

Abstract
BACKGROUND
Diastolic dysfunction is a poorly understood but clinically pervasive syndrome that is characterized by increased diastolic stiffness. Titin is the main determinant of cellular passive stiffness. However, the physiological role that titin’s tandem Ig segment plays in stiffness generation and whether shortening this segment is sufficient to cause diastolic dysfunction needs to be established.
METHODS AND RESULTS
We generated a mouse model in which nine immunoglobulin (Ig)-like domains (Ig3-11) were deleted from the proximal tandem Ig segment of titin’s spring region (IG KO). Exon microarray analysis revealed no adaptations in titin splicing, while novel phospho-specific antibodies did not detect changes in titin phosphorylation. Passive myocyte stiffness was increased in the IG KO and immunoelectron microscopy revealed increased extension of the remaining titin spring segments as the sole likely underlying mechanism. Diastolic stiffness was increased at the tissue and organ levels, with no consistent changes in ECM composition or ECM-based passive stiffness, supporting a titin-based mechanism for in-vivo diastolic dysfunction. Additionally, IG KO mice have a reduced exercise tolerance, a phenotype often associated with diastolic dysfunction.
CONCLUSIONS
Increased titin-based passive stiffness is sufficient to cause diastolic dysfunction with exercise intolerance.
Keywords: Passive stiffness, elasticity, extracellular matrix, exercise, hypertrophy
INTRODUCTION
Although much research has been focused on LV systolic function, understanding normal and pathologic diastolic function is of great clinical significance as well1–4. It has been hypothesized that the giant myofilament titin plays an important role in diastolic function1, 3, 5, 6. Titin spans from Z-disk to M-band of the sarcomere and has an extensible I-band region that functions as a molecular spring that largely defines cardiomyocyte passive stiffness7. Alteration in titin isoform expression is a mechanism that changes the extensibility of titin’s I-band and modulates titin’s passive stiffness in health8–10 and disease11, 12. The extensible I-band region of titin is comprised of the N2B and PEVK segments along with proximal and distal tandem Ig segments composed of serially-linked immunoglobulin(Ig)-like domains13. Mouse models absent of either the N2B or PEVK segments have previously been created and show increased passive stiffness14, 15. However, even though the extension of the tandem Ig segment dominates titin’s elasticity at physiological sarcomere lengths (SL)8, 16, its in-vivo physiologic roles have not been addressed. Hence, we made a genetic model that has a shortened tandem Ig segment and evaluated how this alters diastolic function of the heart. Unlike the N2B and PEVK segments, the tandem Ig segment that was removed has no known phosphorylation sites in cardiac muscle17–20. Thus, shortening of the tandem Ig segment is expected to give rise to a pure model of mechanical stiffness increase that will make it possible to test the effect of an increase in titin-based stiffness on diastolic function.
The mouse model is deficient in titin exons 30-38, which deletes 9 of titin’s 15 Ig domains (Ig 3-11) from the proximal I-band segment (Fig. 1A), a model that is referred to as the IG KO. The IG KO model can be viewed as a ‘mechanical analog’ of the increased titin-based stiffness that is known to occur in HFpEF patients21 and thus might be beneficial in elucidating disease mechanisms in HFpEF. We studied passive stiffness over a wide range of increasing physiologic complexity, including the cardiomyocyte, muscle, and the ex-vivo and in-vivo LV chamber, and we assessed additional adaptations in titin, other sarcomeric proteins, and the extracellular matrix. Because of their association with HFpEF, we also evaluated whether stiffer titin altered cardiac hypertrophy by evaluating trophicity and hypertrophic signaling1, 22, and whether stiffer titin reduced exercise tolerance using treadmill and volunteer running wheel exercise23.
Figure 1.

Basic characterization of the IG KO mouse model. A) Location of Ig 3-11 (deleted in the IG KO) in the spring region of titin (Ig domains are indicated by the rectangular red structures). B) PCR products showing differential gene expression from WT, heterozygous and homozygous IG KO mice. C) Titin exon microarray analysis shows titin exon expression changes only in the 9 deleted exons. D) Titin protein analysis (1% agarose gel). Top: the shortened titin (IG KO) has a higher mobility when compared to the WT titin bands and a doublet can be seen in the HET mice. Bottom: Quantitative analysis shows that titin isoform expression is unchanged (n=6). E) Titin phosphorylation is unchanged in the IG KO. Top: PKA back-phosphorylation and phospho-specific pS26 and pS170 antibodies to PKC Western blotting examples. Bottom: quantification showing unchanged phosphorylation levels in the IG KO (n=4). CB: coomassie blue; AR: autoradiography; PonS: Ponceau S; box denotes 25th and 75th percentile and whiskers display min and max. See Supplemental Figure S7 for dot plots.
MATERIALS AND METHODS
An expanded Methods section is available in the online supplement.
GENERATION OF MICE EXPRESSING SHORTER TITIN IG SEGMENT
A targeting construct was assembled to replace titin exons 30-38 (encoding Ig3-11) with a floxed neomyocin expression cassette, which was subsequently removed (Fig. S1A). Mice were bred on a C57BL/6 background for 8 generations and only males were studied. Animal experiments were approved by the University of Arizona Institutional Animal Care and Use Committee and followed the U.S National Institutes of Health “Using Animals in Intramural Research” guidelines for animal use.
