

Abstract
Peripheral artery disease (PAD) is characterized by chronic muscle ischemia. Compensatory angiogenesis is minimal within ischemic muscle despite an increase in angiogenic factors. This may occur due to the prevalence of angiostatic factors. Regulatory mechanisms that could evoke an angiostatic environment during ischemia are largely unknown. Forkhead box O (FoxO) transcription factors, known to repress endothelial cell proliferation in vitro, are potential candidates. Our goal was to determine whether FoxO proteins promote an angiostatic phenotype within ischemic muscle. FoxO1 and the angiostatic matrix protein thrombospondin 1 (THBS1) were elevated in ischemic muscle from PAD patients, or from mice post-femoral artery ligation. Mice with conditional endothelial cell-directed deletion of FoxO proteins (Mx1Cre+, FoxO1,3,4L/L, referred to as FoxOΔ) were used to assess the role of endothelial FoxO proteins within ischemic tissue. FoxO deletion abrogated the elevation of FoxO1 and THBS1 proteins, enhanced hindlimb blood flow recovery and improved neovascularization in murine ischemic muscle. Endothelial cell outgrowth from 3D explant cultures was more robust in muscles derived from FoxOΔ mice. FoxO1 overexpression induced THBS1 production, and a direct interaction of endogenous FoxO1 with the THBS1 promoter was detectable in primary endothelial cells. We provide evidence that FoxO1 directly regulates THBS1 within ischemic muscle. Altogether, these findings bring novel insight into the regulatory mechanisms underlying the repression of angiogenesis within peripheral ischemic tissues.
Keywords: Endothelium, Ischemia, Angiogenesis, Peripheral artery disease, Skeletal muscle, Capillary
Introduction
Peripheral arterial disease (PAD) involves the narrowing of arteries and results in impaired blood flow, usually to the lower limb, which ultimately leads to tissue necrosis and limb amputation [1-3]. Although a marked reduction of oxygen tension occurs in the peripheral tissue, which is a key stimulus for angiogenesis, the capillary density remains unchanged or even reduced within the ischemic lower leg muscles of PAD patients [4-7]. The molecular mechanisms underlying this paradoxical lack of angiogenesis within the ischemic muscles are not established. This consequence may be explained by the prevalence of intrinsic negative regulatory factors that act to counterbalance the pro-angiogenic signals. Considering that muscle capillarity is a significant determinant of oxygen extraction and muscle performance, and is a strong predictor of exercise tolerance in PAD patients [6, 8, 9], it is crucial to identify at the molecular level the mechanisms that restrain capillary growth within the ischemic muscle.
Previously, we demonstrated that prolonged moderate ischemia increases endogenous forkhead box O1 (FoxO1) protein within the microvasculature of rat skeletal muscle [10]. Forkhead box O (FoxO) transcription factors are established regulators of cell cycle inhibition, stress resistance, metabolism, autophagy and apoptosis [11-14]. FoxO1 is a critical regulator of embryonic vascular development. The loss of FoxO1, via either global or endothelial cell-restricted deletion, resulted in embryonic lethality due to severe defects in vascular network organization [15-17]. Furthermore, a role for FoxO1 in the postnatal maintenance of vascular homeostasis has been demonstrated, as the endothelial cell-directed deletion of FoxO1 in adult mice induced the development of severe hemangiomas as a consequence of excessive endothelial proliferation coupled with reduced apoptosis [18]. These results illustrate that a decrease in FoxO1 protein may promote a pathological pro-angiogenic switch.
In contrast, it remains unknown whether enhanced FoxO1 expression represents a barrier to angiogenesis in the adult, which would potentially contribute to the development or progression of ischemic diseases such as PAD. As of yet, an unequivocal role for FoxO1 in promoting an angiostatic environment within ischemic muscle has not been established. Furthermore, while some direct targets of FoxO have been validated in cultured endothelial cells [18, 19], critical effectors of the FoxO1-driven angiostatic response in vivo remain to be identified.
Here, we hypothesize that enhanced FoxO1 activity triggers an angiostatic phenotype in the endothelium of the ischemic muscle. We provide evidence of a pathological elevation of FoxO1 protein within the ischemic muscles of PAD patients, which correlated positively with an increase in the angiostatic matrix protein thrombospondin 1 (THBS1). FoxO1 and THBS1 also increased in a murine model of muscle ischemia. Directed deletion of FoxO1,3,4 within endothelial cells reduced THBS1 levels and significantly enhanced hindlimb blood flow recovery as well as angiogenesis within ischemic muscle. We establish that FoxO1 is a direct transcriptional regulator of THBS1 within microvascular endothelial cells. Our results provide mechanistic evidence that FoxO1, through promoting an angiostatic phenotype, plays a significant role in the pathophysiology of tissue ischemia.
Materials and methods
Human PAD and control biopsies
Human muscle biopsies were collected in accordance with a protocol approved by the Institutional Review Board of the Omaha Veterans Affairs Medical Center, Omaha, Nebraska and of the University of Nebraska Medical Center, Omaha, Nebraska. All subjects gave informed consent. Gastrocnemius muscle biopsies from male PAD and control subjects were collected at the University of Nebraska Medical Center (n = 14 per group). PAD patients presented with Stage II–IV according to the Fontaine classification system [20] (symptoms ranging from intermittent claudication to skin ulcers and gangrene). Non-PAD control subjects were age-matched to the PAD patients and the majority had one or more cardiovascular risk factors (cigarette smoking, diabetes mellitus, hypertension), but none of them had indication of PAD (as assessed by clinical history, physical examination and an ankle brachial index of no less than 0.9). See Table 1 for a summary of subject characteristics. The biopsies, obtained from the anteromedial region of the muscle belly (10 cm distal to the tibial tuberosity) while subjects were under general anesthesia, were snap-frozen in liquid nitrogen immediately upon collection. Western blotting of muscle homogenates was conducted as described below.
Table 1.
Demographics for control and PAD subjects
| Control | PAD | |
|---|---|---|
| n | 14 | 14 |
| Age (years)a | 62 ± 6 | 64 ± 7 |
| Body mass indexa | 28.6 ± 4.5 | 24.3 ± 4.9§ |
| Smoking (never/former/current)b | 7/2/5 | 5/1/8 |
| Coronary artery diseasec | 2 (14 %) | 10 (71 %) |
| Obesity | 6 (43 %) | 2 (14 %) |
| Dyslipidemia | 5 (36 %) | 10 (71 %) |
| IDDM | 1 (7 %) | 6 (43 %) |
| Hypertension | 7 (50 %) | 12 (86 %) |
| Brachial systolic pressurea (mmHg) | 132 ± 12.6 | 142 ± 16 |
| Ankle brachial index | 1.13 ± 0.11 | 0.31 ± 0.25∥ |
Mean ± SD
Number of subjects in each category
Number of subjects (% of sample)
p = 0.027 versus control
p < 0.0001 versus control
Mouse model of femoral artery ligation
All mouse protocols were approved by the York University Animal Care Committee and conducted in accordance with the American Physiological Society’s “Guiding Principles for the care and use of animals in research”. MxCre+;-FoxO1,3,4flox/floxor MxCre−, FoxO1,3,4flox/flox on a FVB/n background [18] were housed in pathogen-free conditions at the York University Vivarium. All mice received a series of three injections of polyinosine:cytidine (pI:pC) (400 μg per injection; Invivogen, San Diego, CA) at 4 weeks of age to transiently activate the Mx1 promoter, inducing Cre expression and resulting in the deletion of floxed FoxO alleles [21]. In all experiments, MxCre+ (FoxOΔ) mice were compared to control age-matched littermate MxCre− (FoxOL/L) mice. At 10–12 weeks of age, restriction of hindlimb blood flow was induced by unilateral ligation of the right femoral artery distal to the epigastric artery. Briefly, hair was removed from the lower limbs using depilatory cream. An incision was made in the medial thigh of the right hind limb to expose the femoral artery. The artery was separated from nerve and vein, and then was ligated tightly using double knots of 7-0 silk suture thread. The incision was closed with 5-0 silk sutures. Sham surgery consisted of all steps except for ligation of the artery. Ligation and sham surgeries were conducted under isoflurane inhalation anesthesia and mice were administered buprenorphine (0.05 mg/kg) at the time of surgery for pain relief. Hindlimb blood flow was assessed prior to and immediately after surgery and on days 4, 7 and 10 after ligation, by non-invasive laser Doppler imaging (LDI2-HR; Moor Instruments, Wilmington DE). Two or three lower limb perfusion images were recorded in each mouse at each time point, while the mouse was maintained at 37 °C under isoflurane inhalation anaesthesia. Software was used to delineate a region of interest (ROI) that encompassed the lower limb of the ligated limb, and an ROI of equivalent area for the contralateral non-ligated limb. At each time point, a perfusion ratio was generated by calculating the ratio of the ROI average pixel intensity in the ligated limb to that of the non-ligated limb. In some animals, tibialis anterior (TA) muscle blood flow also was assessed simultaneously in ligated and non-ligated limbs using dual laser Doppler needle probes (Probe 403, Peri-Flux, PeriMed Instruments, Ardmore, PA, USA).
