward fibronectin (FN) (Figures 7C and 7F). We determined the mean square displacement (MSD) over time from cell motion tracks and fitted it to a random movement model (MSD(τ) = 4D*τα; Suraneni et al., 2012) (Figures S7A and S7B) in which the greater the exponent α, the more directionally persistent the movement (Figures 7D and 7G). We also determined the “compass index,” measured as the angle of each turn in the track relative to the direction of the gradient (Arrieumerlou and Meyer, 2005). This showed that cells migrated randomly in uniform distributions of PDGF or FN and directionally toward gradients of these cues, independent of expression of mutant paxillins (Figures 7D, 7E, 7G, and 7H). Similar results were obtained in Boyden chamber assays (Figures S7D and S7E). This suggests that neither paxillin phosphoregulation nor vinculin binding is required for chemotaxis or haptotaxis on stiff ECMs.
To assess the role of FA traction dynamics in durotaxis, we utilized the assay of Wang et al. (2001). Cells were plated on FN-coupled PAA substrates, and a tangential strain in the direction away from a cell was applied to the substrate with a microneedle (10 μm from the cell edge) to locally pull the PAA to its elastic limit (Figure S7F). Because strain decays with distance from the point of application (Landau and Lifshitz, 1970), nonlinear strain stiffening of the PAA only occurs close to the needle, creating a local rigidity gradient toward the needle. For controls, quantification of either MSD or compass index showed that cells migrated directionally up the stiffness gradient independent of the bulk ECM rigidity (Movies S3 and S4; Figures 7I, 7J, S7C, and S7F). In contrast, cells expressing paxillinY31/118E or paxillinE151Q mutants migrated toward the rigidity gradient when plated on soft ECMs (4.1 kPa), but had significantly reduced migration persistence and randomized migration directionality on more rigid (32 kPa) ECM (Movie S4; Figures 7I, 7J, S7C, and S7F). This indicates that paxillin phosphoregulation and a paxillin-vinculin interaction are not required for durotaxis per se, but that these perturbations narrow the range of ECM rigidities to which cells respond. Interestingly, directional migration up the ECM-rigidity gradient was not due to a bias in the direction of membrane protrusion, as we found no difference in the area of protrusion extending toward either stiffer or softer ECM (Figure 7K). Together, these results suggest that tugging traction dynamics within FA slow random cell migration and promote durotaxis, whereas FA maturation, chemotaxis, and haptotaxis occur independent of the dynamic state of FA traction.
Discussion
We used high-resolution time-lapse TFM to characterize the distribution and dynamics of cell-generated forces on the ECM at the nanoscale within mature FAs. We found that maximal cytoskeleton-ECM force transmission is not at the site of stress fiber-FA attachment as predicted by modeling (Nicolas et al., 2004; Raz–Ben Aroush and Wagner, 2006) but rather near the FA tip toward the leading edge. Surprisingly, steady-state mature FAs that appear static by other methods of microscopy can actually possess internal fluctuations in mechanics. When traction is dynamic within FAs, it exhibits a pattern that is reminiscent of repeated, centripetal tugging on the ECM. Furthermore, the traction dynamics of neighboring FAs in a single cell are not correlated, indicating that individual FAs act autonomously. A FAK/ phosphopaxillin/vinculin pathway is essential for cells to exert high traction and to enable FA tugging over a broad range of ECM rigidities. Tugging traction in FA is dispensable for FA maturation, chemotaxis, and haptotaxis but is critical to direct cell migration toward rigid ECM.
We find that FAs exhibit tugging traction fluctuations on a wide range of ECM rigidities, but the choice of tugging versus stable traction states is tension dependent and is regulated by a specific signaling pathway. Reduction of tension on FAs by soft ECM or inhibition of ROCK-mediated contractility shifts FAs from stable to tugging traction states. ECM-rigidity-regulated traction fluctuations within FA have been predicted by a mechanical model of an FA molecular clutch (Chan and Odde, 2008), although the identity of the clutch molecules(s) was not determined. Fluctuation of talin length in mature FAs was also recently reported (Margadant et al., 2011), although its relation to ECM traction was not tested. We find that disruption of FAK activity, phosphoregulation of paxillin, or a vinculin-paxillin interaction reduces traction stress and lowers the threshold of rigidity that promotes tugging traction to softer ECM. We suggest that FAK-mediated phosphorylation of paxillin could induce FA recruitment of vinculin to locally strengthen the molecular clutch (Mierke et al., 2010; Fabry et al., 2011). Indeed, local variation in paxillin phosphorylation state (Zaidel-Bar et al., 2007) and vinculin-FA binding affinity (Wolfenson et al., 2009) have been observed within a single FA, and this could locally modulate FA traction. Our results suggest that strengthening the molecular clutch via the FAK/phosphopaxillin/vinculin pathway broadens the range of rigidities over which dynamic ECM-rigidity sampling operates.