PROTEIN EXPRESSION, PHOSPHORYLATION AND GENE EXPRESSION
Titin and sarcomeric protein expression analysis was performed using standard SDS-PAGE methods24, 25. Phosphorylation was studied using ProQ diamond staining and phosphor-specific antibodies14–17, 19, 26. Quantitative Real Time-PCR (qPCR) was used to study gene expression27, 28.
IMMUNOELECTRON MICROSCOPY AND HISTOLOGY
Immunoelectron microscopy was used to measure titin segment extension. Picrosirius red (PSR) staining was used to measure the collagen volume fraction in LV cross-sections29.
CARDIAC MECHANICS
Using previously published techniques, we measured passive stress-SL relationships in skinned LV cardiomyocytes30 and muscle strips31. LV diastolic wall stress volume and wall stress-SL relationships were determined via an isolated heart method16.
IN-VIVO CHARACTERIZATION
In-vivo pressure-volume measurements were obtained using an admittance-based system in anesthetized mice14. Exercise tolerance was evaluated using both treadmill and voluntary running wheel tests.
STATISTICS
Mann-Whitney (Wilcoxon rank sum) was used to assess differences between genotypes. Results are shown as mean ± SEM, with p<0.05 taken as significant.
RESULTS
Generation of a mouse model with a shortened proximal tandem Ig segment (IG KO)
Exons 30-38 were constitutively deleted from the mouse titin gene and replaced with a neomycin resistance cassette that was subsequently removed (Fig. S1A). These exons code for 9 immunoglobulin-like domains (Ig 3-11) that are part of the proximal tandem Ig segment (Fig. 1A). Homozygous IG KO mice are fertile and survive to old age. Our custom titin exon microarray9 further validated that a loss of expression of exons 30-38 occurred without adaptive changes in splicing elsewhere in titin (Fig. 1C). We studied whether shortening of the proximal Ig segment affects titin protein expression and posttranslational modifications on titin. The deleted 9 immunoglobulin-like domains comprise 783 amino acids, representing an 88kDa polypeptide. Using a 1% agarose gel electrophoresis system, the mutant titin is resolved as a slightly higher mobility band compared to wildtype (WT) titin (Fig. 1D). Changes in isoform splicing and phosphorylation can also modulate passive stiffness and we evaluated whether such compensatory changes occurred in the IG KO mice. Quantification of titin isoform expression (N2BA:N2B ratio), titin cleavage product (T2), and the total titin (TT) to myosin heavy chain (MHC) ratio indicates no changes in titin expression in the IG KO mice at the studied ages (3–12 months) (Fig. 1D, bottom panel show results from 3 mo old mice). PKA back-phosphorylation assays19 and Western blots using antibodies to the PKC phosphorylation sites in the PEVK region (Fig. 1E)26, along with ProQ Diamond staining reveal no change in posttranslational modifications of titin (Fig. S1B,C). Similarly, quantification of myosin isoform ratios along with thin and thick filament protein expression (cMyBP-C, Desmin, Actin, TnT, Tm, TnI, MLC2v) and phosphorylation reveal no changes (Fig. S1B,C). In summary, we made an IG KO mouse that expresses a shortened proximal tandem Ig segment with only 6 of the 15 proximal Ig domains in WT titin, without changes elsewhere in the titin molecule or in other any of the other sarcomeric proteins analyzed.
Cellular stiffness
To investigate the effect of shortening the proximal tandem Ig segment on passive stiffness, we performed experiments on LV skinned cardiac myocytes. Stretch-hold-release protocols on passive skinned cardiomyocytes (Fig. 2A, inset shows representative results) revealed that peak and steady state stress are increased in the IG KO by 60% and 65%, respectively (Fig. 2B,C). Passive stress measured during the stretch was converted to stiffness (slope of stress-SL relationship); stiffness was increased in the IG KO at all SLs (Fig. 2D). The increase was 26±21% at 2.0 μm and 95±22% at 2.3 μm (average increases in the SL range of 2.0–2.3 μm is 63%). We also performed a dynamic stiffness analysis using small amplitude sinusoidal length oscillations to determine the elastic and viscous moduli at multiple SL (see Supplemental Methods for details). The elastic moduli were several-fold larger than the viscous moduli in both genotypes (Fig. 2E,F). In the IG KO cells, the elastic moduli were higher than in WT cells and the mean value across the frequency range that we probed was increased ~85% at each of the 4 SLs that were studied (Fig. 2E). In contrast, the viscous moduli were increased 50% at short SLs but >85% at SLs longer than 2.2 μm (Fig. 2F). These myocyte data indicate that shortening the proximal tandem Ig segment of titin greatly increases cellular passive stiffness mainly due to increases in elastic stiffness.
Figure 2.

Cardiomyocyte mechanical properties in relaxing solution. A) Passive tension is higher in the IG KO cell (n=5 for WT and IG KO). Inset: Representative stretch-hold-release experiment. From these experiments, both peak stress (B) and steady state stress (C) is increased in the IG KO cells. D) Passive stiffness measured during the ramp stretch is also increased in the IG KO mouse at SLs>2.0 μm. Dynamic stiffness analysis using small amplitude sinusoidal length oscillations to determine elastic (E) and viscous (F) moduli of the myocytes. Elastic stiffness is increased at all SLs>2.0μm but viscous properties are increased only at SLs≥2.2 μm (n=6). Box denotes 25th and 75th percentile and whiskers display min and max, * p<0.05; **p<0.01. See Supplemental Figure S8 for dot plots.