Mouse muscle histology and capillary to fiber ratio
Capillary to muscle fiber ratio, an indicator of angiogenesis, was assessed on cross-sections of tibialis anterior (TA) and extensor digitorum longus (EDL) muscles, as described previously [10]. FoxO1, F4/80 and THBS1 immunostaining was conducted on 4 day post-ligation muscle cross-sections. Frozen muscle cross-sections were incubated with anti-FoxO1 (1:200, #2880 Cell Signaling, New England Biolabs, Pickering, ON, Canada), anti-F4/80 (1:100; #MF48000, Invitrogen Canada) or anti-THBS1 (1:100, #CLSG36856 Cedarlane Corp., Mississauga, ON, Canada), or the appropriate non-immune sera (normal rabbit IgG, #2729, Cell Signaling Technology; normal mouse IgG, #sc-2025, Santa Cruz). Primary antibodies were detected with secondary antibody conjugated to Alexa Fluor568 goat anti-rabbit or goat anti-mouse (#A11011 or A11004, respectively, Invitrogen, Burlington, ON, Canada) or Alexa Fluor488 goat anti-rat (112-545-003, Jackson ImmunoResearch, West Grove, PA, USA). Sections were counter-stained with FITC or rhodamine-conjugated Griffonia simplificolia isolectin-B4 (Vector #FL-1201 Burlington, ON, Canada) to detect capillaries. Staining was visualized using a Zeiss Inverted microscope (M200) equipped with a 40× oil immersion objective, a cooled digital CCD camera and Metamorph imaging software. Exposure settings were set manually and were identical for all specimens. Representative images were selected following examination of five to six tissue sections from each mouse (n = 3 per group). Multi-wavelength exposures were merged using Metamorph software.
Protein and RNA analyses
Murine primary microvascular endothelial cells and gastrocnemius muscle tissues from mice or human biopsies were lysed for protein or RNA extraction, and analyzed by Western blotting or qRT-PCR, as previously described [10].
Muscle explant cultures
Small segments of freshly isolated plantaris or soleus (approximately 0.1 mg) from FoxOΔ and FoxOL/L mice were embedded in type I collagen matrix, as described previously [22]. After 6 days of culture, the explants were fixed and migrating endothelial cells were stained for alkaline phosphatase reactivity to detect endothelial cells. The area of migration and the alkaline phosphatase intensity were calculated using Carestream software.
Endothelial cell culture
Primary microvascular endothelial cells were isolated from a combination of gastrocnemius, EDL and TA muscles of FoxOL/L or FoxOΔ mice, and maintained in culture as described previously [10]. Cells were used for experiments between passages 3 and 6. In some cases, transduction of FoxOL/L cells was carried out using adenoviruses encoding Cre recombinase (Adeno-Cre; Vector Biolabs, Philadelphia, PA, USA) to induce FoxO deficiency; wildtype FoxO1 (Adeno-WTFoxO1) or constitutively active FoxO1 (Adeno-CAFoxO1; both generously provided by Dr. Robert Gerard, University of Texas Southwestern) to over-express FoxO1 levels. Adeno-β-gal transduction was used as a negative control. All transductions were performed using 100 pfu per cell. For experiments, cells were serum-starved overnight (1 % FBS), then lysed for subsequent analysis of protein (RIPA lysis buffer) by Western blotting and mRNA (Cells to cDNA lysis buffer; Invitrogen Canada) by qRT-PCR.
Western blotting
20–40 μg of total cellular protein were denatured and equivalent amounts of protein were separated using SDS-PAGE, then transferred to polyvinylidene difluoride membranes (Immobilon P; Fisher Thermoscientific). After blocking with 5 % fat-free milk, membranes were probed overnight at 4 °C using antibodies as follows: FoxO1 (1:1,000, #2880 Cell Signaling, New England Biolabs, Pickering, ON, Canada); p27KIP1 (1:200, #sc-1641 Santa Cruz); thrombospondin 1 (1:400, clone A6.1; #CLSG36856, Cedarlane Corp.); β-actin (1:1,000, #4967 Cell Signaling), αβ-tubulin (1:1,000, #2148 Cell Signaling). After detection with appropriate secondary antibody conjugated with horseradish peroxidase, membranes were incubated with Super West Pico (Pierce; Fisher Thermoscientific, Whitby, ON, Canada) or Immobilon Western ECL (Millipore; Fisher Thermoscientific) as per manufacturer’s instructions and signals were detected using an imaging station (Kodak 4000MM Pro; Carestream Molecular Imaging; Woodbridge, CT, USA). Bands were quantified using Carestream or Image J software.
qRT-PCR
cDNA samples were analyzed by qPCR using Taqman probes using the ABI 7500 Fast PCR System (Invitrogen Canada; Burlington, ON, Canada) with qPCR mastermix (P/N 11743; Invitrogen Canada) and Taqman probes (Invitrogen Canada) as follows: GAPDH (Mm03302249_g1), HPRT-1 (Mm00446968_m1), p27KIP1 (Mm00438168_m1), FoxO1 (Mm00490672_m1), FoxO3 (Mm01185722_m1), mEMR1 (Mm00802529_m1), THBS1 (Mm01335418_m1). The comparative Ct method was used to determine relative quantification of mRNA expression, using HPRT or GAPDH to normalize for amount of RNA per sample.
Chromatin immunoprecipitation
Murine primary microvascular endothelial cells isolated from FoxOL/L mice (as described above) were serum starved overnight in DMEM containing 1 % FBS. Chromatin was crosslinked by formaldehyde incubation and cells were scraped then pelleted by centrifugation followed by resuspension in SDS lysis buffer. Crosslinked chromatin was sheared using a Bioruptor (15 cycles, 15 s shearing, 45 s at rest, 4 °C) to obtain DNA fragments to an average size 400–600 bp. One tenth of lysate was used for input and the rest was used for chromatin immunoprecipitation. Samples were pre-cleared (60 min, 4 °C) with protein A/G agarose beads (Upstate Biotechnology) then incubated with 6 μl of FoxO1 antibody (#2880 C29H4, Cell Signaling Technology) or normal rabbit IgG (#2729, Cell Signaling Technology) for 24 h. The immune complexes were collected by adsorption to protein A/G agarose beads (2 h, 4 °C). Beads were washed once with immune complex wash buffer (20 mM Tris–HCl pH 8.1, 0.1 % SDS, 1 % Triton X-100, 2 mM EDTA) containing 150 mM NaCl (10 min) and once with the immune complex wash buffer containing 500 mM NaCl (15 min). Beads were washed with a final buffer containing 10 mM Tris–HCl pH 8.1, 1 mM EDTA, 1 % deoxycholic acid, 1 % IGEPAL-CA630, 0.25 M LiCl for 30 min. After two washes with TE buffer (2 × 5 min), the immune complexes were eluted with 0.1 M NaHCO3 containing 1 % SDS (2 × 15 min at room temperature). Elutes were adjusted to 200 mM NaCl and incubated at 65 °C with 3 μg of RNase overnight. After purification using the QiaAmp DNA Micro kit (#56304, Qiagen, Toronto, ON, Canada), DNA fragments were amplified by PCR reaction. Primers (based on the murine THBS1 or Cdkn1b sequences (Genbank #M62449.1 and NC_000072, respectively)) were selected using the Primer Blast program (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Details of primer sequences, location and product sizes are listed in Online Resource Table 1. Products were analyzed by 2 % TAE agarose gel containing ethidium bromide. Grayscale images were inverted to enhance visualization of the bands.