The requirement for tugging FA traction in durotaxis suggests that tugging is a means of repeatedly sensing the local ECM-rigidity landscape over time. Individual FAs within a single cell all tugging autonomously thus could mediate dynamic ECM-rigidity sensing at the spatial resolution of FA density in cells (∼1–5 μm spacing). Cells migrating in a physiological three-dimensional (3D) ECM probe and sample a range of fibrils of different sizes and rigidities. High-density, dynamic ECM-rigidity sensing by individual tugging FAs could allow tight control of directional migration to guide cells along highly localized or dynamically changing ECM-rigidity gradients during durotaxis. Whether nascent adhesions at the leading edge contribute to durotaxis and how dynamic ECM-rigidity sampling by mature FAs is translated by the cell into spatial control of the cytoskeleton and adhesion dynamics to guide directed migration toward stiff ECMs remain to be determined. We suggest that FA-mediated sensing of local stiffness cues may be utilized in addition to biochemical gradient sensing of diffusible and immobilized cues to fine-tune cell pathfinding during development, morphogenesis, and pathological processes such as metastasis.
Experimental Procedures
Cell Culture, Transfection, and Reagents
MEFs were maintained and transfected with cDNAs encoding fluorescent fusion proteins, as in Pasapera et al. (2010). cDNA encoding paxillin-mApple was provided by Dr. M. Davidson (Florida State University). PaxillinE151Q was created by site-directed mutagenesis (QuikChange II, Agilent Technologies) with the following primers purchased from MWG-Biotech:
forward 5′-ggcttctcctgcaactgaatgctgttcaacataatcccccc-3′
reverse 5′ -ggggggattatgttgaacagcattcagttgcaggagaagcc-3′.
SmartPool siRNAs for paxillin, hic-5, and vinculin, as well as nontargeting siRNAs were purchased from Thermo Scientific. The following pharmacological inhibitors were used: 1 μM Y-27632 (EMD Millipore) and 10 μM PF-228 (PF-573228 Sigma-Aldrich).
High-Resolution TFM
High-resolution TFM was performed as described previously (Gardel et al., 2008; Sabass et al., 2008; also see the Extended Experimental Procedures).
For simultaneous measurement of eGFP-paxillin intensity and traction magnitude across individual FAs, we used a custom Matlab script. A line was drawn manually along the major axis of an FA, and the means of traction stress and eGFP-paxillin intensity were calculated within a 0.94 × 0.94 μm window centered at every pixel along the line. Because precise segmentation of FAs is challenging due to its sensitivity to local image intensity, we detected the pixel along this line with maximal eGFP-paxillin intensity and considered it as the center of the FA. The position of peak traction from the FA center was considered positive if it was skewed from center toward the distal tip of the FA (toward the leading edge).
To analyze stress vectors within specific regions of interest (ROI, either individual FAs or whole cells), a binary region mask was created by automated intensity thresholding of eGFP-paxillin images and dilated by 20 pixels to include vectors that enter the ROI but whose origins lay just outside the ROI. Traction vectors outside the cell were defined as background, and only vectors whose magnitudes ≥ 2× greater than background were included in the analysis. The sum of traction stresses per unit area was calculated and multiplied by the area of either an FA or cell.
Immunofluorescence, Immunoprecipitation, and Western Blot
Indirect immunofluorescence, immunoprecipitation, and western blot were performed as described (Pasapera et al., 2010) with mouse anti-paxillin (BD Bioscience) and anti-vinculin clone 4505 (Sigma-Aldrich) and rabbit anti-GFP (Abcam), anti-MLC, and anti-phospho-MLC (Cell Signaling).
Measurement of Random and Directed Cell Migration
Random cell migration on FN-coupled PAA substrates was assessed by time-lapse phase-contrast microscopy on PAA substrates lacking fluorescent beads. Cells were imaged for 12 hr at 5 min intervals with a 10× 0.3NA Plan Fluor objective lens (Nikon Instruments). Displacement of a cell between sequential frames was quantified by tracking the position of its nucleus with MetaMorph software (Molecular Devices).