Immunoelectron microscopy
To examine the effect of reducing the length of the proximal tandem Ig segment on the extensibility of the remaining I-band segments, we performed immunoelectron microscopy. The antibodies that were used to demarcate the ends of the spring segments are shown in Fig. 3A along with a schematic of titin’s I-band region. Examples of labeled sarcomeres are in Fig. 3B, and scattergrams of mid Z-disk to epitope distances are shown as a function of SL in Fig. 3C,D. A large decrease in the end-to-end length of the proximal tandem Ig segment length in the IG KO across the measured sarcomere length range is readily apparent. At a SL of 2.3 μm, extension of the proximal tandem Ig segment was reduced by ~60% in the IG KO; the remaining I-band segments extended to a higher degree. Extension of the PEVK and N2B segments increased, with the largest increase in the N2B segment (Fig.3F). At a SL of 2.3 μm, which is near the upper limit of the physiologic SL range16, the proximal tandem Ig extends ~25 nm less than in the WT and the N2B segment accommodates ~75% of the ‘missing extension’ and the PEVK ~25% (Fig. 3E,F). The difference between the N2B and PEVK extension is consistent with their different contour lengths (~200nm and 70nm, respectively) and persistence length (~0.65 and ~1.0 nm, respectively) that results in a longer end-to-end length for a given force level in the N2B segment than in the PEVK segment (see also discussion). The extension of the distal tandem Ig segment increased minimally in the IG KO, in line with the relatively high stiffness of this segment32. Thus, the deletion of titin Ig domains reduces the extension in the proximal Ig segment and increases the extension of the N2B and PEVK segments.
Figure 3.

Titin I-band segment extension. A) Antibodies were used to demarcate the tandem Ig, N2B and PEVK segments of titin and B) labeled skinned fibers from 6 WT and IG KO cardiac sarcomeres were studied using immunoelectron microscopy. Z-disk to epitope distances were calculated for WT (C) and KO (D) sarcomeres across a range of SLs. Black lines indicate the lengths of N2B and Proximal Ig segments. E) Segment extension estimated via monoexponential fits indicate that the proximal Ig segment is shortened by nearly 30nm at 2.3 μm and that the extension of the remaining I-band segments is increased with N2B segment extension dominating (F).
Integrative physiology and diastolic dysfunction
Myocardial passive stiffness is determined by titin and the extracellular matrix (ECM)7. To study if the extracellular stiffness adapts in response to the increased cellular passive stiffness of the IG KO, we studied skinned myocardial muscle. We determined titin-based and ECM-based stiffness following myofilament extraction7. Titin-based stiffness of skinned myocardium, calculated from the extraction sensitive stress, was significantly increased in the IG KO tissues at SLs of 2.05 μm and greater (data not shown), similar to the increase measured in cells. The ECM-based stress was not significantly different in the IG KO (Fig. 4A). Importantly, ECM-based stiffness, calculated from the extraction insensitive stress-SL relation, was not different in the IG KO compared to the WT (Fig. 4B). qPCR revealed a significant decrease in the expressions of collagen Iα1 and IIIα1 at 3 mo, which normalized at 12 mo (Fig. S2A–C). Expressions of matrix metalloproteinases (MMP), tissue inhibitors of metalloproteinases (TIMP, and LOX were unchanged (except for a decrease in TIMP2 at 3 mo), suggestive of similar rates of ECM turnover and crosslinking (Fig. S2D–H). PSR staining of hearts suggested no differences in the collagen-volume fraction (Fig. 4C, Fig. S2I). Thus, the ECM is unlikely to change the stiffness in IG KO cardiac muscle compared to WT, and only titin-based stiffness is increased.
Figure 4.

Integrative Physiology and Diastolic Dysfunction at 3 mo. A) ECM based stress-sarcomere length relationships are unchanged in the IG KO (n=8, WT open diamonds; n=10 IG KO gray diamonds). ECM based stiffness (B), as well as collagen volume fraction by PSR staining (n=6) (C) is not changed. Ex-vivo isolated heart data provides wall stress-sarcomere length relationships (D) that show increased stiffness in the IG KO at physiologic sarcomere lengths (2.0–2.3 μm) (E) (n=12). In-vivo end diastolic stress volume relationships (EDSVR) exhibits a 37% increase in stiffness (F) (n=10). Horizontal lines denote mean±SEM, * p<0.05.
To examine if the increased titin-stiffness manifests itself in the intact left ventricle, we utilized an ex-vivo isolated heart system to determine the diastolic stress-SL relationship of the LV14–16. The baseline function of the WT and IG KO hearts was similar–including their developed wall stress and relaxation parameters (Table S1). The diastolic stress-volume relationship (DSVR) calculated using a spherical LV model showed an increase in the IG KO (Fig. S3C–D). To exclude possible differences in SL, we determined the diastolic SL of circumferentially aligned mid-myocardial fibers of hearts chemically fixed at known volumes16. The obtained SL-volume relationship (Fig. S3E) was used to convert the diastolic stress-volume relations into diastolic stress-SL relations (Fig. 4D) and a significant 78% increase in stiffness derived from the diastolic stress-SL relationship was revealed (Fig. 4E). Finally, we also performed an in-vivo pressure (P)-volume (V) analysis with IVC occlusion to determine diastolic stiffness (see Methods for details and Table S1 for baseline functional measurements). The end-diastolic wall stress volume relationship (EDSVR) was significantly increased by 36% in the IG KO (Fig. 4F, Table S1). Thus IG KO mice have increased diastolic stiffness from the cell to the in-vivo LV chamber levels.