Statistical analyses
Results were expressed as mean ± SEM, and analyzed by two-tailed Student’s t test, one-way ANOVA or two-way ANOVA and Bonferroni post-tests (Prism4; Graphpad Software Inc; La Jolla, CA, USA). Pearson r correlation coefficients were calculated using Prism 4. In all cases, p values of less than 0.05 were considered statistically significant.
Results
Concomitant upregulation of FoxO1 and thrombospondin 1 proteins occurs within human and rodent ischemic muscle
Previously, we reported significant increases in the protein level of FoxO1 within the endothelial cells of ischemic rat muscle [10]. It remains unknown whether expression of FoxO proteins is altered in the pathological context of PAD. To address this, we assessed FoxO protein levels within gastrocnemius muscle biopsies obtained from PAD patients or age-matched control subjects. PAD patients had moderate to severe stages of the disease, and a significant reduction of the ankle brachial pressure index (0.31 ± 0.25 vs. 1.13 ± 0.11 in the control group, p < 0.05). Characteristics of the PAD and control populations are summarized in Table 1. We analysed protein levels of FoxO1 and FoxO3a, because these FoxO proteins are most abundantly expressed by endothelial cells [23]. FoxO1 protein levels were significantly higher in PAD subjects compared to the control subjects (p = 0.013, 0.41 ± 0.05 vs. 0.62 ± 0.05, respectively), while FoxO3a protein levels were not different between control and PAD subjects (p = 0.73, 1.02 ± 0.09 vs. 0.93 ± 0.21, respectively, Fig. 1a). Similarly, FoxO1, but not FoxO3a levels correlated inversely with ankle-brachial index, a measure of disease severity (Online Resource 1). Levels of the FoxO target gene, p27KIP1, were increased in the PAD muscle biopsies (p = 0.047, 0.34 ± 0.05 vs. control 0.26 ± 0.02, Fig. 1a). We also observed an increase in the amount of the angiostatic matrix protein thrombospondin 1 (THBS1) in the muscle of PAD patients compared to control subjects (0.22 ± 0.10 vs. 0.10 ± 0.03, respectively, Fig. 1a). A significant positive correlation was observed between expression of FoxO1 and p27KIP1 protein (Fig. 1b), which may be interpreted as an indication of increased FoxO1 transcriptional activity within the ischemic tissue. Notably, FoxO1 protein level also correlated significantly with THBS1 levels (Fig. 1b), leading us to postulate that a regulatory link may exist between these proteins.
Fig. 1.
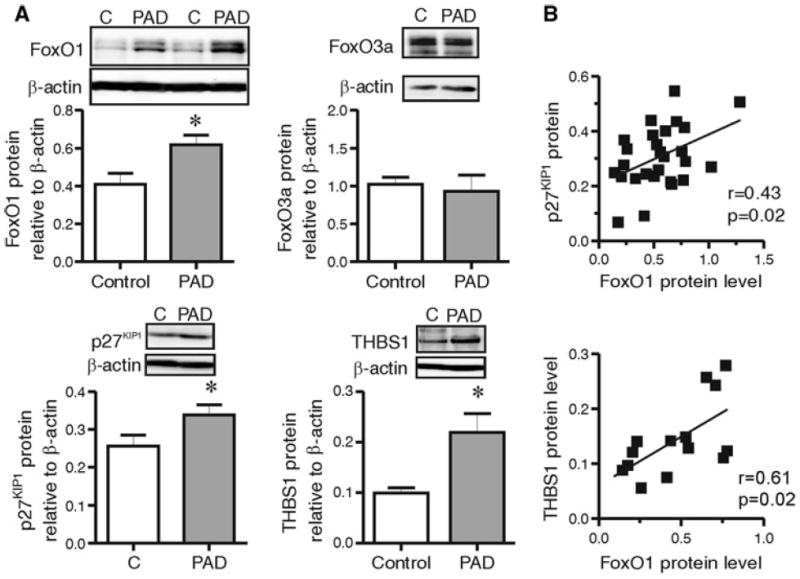
FoxO1, but not FoxO3a, protein level increases in response to PAD and is associated with enhanced p27KIP1 and THBS1 protein expression. Gastrocnemius muscles from control and PAD subjects were assessed by Western blot a for FoxO1 and FoxO3a, p27KIP1 and THBS1 protein levels, and quantified relative to β-actin. Data are mean ± SEM; * indicates significant differences: p = 0.01 for FoxO1; p = 0.047 for p27KIP1; p = 0.01 for THBS1 (Unpaired two tailed Student’s t tests, n = 8–13 per group). b FoxO1 relative protein levels were compared with protein levels of p27KIP1 (top; N = 26) and THBS1 (bottom; N = 16). The Pearson’s correlation coefficient (r) was calculated to assess the relationships between FoxO protein levels and each of the specified parameters
FoxO and THBS1 expression levels also were examined in a mouse model of moderate hindlimb ischemia induced by unilateral femoral artery ligation. We found that FoxO1 protein levels increased significantly in gastrocnemius muscle after 4 and 7 days of ischemia (Fig. 2), consistent with our previous observation in a rat model of moderate muscle ischemia [10]. In contrast, FoxO3a protein levels were not altered in response to ischemia (Fig. 2). Levels of the angiostatic protein THBS1 also increased in response to ischemia, with a time course that corresponded to the increase in FoxO1 (Fig. 2). The observation of an up-regulation of FoxO1 and thrombospondin 1 protein expression both in the murine model of hindlimb ischemia and in the muscle from PAD patients provides evidence that this is a conserved phenomenon occurring within ischemic muscle.
Fig. 2.