Durotaxis assays were performed essentially as described by Wang et al. (2001), as follows. Cells expressing either wild-type or mutated paxillin tagged with eGFP were plated on 4.1 or 32 kPa FN-coupled PAA substrates, and cell behavior on the unstrained substrate was visualized for 10 min by time-lapse phase-contrast microscopy at 15 s intervals. To create a local rigidity gradient in the PAA gel, external strain was locally applied to the substrate at a location 10 μm away from a quiescent edge of a cell by pulling on the substrate with a microneedle mounted on a PatchMan NP2 micromanipulator (Eppendorf), and images were acquired for an additional 60 min. The substrate was strained to just within its elastic limit, i.e., to the point when increasing applied stress no longer changed substrate deformation but induced tearing of the PAA gel. With practice, this distance was simple to reach without damaging the PAA. The effect of substrate strain on the direction of cell migration was quantified by tracking the cell nucleus with MetaMorph (Molecular Devices).
Chemotaxis and haptotaxis were assayed with μ-Slide Chemotaxis microfluidic chambers according to manufacturer's instruction (Ibidi, further details in the Extended Experimental Procedures).
Analysis of Directional Migration
Two metrics were utilized to determine the directional persistence of cell movement. First, we determined the MSD over time from cell motion tracks and fitted it to a random movement model with the Matlab curve-fitting tool (MSD(τ) = 4D*τα; Suraneni et al., 2012). Second, we determined the “compass index,” measured as the angle of each turn in the track relative to the direction of the gradient (Wu et al., 2012).
Quantification of asymmetry in membrane protrusion relative to the direction of the ECM stiffness gradient was performed by a custom Matlab script. Briefly, bitmap images of cell edge were created by automated thresholding of cell migration movies acquired at 10 min intervals, and location of cell centroid was determined for every frame. Pairs of images were subtracted sequentially to segment cell protrusions, and the number of pixels in protrusion areas extending toward stiffer ECM was calculated and normalized to the total area of all protrusions.
Statistical Analysis
All measurements of peak traction position are shown as mean ± standard deviation (SD) and compared with Mann-Whitney tests. Box plots in all figures indicate the 25th percentile (lower bound), median (middle line), and 75th percentile (upper bound), with nearest observations within 1.5 times the interquartile range (whiskers), 95% confidence interval of the median (notches) and outliers (+), and means indicated above each plot.
Supplementary Material
Acknowledgments
We thank Mary Beckerle for mouse embryonic fibroblasts, Michael Davidson for mApple paxillin, William Shin for support of the Waterman Lab microscopes, Sharon Campbell and Robert Fischer for helpful suggestions, and the NHLBI flow cytometry core facility. This research was supported by the Division of Intramural Research, National Heart Lung and Blood Institute.
Footnotes
Supplemental Information: Supplemental Information includes Extended Experimental Procedures, seven figures, and four movies and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2012.11.034.
References
- Arrieumerlou C, Meyer T. A local coupling model and compass parameter for eukaryotic chemotaxis. Dev Cell. 2005;8:215–227. doi: 10.1016/j.devcel.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol. 1996;135:1109–1123. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CE, Odde DJ. Traction dynamics of filopodia on compliant substrates. Science. 2008;322:1687–1691. doi: 10.1126/science.1163595. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol. 2005;171:153–164. doi: 10.1083/jcb.200506152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld DW, Michael KE, Hanks SK, García AJ. Focal adhesion kinase-dependent regulation of adhesive forces involves vinculin recruitment to focal adhesions. Biol Cell. 2010a;102:203–213. doi: 10.1042/BC20090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld DW, Shin H, Gallant ND, Michael KE, Radhakrishna H, García AJ. Contractility modulates cell adhesion strengthening through focal adhesion kinase and assembly of vinculin-containing focal adhesions. J Cell Physiol. 2010b;223:746–756. doi: 10.1002/jcp.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Fabry B, Klemm AH, Kienle S, Schäffer TE, Goldmann WH. Focal adhesion kinase stabilizes the cytoskeleton. Biophys J. 2011;101:2131–2138. doi: 10.1016/j.bpj.2011.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan LA, Ju YE, Marg B, Osterfield M, Janmey PA. Neurite branching on deformable substrates. Neuroreport. 2002;13:2411–2415. doi: 10.1097/01.wnr.0000048003.96487.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J Cell Biol. 2008;183:999–1005. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo WH, Frey MT, Burnham NA, Wang YL. Substrate rigidity regulates the formation and maintenance of tissues. Biophys J. 2006;90:2213–2220. doi: 10.1529/biophysj.105.070144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber DE, Folkman J. How does extracellular matrix control capillary morphogenesis? Cell. 1989;58:803–805. doi: 10.1016/0092-8674(89)90928-8. [DOI] [PubMed] [Google Scholar]