Exercise intolerance is considered a global manifestation of diastolic dysfunction. Accordingly we investigated exercise tolerance in 3 mo old WT and IG KO mice. Running mice on a treadmill at progressively higher speeds (see Methods for details) revealed a ~24% reduced exercise tolerance in IG KO mice (Fig. 5A). Similarly, using a voluntary running wheel exercise test, we found IG KO mice had a 33% decrease in distance obtained compared to WT (Fig. 5B).
Figure 5.
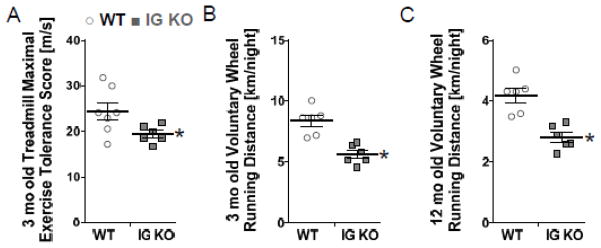
At 3 mo, maximal running speed on a treadmill test revealed the IG KO had a 25% reduced exercise tolerance (A), while volunteer running showed that the IG KO ran a 33% shorter distance each night (B) (n=6). A further reduction in exercise intolerance was found at 12mo (n=6). Horizontal lines denote mean±SEM, * p<0.05.
Cardiac trophicity
Diastolic dysfunction has been linked to changes in LV trophicity1. Therefore, we determined the left ventricular weight (LVW, mg) and the LVW: Tibia length (TL, mm) ratio in neonatal (5 day), young adult mice (3 mo), and middle-aged mice (12 mo) (Table S2). At 5 days we found smaller LVW:TL in the IG KO, no difference at 3 mo of age, but a significantly larger LVW:TL value in IG KO mice at 12 mo (Fig. 6A). These results indicate that the IG KO mice have alterations in trophicity from WT and exhibit age-dependent cardiac hypertrophy.
Figure 6.

Cardiac Trophicity. LVW:Tibia Length (TL) ratio is increased in older mice (A, n=6 for 5d day, n=28 for 3 mo; n=8 for 12 mo). Representative Western blots from 3mo mice (B). Expression of titin binding proteins FHL1, FHL2, and CARP were normalized to the 3mo WT (C). (n=4 for 5 day, n=8 for 3, 12 mo) Box denotes 25th and 75th percentile and whiskers display min and max, * p<0.05; ** p<0.01; ***p<0.001. See Supplemental figure S9 for dot plots.
We next studied expression of cardiac failure and hypertrophic markers in 3 mo and 12 mo old IG KO and WT mice. While gene products for ANP, BNP, MAPKAPK2, skeletal actin, αMHC, and βMHC are expected to increase in failure or hypertrophy14, we found no change in most markers with significant decreases in skeletal actin at 3 mo, and ANP at 3 mo (Fig. S4). Considering that several of titin’s binding proteins have been linked to hypertrophy signaling14, 33, we measured expression of titin-binding proteins. Proteins that bind to the M-line and Z-disk regions of titin showed no changes between WT and IG KO hearts (Fig. S5A). FHL1, FHL2, and CARP expressions, proteins that interact with titin’s extensible I-band region, had severalfold increased expression levels in the IG KO at both 3 and 12mo, but only FHL1 continuously increased with age (Fig. 6B–C). Because FHL2 was recently implicated in suppression of hypertrophy, we also investigated the calcineurin/NFAT pathway, which might be a downstream target28; however, we found no clear correlation between these markers and the hypertrophy observed (Fig. S5B–F)
Age-Related Diastolic Dysfunction
Since diastolic dysfunction is observed disproportionately in elderly patients, we also evaluated exercise tolerance in 12 mo old WT and IG KO mice using voluntary wheel running. IG KO mice showed a 48% decrease in running distance (Fig. 5C), much larger than in the young mice (above) suggesting that increasing age exacerbates exercise intolerance. Echocardiography in these mice revealed a significant increase in LA diameter in IG KO mice that was consistent with post-mortem increases in LA mass, as well as a trend (p=0.14) to shorter E-wave deceleration time (Fig. 7A–C, Table S3). PV studies revealed normal systolic function, but significant increases in EDPVR and EDSVR in KO mice consistent with a stiffer LV chamber (Fig. 7E,F, Table S3).
Figure 7.

Age-Related Enhancement of Diastolic Dysfunction in 12 mo old mice. Left atrial enlargement is noted in the IG KO in vivo (A) and mass is increased upon sacrifice (B). Diastolic stiffness, measured via mitral deceleration time is increased(C). Representative PV loop from 12mo WT (D). EDPVR (E) and EDSVR (F) are increased in the IG KO. Horizontal lines denote mean±SEM, n=6 * p<0.05; ** p<0.01.
DISCUSSION
Diastolic dysfunction occurs in approximately 50% of heart failure cases and is characterized by increased stiffening of the LV despite seemingly normal systolic function. This study focused on a novel titin model that has a shortened proximal tandem Ig segment obtained by deleting domains Ig 3-11 (IG KO). Studies from myocytes to the LV chamber levels showed that the primary phenotype of the model is increased diastolic stiffness. Titin exon expression analysis and protein studies did not detect compensatory changes in isoform expression or phosphorylation of titin. Immunoelectron microscopy revealed that the titin spring segments that remain in the IG KO extend to a higher degree than in WT mice, providing an explanation for the stiffness increase in the IG KO. IG KO mice exhibited age-dependent LV hypertrophy and importantly, IG KO mice exhibited exercise intolerance and diastolic dysfunction that were exacerbated with age; phenotypes that are associated with diastolic dysfunction in patients.