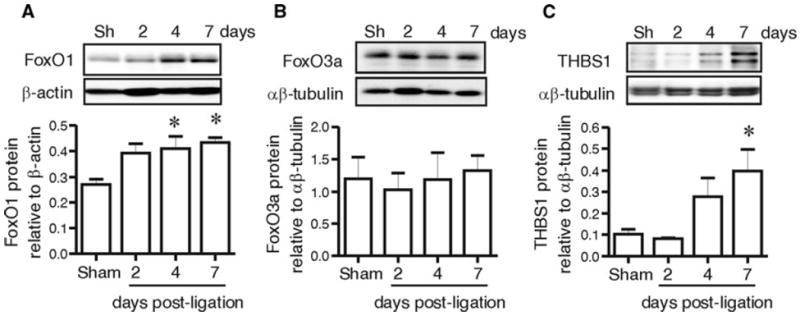
FoxO1, but not FoxO3a, protein level increases within ischemic mouse muscle, corresponding with an increase in THBS1 protein expression. FoxO1, FoxO3a and THBS1 (a, b, c) protein levels were assessed in murine gastrocnemius muscle 2, 4 or 7 days following femoral artery ligation and corresponding sham (Sh) operations and quantified relative to αβ-tubulin or β-actin. Data are mean ± SEM; n = 4–6. One-way ANOVA shows a main effect of arterial ligation on the levels of FoxO1 and THBS1 but not on FoxO3a (p = 0.02, p = 0.015 and p > 0.05, respectively, one way ANOVA). * indicates differences versus Sham (Bonferroni post hoc analysis, p < 0.05)
Endothelial cell–directed deletion of FoxO proteins prevents the up-regulation of FoxO1 in the ischemic muscle, and increases blood flow recovery and angiogenesis
A murine genetic model of conditional combined deletion of FoxO1,3,4, in which Cre expression is under the control of the inducible Mx1 promoter, was employed to assess the role of FoxO1 within ischemic tissue. The Mx1 promoter confines Cre expression to cells that exhibit interferon α/β responsiveness, which includes the endothelium, hematopoietic cells, as well as hepatocytes, and notably, not the skeletal myocytes [18, 24]. This model is useful particularly because it circumvent the embryonic lethality observed with constitutive endothelial cell specific deletion of FoxO1 [17]. Furthermore, targeting all three FoxO members avoided possible functional compensation by FoxO3 or FoxO4 [18]. Conditional deletion of FoxO alleles was confirmed by analysis of genomic DNA (Online Resource 2). Protein analysis of gastrocnemius homogenates indicated a partial reduction of FoxO1 and 3 levels (24 and 35 % reduction compared to controls, respectively) (Fig. 3a), which is consistent with FoxO deletion only within the vascular compartment of the muscle tissue. To directly assess the extent of endothelial deletion of FoxO proteins, we analyzed the endothelial cell fraction isolated from the muscles of FoxO-deleted (FoxOΔ and non-deleted (FoxOL/L) mice. As expected, there was a substantial reduction of FoxO1 and 3 protein levels in the endothelial fraction derived from the muscles of FoxOΔ mice (63 and 70 % reduction compared to control cells, respectively) (Fig. 3b). This is in agreement with previous report of Mx1Cre-mediated deletion of FoxO proteins within endothelial cells isolated from liver [18]. FoxO4 protein was undetectable within the endothelial cell fraction from FoxOL/L mice. Co-immunostaining of FoxO1 and Isolectin B4 (to localize capillaries) within TA/EDL muscles indicated the presence of FoxO1 within both capillaries and muscle fibers of FoxOL/L mice. In accordance with western blotting data, minimal capillary-associated FoxO1 staining was observed within the muscle of FoxOΔ mice (Fig. 3c).
Fig. 3.
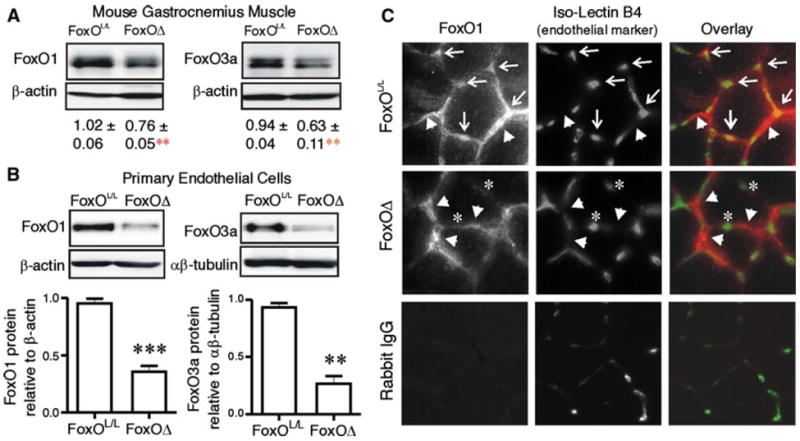
FoxO1 and FoxO3a deletion occur in skeletal muscle microvascular endothelial cells of FoxOΔ mice. FoxO1 and FoxO3a levels were measured by Western blotting on protein extracts of gastrocnemius muscles a harvested from 6 week old FoxOL/L and FoxOΔ mice. Protein level relative to β-actin is expressed as mean ± SEM. Differences vs. FoxOL/L are **p = 0.01 (n = 4–7 per group). FoxO1 and FoxO3a levels were assessed by Western blotting on protein extracts of microvascular endothelial cells b isolated from skeletal muscle of FoxOL/L and FoxOΔ mice. ***p < 0.0001 and **p < 0.01 (n = 3 to 4 independent cell isolations from a total of 9 mice per group). Cross-sections of TA/EDL muscles (4 days post femoral artery ligation) were immuostained c for FoxO1 (red) and Isolectin B4 (green). Arrows indicate capillaries positive for FoxO1 staining and arrowheads indicate myocyte/interstitial cell-associated FoxO1 immunoreactivity. Notably, FoxO1 staining did not overlap with Isolectin B4 staining in muscle cross-sections of FoxOΔ mice; asterisks highlight representative capillaries that do not exhibit FoxO1 immunoreactivity. Immunostaining of a FoxOL/L muscle section with normal rabbit IgG as a negative control for the FoxO1 antibody is shown in the bottom left hand panel
To further interrogate the involvement of FoxO proteins in promoting an angiostatic phenotype, we assessed the angiogenic process in skeletal muscle biopsies derived from non-ischemic soleus (oxidative) or plantaris (glycolytic) muscles of FoxOΔ and FoxOL/L mice. After 6 days of culture within a 3D collagen matrix, endothelial cells have proliferated and migrated out of the explant into the surrounding collagen matrix (Fig. 4a). Independently of the type of muscle, endothelial cell density and outgrowth were significantly greater in FoxOΔ compared to FoxOL/L muscle explants (Fig. 4b). Together, these results confirm the strong angiostatic effect of FoxO proteins on skeletal muscle endothelial cells, and validate the use of FoxOΔ mice to assess the functional contribution of endothelial FoxO proteins in response to muscle ischemia.
Fig. 4.
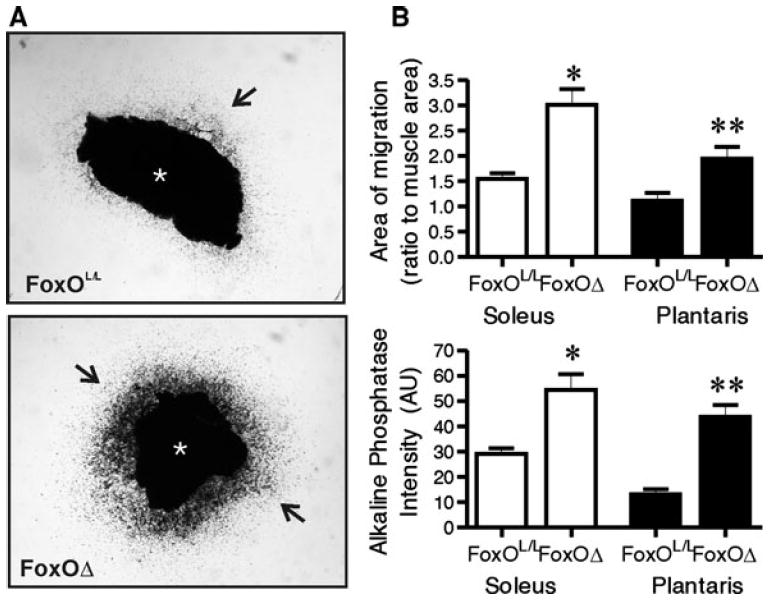
FoxO deletion promotes endothelial cell outgrowth from ex vivo muscle explants. Muscle explant culture was performed on soleus and plantaris muscle biopsies of non-ischemic FoxOL/L and FoxOΔ mice, and representative images of soleus biopsies are shown (a). The asterisk denotes the original muscle explant; arrows point to endothelial cell outgrowth. The area of migration (upper graph) and the endothelial cell density (lower graph) within the area of migration of explants derived from soleus (white bars) or plantaris (black bars) were calculated (b). Multiple explants from an individual mouse were averaged. Data show mean ± SEM (n = 4–6 mice per condition, *p < 0.05 and **p < 0.01 versus matched FoxOL/L muscle explants, unpaired two-tailed Student’s t test)
Femoral artery ligation was utilized to induce moderate ischemia in FoxOΔ and FoxOL/L mice. There was no indication of inflammation, or influx of monocytes, within ischemic TA/EDL muscle of either FoxOΔ or FoxOL/L mice (Online Resource 3), which is in accordance with our observations of moderately ischemic rat skeletal muscle. Western blotting confirmed that FoxO1 protein increased at 4 days post-ligation in the gastrocnemius muscle of FoxOL/L mice compared to sham-operated mice. In contrast, FoxO1 protein did not increase in the ischemic muscle of FoxOΔ mice following femoral artery ligation (Fig. 5). We next analyzed blood flow recovery and muscle capillarization following the induction of muscle ischemia. An initial reduction in hindlimb blood flow of approximately 50 % was observed in all mice after femoral artery ligation (Fig. 6a). In the subsequent 10 days, there was no significant improvement in limb perfusion in the FoxOL/L mice, while FoxOΔ ischemic limbs showed a recovery to approximately 85 % of pre-surgery levels at days 7 and 10. Significant differences in limb perfusion were observed between FoxOL/L and FoxOΔ mice at days 7 and 10 days post-ligation (Fig. 5a). Flow probe analysis of TA muscle blood flow at days 0, 4 and 10 post-ligation demonstrated a similar pattern of flow recovery as that observed with the blood flow imager (Online Resource 4). Significant differences in relative TA blood flow in FoxOL/L compared to FoxOΔ mice were apparent at day 10 days post-ligation. Furthermore, at 10 days post-ligation, relative blood flow was not significantly different in the ligated versus sham FoxOΔ mice while flow remained significantly different in the ligated compared to sham FoxOL/L mice. The capillary-to-fiber ratio (C:F) was measured as an indicator of angiogenesis or capillary regression within the ischemic muscle. 10 days post ligation, TA/EDL muscle C:F was not significantly different between FoxOL/L and FoxOΔ sham animals. However, C:F was significantly higher in the ischemic hindlimb muscle of FoxOΔ mice compared to FoxOL/L mice (Fig. 5b). Our results bring evidence that FoxO deletion promotes a pro-angiogenic phenotype in the vasculature of ischemic muscle.