- Janmey PA, McCulloch CA. Cell mechanics: integrating cell responses to mechanical stimuli. Annu Rev Biomed Eng. 2007;9:1–34. doi: 10.1146/annurev.bioeng.9.060906.151927. [DOI] [PubMed] [Google Scholar]
- Jiang G, Huang AH, Cai Y, Tanase M, Sheetz MP. Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophys J. 2006;90:1804–1809. doi: 10.1529/biophysj.105.072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch D, Rosoff WJ, Jiang J, Geller HM, Urbach JS. Strength in the periphery: growth cone biomechanics and substrate rigidity response in peripheral and central nervous system neurons. Biophys J. 2012;102:452–460. doi: 10.1016/j.bpj.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega J. Phototoxicity and photoinactivation of blebbistatin in UV and visible light. Biochem Biophys Res Commun. 2004;320:1020–1025. doi: 10.1016/j.bbrc.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Landau LD, Lifshitz EM. Theory of Elasticity. Second. Oxford: Pergamon Press; 1970. [Google Scholar]
- Mandeville JT, Lawson MA, Maxfield FR. Dynamic imaging of neutrophil migration in three dimensions: mechanical interactions between cells and matrix. J Leukoc Biol. 1997;61:188–200. doi: 10.1002/jlb.61.2.188. [DOI] [PubMed] [Google Scholar]
- Margadant F, Chew LL, Hu X, Yu H, Bate N, Zhang X, Sheetz MP. Mechanotransduction in vivo by repeated talin stretch-relaxation events depends upon vinculin. PLoS Biol. 2011;9:e1001223. doi: 10.1371/journal.pbio.1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierke CT, Kollmannsberger P, Zitterbart DP, Diez G, Koch TM, Marg S, Ziegler WH, Goldmann WH, Fabry B. Vinculin facilitates cell invasion into three-dimensional collagen matrices. J Biol Chem. 2010;285:13121–13130. doi: 10.1074/jbc.M109.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A, Geiger B, Safran SA. Cell mechanosensitivity controls the anisotropy of focal adhesions. Proc Natl Acad Sci USA. 2004;101:12520–12525. doi: 10.1073/pnas.0403539101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasapera AM, Schneider IC, Rericha E, Schlaepfer DD, Waterman CM. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J Cell Biol. 2010;188:877–890. doi: 10.1083/jcb.200906012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pelham RJ, Jr, Wang YL. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, Geiger B, Bershadsky AD. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat Cell Biol. 2011;13:1457–1465. doi: 10.1038/ncb2370. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz–Ben Aroush D, Wagner HD. Shear-stress profile along a cell focal adhesion. Adv Mater. 2006;18:1537–1540. [Google Scholar]
- Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabass B, Gardel ML, Waterman CM, Schwarz US. High resolution traction force microscopy based on experimental and computational advances. Biophys J. 2008;94:207–220. doi: 10.1529/biophysj.107.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider IC, Hays CK, Waterman CM. Epidermal growth factor-induced contraction regulates paxillin phosphorylation to temporally separate traction generation from de-adhesion. Mol Biol Cell. 2009;20:3155–3167. doi: 10.1091/mbc.E09-03-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–14852. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- Stricker J, Aratyn-Schaus Y, Oakes PW, Gardel ML. Spatiotemporal constraints on the force-dependent growth of focal adhesions. Biophys J. 2011;100:2883–2893. doi: 10.1016/j.bpj.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–251. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009;69:4167–4174. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Dembo M, Hanks SK, Wang YL. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci USA. 2001;98:11295–11300. doi: 10.1073/pnas.201201198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenson H, Lubelski A, Regev T, Klafter J, Henis YI, Geiger B. A role for the juxtamembrane cytoplasm in the molecular dynamics of focal adhesions. PLoS ONE. 2009;4:e4304. doi: 10.1371/journal.pone.0004304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007;120:137–148. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.