Passive stiffness of cardiac myocytes
The IG KO model was made to study the role of titin-based stiffness in diastolic function and to establish whether an increase in titin-based stiffness per se is sufficient to cause diastolic dysfunction. We targeted the tandem Ig segment for deletion because it dominates the extensibility of titin in the physiological SL range8. This is different from the two existing spring segment deletion models: the N2B KO in which the N2B segment has been deleted15 and the PEVK KO in which the full PEVK segment of the N2B cardiac titin isoform has been deleted14 –both segments dominate titin’s extensibility toward the upper limit of the physiological SL range and beyond8. The earlier models also eliminate known phosphorylation sites that also alter passive stiffness17–19. PKA19, PKG18, ERK234 and CamKII35 phosphorylation sites are deleted in the N2B KO and PKCα and CamKII35 sites are deleted in the PEVK KO17 but no known phosphorylation sites have been detected in these Ig segments in cardiac tissue17, 20. Additionally, changes in titin isoform expression take place in the N2B KO and PEVK KO models with increased expression of the compliant N2BA isoform in the N2B KO15 (this compensates for the stiffness increase caused by the N2B segment deletion) and an opposite expression change occurs in the PEVK KO14 (this exacerbates the passive stiffness increase that is due to the PEVK deletion). In contrast, titin isoform expression is not altered in the IG KO (Fig. 1D). While it is unclear what underlies these differences between the models, it seems unlikely that the titin-based stiffness increase drives changes in isoform expression– given that all models have increased stiffness a change in titin expression in the IG KO would also be expected. It is fortuitous that changes in phosphorylation and isoform splicing are absent in the IG KO, making it possible to attribute the phenotypes directly to the shortened titin.
Passive stiffness of skinned cardiac myocytes at physiological sarcomere lengths (~1.9–2.3 μm) is known to be primarily titin-based7 and the increased passive stiffness in the IG KO (Fig. 2D) is therefore likely due to titin. The only difference in titin that was found via immunoelectron microscopy in the IG KO (in addition to the absence of Ig3-11) is the higher extension of the distal tandem Ig, N2B and PEVK spring segments (Fig. 3E,F). Whether this increased extension explains the measured stiffness increase can be assessed from a wormlike chain (WLC) entropic spring model of titin36, in which force increases non-linearly with the molecule’s fractional extension (end-to-end length divided by the contour length) and is inversely proportional to persistence length (a measurement of the bending rigidity of the molecule). Using a serially-linked WLC model with tandem Ig, the N2B and the PEVK segments each represented by a distinct WLC with contour length and persistence length as determined from single molecule experiments37, the force-SL relation can be calculated for WT and IG KO molecules. Calculations show that the shortened tandem Ig segment and the increased strain of the remaining spring segments in the IG KO results in higher forces in the IG KO molecule than in the WT molecule (Fig. S6), and the degree of increase is similar to that measured. Thus, the passive stiffness increase of IG KO myocytes can be explained by increased extension of titin’s spring segments.
Although the main difference in the passive myocyte properties of IG KO mice is their elastic stiffness, the viscous moduli were also found to be increased (Fig. 2F). Various sources could underlie this increase (e.g., weak crossbridge interaction, friction between thin and thick filaments) but none naturally explain why viscosity is increased only at long SLs. Instead, the SL dependence suggests that Ig domain unfolding may play a role. Ig domains form beta-barrel structures that resist unfolding under physiological conditions which minimizes energy loss during repeated loading cycles38. Of the proximal and distal segments that are expressed in the N2B isoform, the proximal Ig domains have a lower stability (lower unfolding force and higher unfolding rate)39–42 and we therefore removed these to minimize Ig domain unfolding events. However, the increased passive forces in the IG KO will increase the likelihood of unfolding (unfolding rates are force dependent43), especially at long SLs where the force increase is the highest. Thus, increased viscosity at long SLs in the IG KO may be due to increased Ig domain unfolding.
Ventricular diastolic dysfunction
Ex-vivo isolated heart experiments and in-vivo pressure-volume analysis revealed steeper diastolic wall stress – volume relations in 3 mo old IG KO and higher stiffness values derived from these relationships. We expected that the IG KO mouse would also exhibit increased end diastolic pressures but only found a trend towards an increase (Table S2), which is a limitation of this study. (It is possible that the need for anesthesia during in vivo characterization influenced end diastolic pressures.) We also found a significant reduction in mRNA expression levels of collagen Iα1 and IIIα1 at 3 mo (Fig. S2A–C) reflects an attempt to compensate for increased titin-based passive stiffness in IG KO. However, these mRNA changes appear not to cause functional effects as there were no differences found in the ECM-based passive stiffness or collagen-volume fraction between IG KO and WT mice (Fig. 4A–C). With the exception of a decrease in TIMP2 expression at 3mo, this is supported by the absence of expression differences in MMPs, TIMPs, and LOX (Fig. S2D–H). The summation of these results suggests that the ECM contributes little to the increased diastolic wall stiffness of IG KO mice; instead the increase is likely to reflect increased myocyte stiffness. The magnitude of the stiffness increase was less in the in vivo study (37%) than in the cell and muscle study (~85%) and various factors might be involved in this difference (e.g., anesthesia effects). An interesting possibility to consider is the reduced sarcomere strain amplitude in the IG KO that was found in the ex vivo isolated heart experiments (Fig. S3E). While we utilized a single SL-LV volume relationship to derive diastolic stress-SL relations in both genotypes (Fig. 4D), separate fits would result in steeper stress-SL relationships in the IG KO (relative to WT). Thus the single SL-LV volume relationship could reduce the stiffness difference between WT and KO hearts and this might explain why the in vivo stiffness difference is only 37%. In summary, elevated diastolic stiffness is consistently found at all levels in the IG KO, with increased titin stiffness as the likely mechanistic basis.