Fig. 5.
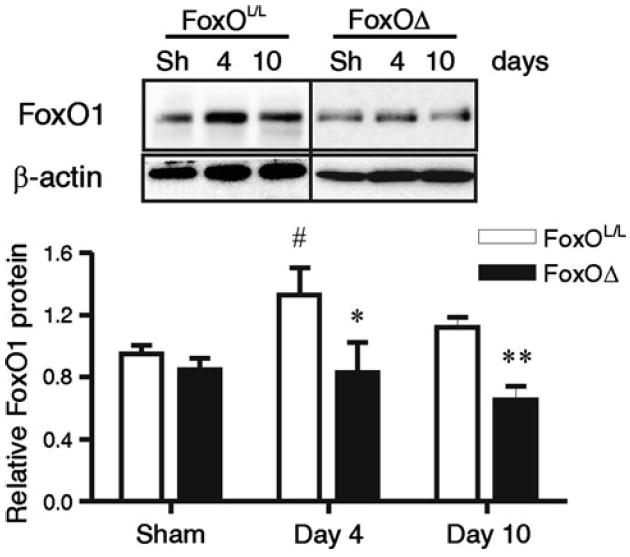
Ischemia-dependent increases in FoxO1 protein do not occur in FoxOΔ mice. FoxO1 protein levels were assessed at 4 and 10 days post-femoral artery ligation in gastrocnemius muscles of FoxOL/L and FoxOΔ mice. Protein levels were expressed relative to β-actin. Vertical line delineates two different blots. Data are mean ± SEM. Two-way ANOVA indicates a main effect of FoxO deletion (p < 0.0001, n = 4–18 per group). Significant differences between FoxOL/L and FoxOΔ are *p < 0.05 and **p < 0.01 (vs. time matched group), and #p < 0.05 (vs. corresponding sham, Bonferroni post hoc)
Fig. 6.
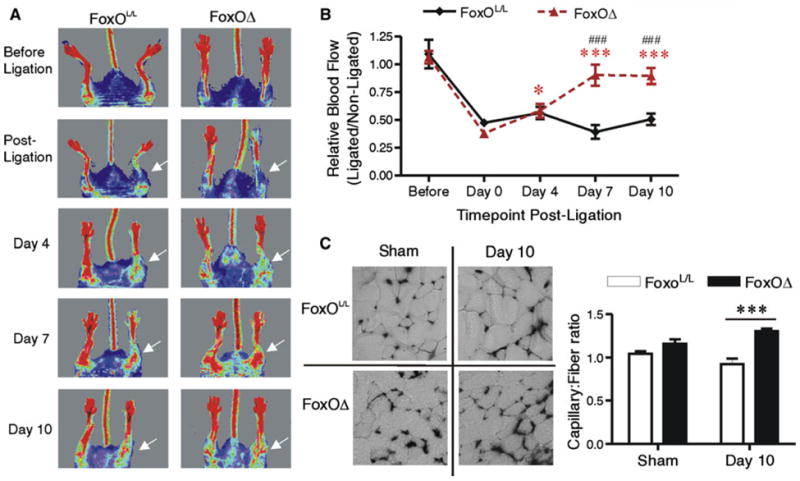
FoxO deletion promotes blood flow recovery and neovascularization following femoral artery ligation. Hindlimb blood flow of FoxOL/L and FoxOΔ mice a was assessed by Laser Doppler imaging before surgery, and 0, 4, 7, and 10 days after femoral artery ligation. A representative time course of images is shown for each mouse phenotype. White arrows indicate the ischemic limb in post-ligation time-points. Data are mean ± SEM of the ischemic/non-ischemic flow ratio. Two-way ANOVA shows main effects of both time and FoxO deletion (p < 0.0001 and p = 0.0008, respectively; n = 5–12 per group). Post hoc analysis indicated that blood flow ratios in FoxOΔ mice were significantly different at days 4, 7 and 10 compared to day 0 (*p < 0.05 and ***p < 0.001 vs. corresponding day 0). Furthermore, blood flow in FoxOΔ mice was significantly different from that of FoxOL/L mice at days 7 and 10 (###p < 0.001 vs. FoxOL/L mice at the corresponding time point). Capillary to fiber ratio b was assessed in TA/EDL muscles 10 days post-ligation. There was a main effect of FoxO deletion (p < 0.0001, n = 4–8 per group) and significant differences between FoxOL/L and FoxOΔ ***p < 0.001 (Bonferroni post hoc analysis)
Thrombospondin 1 is a downstream effector of FoxO proteins in ischemic muscle
Based on our observation of the co-ordinated expression pattern of FoxO1 and THBS1 in ischemic muscle (Figs. 1, 2), we tested whether FoxO deletion would result in altered THBS1 expression. First, we analyzed the expression of THBS1 in the endothelial cell fraction isolated from mouse skeletal muscle, and found that THBS1 mRNA and protein levels were reduced significantly in endothelial cells originating from FoxOΔ compared to FoxOL/L mice (Fig. 7a). Extending this observation, we reasoned that THBS1 expression also would be repressed within the ischemic muscle of FoxOΔ mice. THBS1 mRNA increased significantly 4 days post-ligation within the gastrocnemius muscle of FoxOL/L mice (Fig. 7b), which corresponded temporally with upregulation of FoxO1 protein and slightly preceded the increase of THBS1 protein (Fig. 2a, b). In contrast, no increase in THBS1 mRNA occurred in the ischemic muscles of FoxOΔ mice (Fig. 7b). Immunostaining of TA/EDL muscles showed high levels of peri-capillary THBS1 protein in FoxOL/L mice 4 days following femoral artery ligation, which was minimal in the FoxOΔ mice (Fig. 7c). These data provide evidence that Mx1 promoter-induced deletion of FoxO proteins acts to restrain the expression of THBS1 during ischemia.
Fig. 7.