Ventricular hypertrophy is also associated with diastolic dysfunction and HFpEF4, 21, 22, 44. After birth IG KO mice begin with atrophic LV weights but as they age LVW increases more than in WT resulting in significantly increased LVW and LVW:TL ratio at 1 year (Fig. 6A and Table S2). A recent hypothesis suggests that, in response to increased N2B strain, four-and-half LIM protein 1 (FHL1) expression and binding to the N2B segment increases, causing increased anchoring and signaling of members of the MAPK hypertrophy pathway33. The hypothesis is in agreement with results in the PEVK KO, which has increased N2B strain and is hypertrophied14 and with the N2B KO where the strain sensing mechanism is absent and the LV is atrophied15. We observed an increase in FHL1 expression that intensified with age (Fig. 6B,C). CARP and FHL2 are also upregulated in the IG KO (Fig. 6B,C). CARP is upregulated in a wide range of diseases and conditions45–47, including in the N2B KO that has atrophy15, and is thus unlikely to be a primary factor in the cardiac hypertrophy phenotype of the IG KO. FHL2 is of interest because it has recently been shown to suppress the calcineurin and NFAT hypertrophy pathway28. This pathway does not appear to be altered in the IG KO as expression of calcineurin and NFAT along with downstream RCAN signaling proteins revealed no consistent change in the IG KO (Fig. S5B–F). Given that FHL1 is thought to induce hypertrophy33 and the IG KO model shows increased N2B strain (Fig. 2F), it is tempting to speculate that titin signaling is involved in the hypertrophy, but the mechanism requires additional study and it remains to be explained why FHL1 is upregulated at young age when the LV is atrophied or normal. Investigation of fetal genes commonly associated with failure and hypertrophy revealed no increases (Fig. S4), which might be explained by the fact that these mice are not exhibiting a “failure” phenotype.
Exercise intolerance is a hallmark feature of HFpEF4, 21, 44. Although sedentary IG KO mice appear normal, reduced involuntary treadmill running tolerance and voluntary running wheel activity (Fig. 5A,B) both indicate that their exercise tolerance is compromised. The exercise tolerance and diastolic phenotype of 12 mo animals undergoing exercise testing is even more compelling. Compared to 3 mo old mice, 12 mo old IG KO mice showed a greater reduction (48% vs 33%) in voluntary wheel running (Fig. 5B and C). Various factors may reduce exercise capacity A decrease in systolic reserve is unlikely to have contributed to the decreased exercise tolerance as the IG KO mice appear to have normal systolic function (Supplemental Table S1). It is likely that increased titin-based myocardial stiffness is a contributing factor, as it will limit the ability to enhance ventricular filling by limiting cardiac reserve6, 44, 48. Additional evidence supporting a diastolic stiffness phenotype is revealed in the IG KO mice as evidenced by LA dilation and hypertrophy (Fig. 7A,B)22. Echocardiography and in vivo pressure volume analysis indicate increased diastolic stiffness (Fig. 7E,F) and provide further support that LV dysfunction is present in IG KO mice. Our study suggests that diastolic dysfunction and exercise intolerance are present in IG KO mice, which is similar to conditions observed in HFpEF patients23, 49
In summary, our work on the IG KO shows that shortening the tandem Ig segment leads to a primary diastolic dysfunction phenotype with increased LV stiffness, age-dependent hypertrophy and exercise intolerance. The characteristics of the IG KO overlap with those of HFpEF patients in which increased titin-based stiffness has also been reported and diastolic stiffness is increased21, and thus may elucidate titin’s role in diastolic dysfunction. Although the cause of increased titin-based stiffness is distinct from HFpEF patients (hypo-phosphorylation of titin21) the functional effects that ensue might be similar with increased strain of titin’s spring elements increasing diastolic stiffness and triggering hypertrophy. Thus our novel mouse model with a titin based primary diastolic phenotype might be of great use for elucidating mechanisms that contribute to HFpEF.
Supplementary Material
CLINICAL IMPACT.
To study the role of increased titin-based passive stiffness in diastolic dysfunction we studied a new mouse model in which titin’s elastic I-band has been shortened, as a mechanical analog of increased titin based stiffness in HFpEF (Heart Failure with preserved Ejection Fraction) patients. The model displays increased diastolic stiffness, hypertrophy, and exercise intolerance- all common pathologic findings in HFpEF patients. We propose that titin is a possible therapeutic target for ameliorating diastolic stiffening in HFpEF.
Acknowledgments
We gratefully acknowledge the Arizona Research Labs for assistance with Electron Microscopy, The University of Arizona CMM Histology Service Laboratory for assistance with histology, and are thankful for the many members of our laboratory who contributed to data collection, especially undergraduate researchers Riako Granzier-Nakajima, Shawtaroh Granzier-Nakajima, Amanda Marsh, and Javier Saldana Perez.
Funding Sources: This work was supported by AHA (0825748G, postdoctoral fellowship) to CSC, NIH (T32 HL07249-31) to CSC and KRH; and NIH grant R01HL062881 and a gift from Alan and Alfie Norville to HLG.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: Pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–679. doi: 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halley CM, Houghtaling PL, Khalil MK, Thomas JD, Jaber WA. Mortality rate in patients with diastolic dysfunction and normal systolic function. Arch Intern Med. 2011;171:1082–1087. doi: 10.1001/archinternmed.2011.244. [DOI] [PubMed] [Google Scholar]
- 3.Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- 4.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 5.Granzier H, Wu Y, Siegfried L, LeWinter M. Titin: Physiological function and role in cardiomyopathy and failure. Heart Fail Rev. 2005;10:211–223. doi: 10.1007/s10741-005-5251-7. [DOI] [PubMed] [Google Scholar]
- 6.Borlaug BA, Jaber WA, Ommen SR, Lam CS, Redfield MM, Nishimura RA. Diastolic relaxation and compliance reserve during dynamic exercise in heart failure with preserved ejection fraction. Heart. 2011;97:964–969. doi: 10.1136/hrt.2010.212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Granzier HL, Irving TC. Passive tension in cardiac muscle: Contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027–1044. doi: 10.1016/S0006-3495(95)80278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trombitas K, Redkar A, Centner T, Wu Y, Labeit S, Granzier H. Extensibility of isoforms of cardiac titin: Variation in contour length of molecular subsegments provides a basis for cellular passive stiffness diversity. Biophys J. 2000;79:3226–3234. doi: 10.1016/S0006-3495(00)76555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004;94:505–513. doi: 10.1161/01.RES.0000115522.52554.86. [DOI] [PubMed] [Google Scholar]
- 10.Greaser ML, Warren CM, Esbona K, Guo W, Duan Y, Parrish AM, Krzesinski PR, Norman HS, Dunning S, Fitzsimons DP, Moss RL. Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J Mol Cell Cardiol. 2008;44:983–991. doi: 10.1016/j.yjmcc.2008.02.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. doi: 10.1161/01.CIR.0000135591.37759.AF. [DOI] [PubMed] [Google Scholar]
- 12.Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res. 2004;95:708–716. doi: 10.1161/01.RES.0000143901.37063.2f. [DOI] [PubMed] [Google Scholar]
- 13.Labeit S, Kolmerer B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 14.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschope C, McNabb M, Larson DF, Labeit S, Gotthardt M. Truncation of titin’s elastic pevk region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105:557–564. doi: 10.1161/CIRCRESAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radke MH, Peng J, Wu Y, McNabb M, Nelson OL, Granzier H, Gotthardt M. Targeted deletion of titin n2b region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci U S A. 2007;104:3444–3449. doi: 10.1073/pnas.0608543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung CS, Granzier HL. Contribution of titin and extracellular matrix to passive pressure and measurement of sarcomere length in the mouse left ventricle. J Mol Cell Cardiol. 2011;50:731–739. doi: 10.1016/j.yjmcc.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. Pkc phosphorylation of titin’s pevk element: A novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–638. doi: 10.1161/CIRCRESAHA.109.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase g modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 19.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase a phosphorylates titin’s cardiac-specific n2b domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 20.Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ. Hypophosphorylation of the stiff n2b titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780–786. doi: 10.1161/CIRCRESAHA.108.193326. [DOI] [PubMed] [Google Scholar]
- 22.Zile MR, Gottdiener JS, Hetzel SJ, McMurray JJ, Komajda M, McKelvie R, Baicu CF, Massie BM, Carson PE, Investigators IP. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation. 2011;124:2491–2501. doi: 10.1161/CIRCULATIONAHA.110.011031. [DOI] [PubMed] [Google Scholar]
- 23.Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD, Redfield MM. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2010;56:845–854. doi: 10.1016/j.jacc.2010.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 2003;24:1695–1702. doi: 10.1002/elps.200305392. [DOI] [PubMed] [Google Scholar]
- 25.Reiser PJ, Kline WO. Electrophoretic separation and quantitation of cardiac myosin heavy chain isoforms in eight mammalian species. Am J Physiol. 1998;274:H1048–1053. doi: 10.1152/ajpheart.1998.274.3.H1048. [DOI] [PubMed] [Google Scholar]