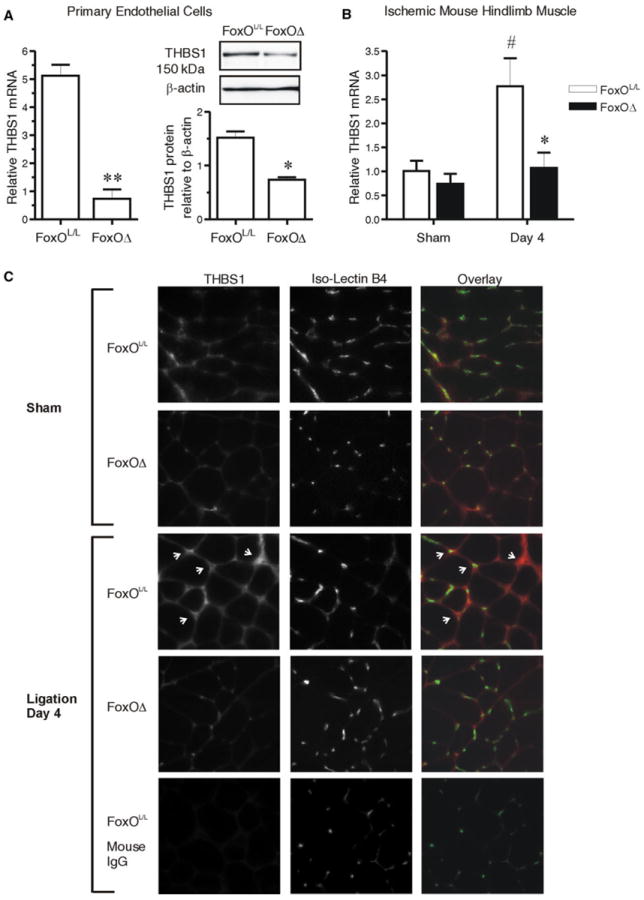
FoxO deletion prevents the ischemia-induced increase in Thrombospondin 1 within the endothelium of mouse muscle. Microvascular endothelial cells were isolated from skeletal muscle of FoxOL/L and FoxOΔ mice. THBS1 mRNA was assessed by q-PCR (normalized to Hprt1) and THBS1 protein a was assessed by Western blot (normalized to β-actin). **p = 0.004 compared to cells from FoxOL/L mice; *p = 0.01, paired Student’s t test, n = 3 independent isolations, from a total of 8 individual mice per condition. THBS1 mRNA gastrocnemius muscles from FoxOL/L and FoxOΔ mice b was assessed by q-PCR (normalized to Hprt1) at 4 days post-femoral artery ligation or corresponding sham operation. Two-way ANOVA shows main effects of FoxO deletion and arterial ligation (p = 0.04 and p = 0.03, respectively). Significant differences are #p < 0.05 versus Sham FoxOL/L, and *p < 0.05 versus day 4 ligated FoxOL/L (Bonferonni Post hoc, n = 4–7 mice). TA/EDL muscle cross-sections from 4 day ligated or sham operated FoxOL/L or FoxOΔ mice were immunostained c for THBS1 (red) and counterstained with Isolectin B4 (green) to identify capillaries. Arrows point to peri-capillary THBS1 immunoreactivity in the ischemic FoxOL/L tissue. Normal mouse IgG was used as a negative control for the THBS1 antibody
The altered phenotype of the FoxOΔ mice is best explained by the reduction in FoxO1 levels, considering that FoxO3a levels were not altered in response to ischemia, and FoxO4 was undetectable in endothelial cells. We therefore focused on investigating the relationship between FoxO1 and THBS1 in cultured primary microvascular endothelial cells. Induced deletion of FoxO proteins in cultured FoxOL/L endothelial cells (via adenoviral transduction of Cre recombinase) resulted in a reduction in THBS1 mRNA levels (Fig. 8a), consistent with the lower mRNA and protein levels of thrombospondin 1 in cells isolated from FoxOΔ mice (Fig. 7a). Conversely, over-expression of wild-type FoxO1 or its constitutively active mutant significantly increased THBS1 mRNA (Fig. 8b). Based on these data, we assessed whether FoxO1 interacts directly with the THBS1 promoter. Alignment analysis of human, canine and murine THBS1 genomic sequences utilizing ECR Browser/RVISTA identified one Forkhead binding element that was highly conserved between species, located within a 600 bp evolutionarily conserved region (ECR) at ~−800 bp relative to the transcription initiation site (Fig. 8c). A second putative Forkhead binding element was identified within the murine genomic sequence (~−1,990 bp), which also shared high homology with the human sequence. We conducted ChIP assays using primer sets to amplify each of these regions. FoxO1 ChIP of primary endothelial cells was validated by assessing a previously documented Forkhead binding element within the 5′ promoter region of Cdkn1b (Fig. 8d) [25, 26]. FoxO1 immunoprecipitation resulted in the amplification of both regions 1 and 2 within the THBS1 locus, but not of the coding region within THBS1 exon 3 (Fig. 8d), thus demonstrating the capacity for FoxO1 to interact directly with both binding elements in the THBS1 promoter region.
Fig. 8.
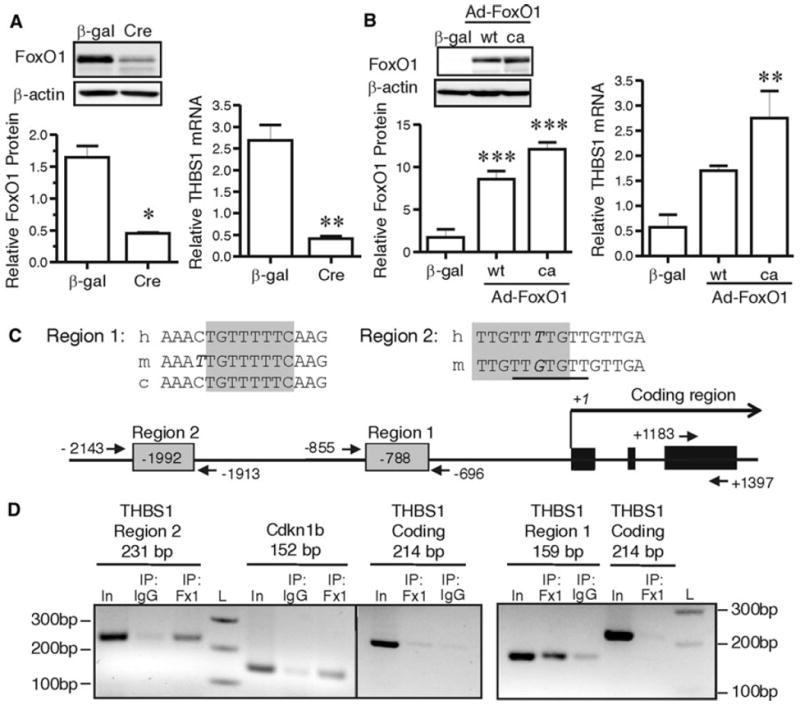
FoxO1 is a direct regulator of Thrombospondin-1. Deletion of FoxO gene products in primary skeletal muscle microvascular endothelial cells was induced by adeno-Cre infection (a). FoxO1 protein and THBS1 mRNA were measured after serum starvation. β-actin and Hprt-1 were used as loading control and housekeeping gene, respectively. *p = 0.02 and **p = 0.009 compared to control adeno-β-gal control transduction, paired Student’s t test, n = 3, 4 independent experiments. Overexpression of FoxO1 in microvascular endothelial cells was induced by Adeno-wt-FoxO1 (wt) or Adeno-ca-FoxO1 (ca) infection (b) and confirmed by Western blotting (***p < 0.0001 vs. adeno-β-gal control transduction, one way ANOVA and Bonferroni post hoc analysis, n = 4). THBS1 mRNA was measured by q-PCR (p = 0.005, one way ANOVA; **p < 0.01 compared to the β-gal transduced condition, Bonferroni post hoc analysis, n = 4). Alignment of conserved FoxO consensus elements within the THBS1 promoter (h human, m mouse, c canus) (c). The shaded boxes indicate the core consensus elements within each region. Base mismatches are denoted by italics. The underlined sequence in Region 2 indicates a second consensus site within the murine sequence. Consensus sites and primer locations within the THBS1 locus are illustrated. ChIP was performed using FoxO1 antibody or normal rabbit IgG (d). Primer sets specific for either Regions 1 and 2 within the THBS1 promoter, the Cdkn1b promoter, or the THBS1 coding sequence (exon 3) were used to do PCR on precipitated DNA. In, Input DNA; Fx1, α-FoxO1 antibody; IgG, normal rabbit IgG; L, 100 bp DNA ladder
Discussion
Here, we identify a regulatory mechanism that promotes an angiostatic environment within ischemic skeletal muscle. We provide novel evidence that the transcription factor FoxO1 is elevated within ischemic gastrocnemius of patients with moderate to severe PAD. Endothelium-directed deletion of FoxO1,3,4 in mice abrogated completely the increases in FoxO1 protein and improved both blood flow recovery and angiogenesis within ischemic hindlimb muscles. FoxO deletion prevented the ischemia-induced increase in the angiostatic matrix protein, THBS1. In primary cultured skeletal muscle endothelial cells, over-expression of FoxO1, and chromatin immunoprecipitation of endogenous FoxO1, demonstrated that FoxO1 is a regulator of THBS1 expression and that it binds to the THBS1 promoter. The upregulation of THBS1 within the ischemic muscle of PAD patients, together with the significant positive correlation between FoxO1 and THBS1 protein levels in human biopsies, provide further evidence that FoxO1 dependent regulation of THBS1 expression occurs in the context of PAD. Together, these findings suggest that the endothelial upregulation of FoxO1/THBS1 is an intrinsic signalling pathway that restrains angiogenesis within ischemic muscle.
Peripheral artery disease is a complex disease that is increasing in prevalence, and to date, there are few treatment options that have demonstrated the capacity to restore vascularization of the affected muscles [1-3]. Understanding the role of negative regulators of vascularization in ischemic muscle represents an alternative approach that may lead to development of more effective therapies. Here, we identify a novel link between PAD and the upregulation of the transcription factor FoxO1. This relationship was observed in muscle biopsies of PAD patients, as well as in a murine model of muscle ischemia. In fact, a significant negative correlation was seen between FoxO1 protein levels and either the ankle-brachial pressure index or the absolute ankle systolic pressure in PAD patients, which are used often as indicators of disease severity (Online Resource 1).
Previous studies have not assessed the role of FoxO1 in muscle ischemia, as both global and endothelial cell specific knock-out of FoxO1 results in embryonic lethality. We utilized the murine model of endothelial-directed conditional FoxO1,3,4 deletion [18] as a tool to investigate the potential angiostatic role of FoxO1 protein within ischemic muscle. Post-natal deletion of FoxO gene products, which is achieved using the Mx1-Cre model, is essential in order to avoid the lethality of embryonic FoxO1 deletion. The combined deletion of FoxO1,3,4 also prevents any functional compensation by FoxO3a and FoxO4 that may occur if only FoxO1 is deleted [15, 18]. Following the induction of moderate ischemia, which was characterized by an initial 50 % reduction in hindlimb perfusion, we observed that FoxO deletion greatly improved blood flow recovery and muscle neovascularization. Considering that FoxO3a levels did not change in response to ischemia, it is probable that endothelial FoxO1 is responsible for the functional consequences within the ischemic muscle. The endothelial-directed deletion of FoxO completely hindered FoxO1 up-regulation in the ischemic muscle. Furthermore, the substantial reduction of FoxO1 and FoxO3a within endothelial cells of the skeletal muscle of adult FoxOΔ mice together with the improved angiogenic capacity of these endothelial cells, as shown in the ex vivo migration assay, support the conclusion that Mx1-Cre-induced FoxO deletion resulted in a significant shift in endothelial cell phenotype. It is possible that altered FoxO expression in other cell types could contribute partially to the effects observed given that the Mx1 promoter activity is not exclusive to endothelial cells. The absence of infiltrating immune cells within the ischemic muscle indicates that this cell population is unlikely to play a role in the altered angiogenic response observed in the FoxOΔ mice.
Functional effects of FoxO upregulation occur via the modified transcription of downstream effectors, such as the cell cycle inhibitor, p27KIP1, which is an established transcriptional target of FoxO proteins [25, 26]. We previously observed a concomitant increased expression of FoxO1 and p27KIP1 proteins in rat ischemic skeletal muscle [10], and now we report a positive correlation between FoxO1 and p27KIP1 protein levels in human skeletal muscle biopsies. From these data, we infer that increased FoxO1 protein level corresponds with enhanced transcription of FoxO target genes. It is probable that FoxO1 orchestrates an angiostatic phenotype within the ischemic muscle through the regulation of an extended network of downstream targets. To date, many distinct FoxO targets have been identified in vitro in endothelial cells, such as eNOS, Sprouty2 and Ang2 [18, 23, 27]. However, not all FoxO1 targets identified by in vitro studies may be relevant in the context of muscle ischemia. For example, we did not observe changes in Sprouty2 mRNA expression within rat ischemic muscle [10]. Thus, we resolved to identify additional effectors of FoxO1 relevant to the vasculature of ischemic muscle.
The positive correlation between expression of FoxO1 and THBS1 led us to investigate a potential FoxO-dependent regulation of THBS1 within the ischemic muscle. THBS1 is known to exert strong angiostatic properties through the induction of anti-proliferative and pro-apoptotic signaling within endothelial cells [28-30]. THBS1 has been reported to be elevated in the plasma and within the ischemic muscle of PAD patients [31]. Increased THBS1 in a mouse model of muscle ischemia is associated with impaired blood flow recovery, while THBS1 deletion enhances blood flow recovery in the mouse ischemic limb [32, 33]. These investigations clearly indicate that THBS1 is a key participant in the skeletal muscle pathophysiology in PAD. However, the mechanisms leading to the upregulation of THBS1 in the context of PAD remained unknown. Here, our results bring new insight into the signalling mechanism responsible for thrombospondin 1 overexpression. Loss of the ischemia-induced expression of THBS1 mRNA in the FoxOΔ mice brings evidence for a FoxO-dependent upregulation of THBS1 within in the vasculature of the ischemic muscle. Direct perturbation of FoxO1 levels in cultured endothelial cells confirmed the FoxO1 dependent regulation of THBS1. We demonstrated FoxO1 binding to two distinct forkhead binding elements within evolutionarily conserved regions of the THBS1 promoter, thus identifying the THBS1 gene as a direct FoxO1 target.
In conclusion, this investigation identifies endothelial FoxO1 as a transcriptional key that unlocks a strongly angiostatic phenotype within ischemic muscle. Our study indicates that upregulation of FoxO1-dependent genes, such as in the muscle of PAD patients, may be sufficient to over-ride compensatory pro-angiogenesis signals. This regulatory influence may underlie the poor angiogenic response often observed in ischemic tissues. Furthermore, we have delineated FoxO1 as a critical regulator of THBS1, which brings new insight into the mechanism responsible for THBS1 overexpression within ischemic tissue. Altogether, these data suggest that the FoxO1-THBS1 signal axis promotes an angiostatic phenotype in the vasculature of ischemic muscle, thus playing a potential role in the pathophysiology of PAD.
Supplementary Material
Acknowledgments
We appreciate the technical assistance of Ms. Justyna Kopycinska and Mr. Emmanuel Nwadozi with animal surgeries and imaging, and Mr. Sammy Liu with endothelial cell isolations. This research was supported by the Canadian Institutes of Health Research (IMH-107537 to T.L.H.), the Heart and Stroke Foundation of Canada (NA7059 to T.L.H.) and the National Institutes of Health Grant (R01 AG034995 to I.P.).
Abbreviations
- PAD
Peripheral artery disease
- THBS1
Thrombospondin 1
- FoxO
Forkhead box O
- C:F
Capillary to muscle fiber ratio
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10456-013-9353-x) contains supplementary material, which is available to authorized users.