- 26.Hudson BD, Hidalgo CG, Gotthardt M, Granzier HL. Excision of titin’s cardiac pevk spring element abolishes pkcalpha-induced increases in myocardial stiffness. J Mol Cell Cardiol. 2010;48:972–978. doi: 10.1016/j.yjmcc.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammed SF, Ohtani T, Korinek J, Lam CSP, Larsen K, Simari RD, Valencik ML, Burnett JC, Redfield MM. Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “nongenomic effects”. Circulation. 2010;122:370–378. doi: 10.1161/CIRCULATIONAHA.109.915215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hojayev B, Rothermel BA, Gillette TG, Hill JA. Fhl2 binds calcineurin and represses pathological cardiac growth. Mol Cell Biol. 2012;32:4025–4034. doi: 10.1128/MCB.05948-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart JA, Jr, Wei CC, Brower GL, Rynders PE, Hankes GH, Dillon AR, Lucchesi PA, Janicki JS, Dell’Italia LJ. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35:311–319. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 30.Muhle-Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H. Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin s1--a possible role in the frank-starling mechanism of the heart. J Mol Biol. 2001;313:431–447. doi: 10.1006/jmbi.2001.5017. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Peng J, Campbell KB, Labeit S, Granzier H. Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. J Mol Cell Cardiol. 2007;42:186–195. doi: 10.1016/j.yjmcc.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 32.Trombitas K, Freiburg A, Centner T, Labeit S, Granzier H. Molecular dissection of n2b cardiac titin’s extensibility. Biophys J. 1999;77:3189–3196. doi: 10.1016/S0006-3495(99)77149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheikh F, Raskin A, Chu PH, Lange S, Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y, Dalton ND, Mahata SK, Dorn GW, 2nd, Brown JH, Peterson KL, Omens JH, McCulloch AD, Chen J. An fhl1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F. A novel mechanism involving four and a half lim domain protein-1 and extracellular-signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem. 2012;287:29273–29284. doi: 10.1074/jbc.M112.372839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hidalgo CG, Chung CS, Saripalli C, Methawasin M, Hutchinson KR, Tsaprailis G, Labeit S, Mattiazzi A, Granzier HL. The multifunctional ca(2+)/calmodulin-dependent protein kinase ii delta (camkiidelta) phosphorylates cardiac titin’s spring elements. J Mol Cell Cardiol. 2013;54:90–97. doi: 10.1016/j.yjmcc.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kellermayer MS, Smith SB, Bustamante C, Granzier HL. Complete unfolding of the titin molecule under external force. J Struct Biol. 1998;122:197–205. doi: 10.1006/jsbi.1998.3988. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe K, Nair P, Labeit D, Kellermayer MS, Greaser M, Labeit S, Granzier H. Molecular mechanics of cardiac titin’s pevk and N2B spring elements. J Biol Chem. 2002;277:11549–11558. doi: 10.1074/jbc.M200356200. [DOI] [PubMed] [Google Scholar]
- 38.Nedrud J, Labeit S, Gotthardt M, Granzier H. Mechanics on myocardium deficient in the n2b region of titin: The cardiac-unique spring element improves efficiency of the cardiac cycle. Biophysi J. 2011;101:1385–1392. doi: 10.1016/j.bpj.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA. Association of the chaperone alphab-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–7924. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Linke WA, Oberhauser AF, Carrion-Vazquez M, Kerkvliet JG, Lu H, Marszalek PE, Fernandez JM. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe K, Muhle-Goll C, Kellermayer MS, Labeit S, Granzier H. Different molecular mechanics displayed by titin’s constitutively and differentially expressed tandem ig segments. J Struct Biol. 2002;137:248–258. doi: 10.1006/jsbi.2002.4458. [DOI] [PubMed] [Google Scholar]
- 42.Zhu Y, Bogomolovas J, Labeit S, Granzier H. Single molecule force spectroscopy of the cardiac titin N2B element: Effects of the molecular chaperone alphab-crystallin with disease-causing mutations. J Biol Chem. 2009;284:13914–13923. doi: 10.1074/jbc.M809743200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kellermayer MS, Smith SB, Granzier HL, Bustamante C. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 1997;276:1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 44.Melenovsky V, Borlaug BA, Rosen B, Hay I, Ferruci L, Morell CH, Lakatta EG, Najjar SS, Kass DA. Cardiovascular features of heart failure with preserved ejection fraction versus nonfailing hypertensive left ventricular hypertrophy in the urban baltimore community: The role of atrial remodeling/dysfunction. J Am Coll Cardiol. 2007;49:198–207. doi: 10.1016/j.jacc.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 45.Buyandelger B, Ng K-E, Miocic S, Piotrowska I, Gunkel S, Ku C-H, Knöll R. Mlp (muscle lim protein) as a stress sensor in the heart. Pflugers Arch. 2011;462:135–142. doi: 10.1007/s00424-011-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witt SH, Labeit D, Granzier H, Labeit S, Witt CC. Dimerization of the cardiac ankyrin protein carp: Implications for marp titin-based signaling. J Muscle Res Cell Motil. 2005;26:401–408. doi: 10.1007/s10974-005-9022-9. [DOI] [PubMed] [Google Scholar]
- 47.Zolk O, Frohme M, Maurer A, Kluxen F-W, Hentsch B, Zubakov D, Hoheisel JD, Zucker IH, Pepe S, Eschenhagen T. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochem Biophys Res Commun. 2002;293:1377–1382. doi: 10.1016/S0006-291X(02)00387-X. [DOI] [PubMed] [Google Scholar]
- 48.Meyer TE, Karamanoglu M, Ehsani AA, Kovács SJ. Left ventricular chamber stiffness at rest as a determinant of exercise capacity in heart failure subjects with decreased ejection fraction. J Appl Physiol. 2004;97:1667–1672. doi: 10.1152/japplphysiol.00078.2004. [DOI] [PubMed] [Google Scholar]
- 49.Maeder MT, Thompson BR, Brunner-La Rocca H-P, Kaye DM. Hemodynamic basis of exercise limitation in patients with heart failure and normal ejection fraction. J Am Coll Cardiol. 2010;56:855–863. doi: 10.1016/j.jacc.2010.04.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.