Conflict of interest None.
Contributor Information
Emilie Roudier, Angiogenesis Research Group, Faculty of Health, York University, Rm. 341 Farquharson Building, 4700 Keele St., Toronto, ON M3J 1P3, Canada.
Malgorzata Milkiewicz, Medical Biology Laboratory, Pomeranian Medical University, Szczecin, Poland.
Olivier Birot, Angiogenesis Research Group, Faculty of Health, York University, Rm. 341 Farquharson Building, 4700 Keele St., Toronto, ON M3J 1P3, Canada.
Dara Slopack, Angiogenesis Research Group, Faculty of Health, York University, Rm. 341 Farquharson Building, 4700 Keele St., Toronto, ON M3J 1P3, Canada.
Andreas Montelius, Division of Clinical Physiology, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden.
Thomas Gustafsson, Division of Clinical Physiology, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden.
Ji Hye Paik, Department of Pathology and Laboratory Medicine, Weill Cornell College, New York, NY 10065, USA.
Ronald A. DePinho, Department of Cancer Biology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
George P. Casale, Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198, USA
Iraklis I. Pipinos, Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198, USA
Tara L. Haas, Email: thaas@yorku.ca, Angiogenesis Research Group, Faculty of Health, York University, Rm. 341 Farquharson Building, 4700 Keele St., Toronto, ON M3J 1P3, Canada.
References
- 1.Criqui MH, Fronek A, Klauber MR, Barrettconnor E, Gabriel S. The sensitivity, specificity, and predictive value of traditional clinical-evaluation of peripheral arterial-disease: results from noninvasive testing in A defined population. Circulation. 1985;71:516–522. doi: 10.1161/01.cir.71.3.516. [DOI] [PubMed] [Google Scholar]
- 2.Criqui MH. Peripheral arterial disease: epidemiological aspects. Vasc Med. 2001;6:3–7. doi: 10.1177/1358836X0100600i102. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, Hiratzka LF, Murphy WRC, Olin JW, Puschett JB, Rosenfield KA, Sacks D, Stanley JC, Taylor LM, White CJ, White J, White RA, Antman EM, Smith SC, Adams CD, Anderson JL, Faxon DP, Fuster V, Gibbons RJ, Halperin JL, Hiratzka LF, Hunt SA, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol. 2006;47:1239–1312. doi: 10.1016/j.jacc.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Henriksson J, Nygaard E, Andersson J, Eklof B. Enzyme activities, fibre types and capillarization in calf muscles of patients with intermittent claudication. Scand J Clin Lab Invest. 1980;40:361–369. doi: 10.3109/00365518009092656. [DOI] [PubMed] [Google Scholar]
- 5.Jansson E, Johansson J, Sylven C, Kaijser L. Calf muscle adaptation in intermittent claudication. Side-differences in muscle metabolic characteristics in patients with unilateral arterial disease. Clin Physiol. 1988;8:17–29. doi: 10.1111/j.1475-097x.1988.tb00258.x. [DOI] [PubMed] [Google Scholar]
- 6.Robbins JL, Jones WS, Duscha BD, Allen JD, Kraus WE, Regensteiner JG, Hiatt WR, Annex BH. Relationship between leg muscle capillary density and peak hyperemic blood flow with endurance capacity in peripheral artery disease. J Appl Physiol. 2011;111:81–86. doi: 10.1152/japplphysiol.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones WS, Duscha BD, Robbins JL, Duggan NN, Regensteiner JG, Kraus WE, Hiatt WR, Dokun AO, Annex BH. Alteration in angiogenic and anti-angiogenic forms of vascular endothelial growth factor-A in skeletal muscle of patients with intermittent claudication following exercise training. Vasc Med. 2012;17:94–100. doi: 10.1177/1358863X11436334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Askew CD, Green S, Walker PJ, Kerr GK, Green AA, Williams AD, Febbraio MA. Skeletal muscle phenotype is associated with exercise tolerance in patients with peripheral arterial disease. J Vasc Surg. 2005;41:802–807. doi: 10.1016/j.jvs.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 9.Duscha BD, Robbins JL, Jones WS, Kraus WE, Lye RJ, Sanders JM, Allen JD, Regensteiner JG, Hiatt WR, Annex BH. Angiogenesis in skeletal muscle precede improvements in peak oxygen uptake in peripheral artery disease patients. Arterioscler Thromb Vasc Biol. 2011;31:2742–2748. doi: 10.1161/ATVBAHA.111.230441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milkiewicz M, Roudier E, Doyle JL, Trifonova A, Birot O, Haas TL. Identification of a mechanism underlying regulation of the anti-angiogenic forkhead transcription factor FoxO1 in cultured endothelial cells and ischemic muscle. Am J Pathol. 2011;178:935–944. doi: 10.1016/j.ajpath.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol. 2002;4:850–858. doi: 10.1038/ncb867. [DOI] [PubMed] [Google Scholar]
- 12.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–2319. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem. 2011;286:7468–7478. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hosaka T, Biggs WH, III, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci USA. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Nakayama K, Ikeda K, Motoyama N, Mori N. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 17.Sengupta A, Chakraborty S, Paik J, Yutzey KE, Evans-Anderson HJ. FoxO1 is required in endothelial but not myocardial cell lineages during cardiovascular development. Dev Dyn. 2012;241:803–813. doi: 10.1002/dvdy.23759. [DOI] [PubMed] [Google Scholar]
- 18.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta. 2007;1775:298–312. doi: 10.1016/j.bbcan.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FGR. Inter-society consensus for the management of peripheral arterial disease (TASC II) J Vasc Surg. 2007;45:S5–S67. doi: 10.1016/j.jvs.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 22.Roudier E, Forn P, Perry ME, Birot O. Murine double minute-2 expression is required for capillary maintenance and exercise-induced angiogenesis in skeletal muscle. FASEB J. 2012;26:4530–4539. doi: 10.1096/fj.12-212720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of FoxO transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EWF, Burgering BMT, Raaijmakers JAM, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27KIP1. Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lees SJ, Childs TE, Booth FW. Age-dependent FOXO regulation of p27Kip1 expression via a conserved binding motif in rat muscle precursor cells. Am J Physiol Cell Physiol. 2008;295:C1238–C1246. doi: 10.1152/ajpcell.00349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, Rudge JS. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes Dev. 2004;18:1060–1071. doi: 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 30.Iruela-Arispe ML, Luque A, Lee N. Thrombospondin modules and angiogenesis. Int J Biochem Cell Biol. 2004;36:1070–1078. doi: 10.1016/j.biocel.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 31.Favier J, Germain S, Emmerich J, Corvol P, Gasc JM. Critical overexpression of thrombospondin 1 in chronic leg ischaemia. J Pathol. 2005;207:358–366. doi: 10.1002/path.1833. [DOI] [PubMed] [Google Scholar]
- 32.Smadja DM, d’Audigier C, Bièche I, Evrard S, Mauge L, Dias JV, Labreuche J, Laurendeau I, Marsac B, Dizier B, Wagner-Ballon O, Boisson-Vidal C, Morandi V, Duong-Van-Huyen JP, Bruneval P, Dignat-George F, Emmerich J, Gaussem P. Thrombospondin-1 Is a plasmatic marker of peripheral arterial disease that modulates endothelial progenitor cell angiogenic properties. Arterioscler Thromb Vasc Biol. 2011;31:551–559. doi: 10.1161/ATVBAHA.110.220624. [DOI] [PubMed] [Google Scholar]
- 33.Brechot N, Gomez E, Bignon M, Khallou-Laschet J, Dussiot M, Cazes A, Alanio-Brechot C, Durand M, Philippe J, Silvestre JS, Van Rooijen N, Corvol P, Nicoletti A, Chazaud B, Germain S. Modulation of macrophage activation state protects tissue from necrosis during critical limb ischemia in thrombospondin-1-deficient mice. PLoS One. 2008;3:e3950. doi: 10.1371/journal.pone.0003950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.