

Abstract
p38α Mitogen-activated Protein Kinase (p38α) is activated by a variety of mechanisms, including autophosphorylation initiated by TGFβ-activated kinase 1 binding protein 1 (TAB1) during myocardial ischemia and other stresses. Chemical genetic approaches and co-expression in mammalian, bacterial and cell-free systems revealed that mouse p38α autophosphorylation occurs in cis by direct interaction with TAB1(371-416). In isolated rat cardiac myocytes and perfused mouse hearts TAT-TAB1(371-416) rapidly activates p38 and profoundly perturbs function. Crystal structures and characterization in solution revealed a bipartite docking site for TAB1 in the p38α C-terminal kinase lobe. TAB1 binding stabilizes active p38α and induces rearrangements within the activation segment by helical extension of the Thr-Gly-Tyr motif that allows auto-phosphorylation in cis. Interference with p38α recognition by TAB1 abolishes its cardiac toxicity. Potentially, such intervention could circumvent the drawbacks seen when pharmacological inhibitors of p38 catalytic activity are used clinically.
INTRODUCTION
The p38 Mitogen-activated Protein Kinase (p38s) family comprises four isoforms; α, β, γ and δ1. Amongst these isoforms, α is ubiquitously expressed and most widely studied. Furthermore p38α is essential for normal development2 whilst the absence of the other isoforms3, even in combination, results in a normal or near normal adult phenotype. Due to the central role of p38α in regulating the immune response, inhibitors of p38α/β kinase activity were among the first relatively selective kinase inhibitors to be developed4, understood at the atomic level5 and clinically tested6 in the treatment of inflammatory diseases. Despite numerous inhibitors with diverse structures and binding modes having been developed, none has progressed beyond Phase II clinical trials6,7. The amount of information in the public domain that would explain the failure of these inhibitors in the clinic is limited but it seems very likely that this attrition is the result of liver and skin toxicity as well as the appearance of bypass or escape mechanisms7,8. These same drawbacks are common to diverse inhibitors with different modes of binding to the p38α active site, suggesting a class effect. For this reason it has been suggested alternative strategies targeting p38α are needed8.
Based on the use of kinase inhibitors and genetic manipulation there is overwhelming evidence that the activation of p38α that accompanies lethal myocardial ischemia aggravates injury9. During myocardial ischemia the activation of p38 is unusual in that catalytic inhibitors prevent the phosphorylation of the Thr-Gly-Tyr (TGY) motif within the activation loop9,10. In subsequent studies this observation was shown to be the result of the likely autophosphorylation initiated by the binding of p38α to the scaffold protein, TGF-β-activated protein kinase 1 (TAK1) binding protein 1 (TAB1)10-14. TAB1 was instrumental in the first described signaling pathway by which p38α becomes activated independent of its usual archetypal, canonical upstream MAP kinase kinases (MAPKK)15. Furthermore, TAB1 phosphorylation by p38 dimishes its ability to activate TAK1, closing a feedback loop16. Subsequently other circumstances enabling p38α autophosphorylation have been described17,18. Nonetheless p38α activation by autophosphorylation remains controversial since there are a number of conceptual obstacles that must be overcome19. In canonical MAP kinase signaling, p38α is activated by the transphosphorylation of the TGY motif by the MAPKKs. This event causes the activation loop to clear the substrate binding cleft and results in the reorientation of the N- and C-terminal lobes needed to bring Lys53, Glu71 and Asp168 into the necessary alignment to coordinate ATP binding20. The macroscopic consequence is a marked increase in the binding affinity of p38α towards ATP21. Thus, ATP binding follows TGY motif transphosphorylation. However, for autophosphorylation to occur the binding of ATP needs to precede TGY phosphorylation, requiring the prior alignment of residues for high affinity ATP interaction. This circuitous paradox has been aptly likened to a Catch-22, and is relevant to at least 4 other mammalian kinases19,22.
Since the TAB1-initiated form of p38α activation has been shown to aggravate myocardial injury by numerous independent groups10-14 it may be therapeutically relevant. Moreover therapies that exploit this relatively unusual mechanism of p38 activation could avoid the drawbacks seen with inhibitors of p38 catalytic activity since the observed toxicity is likely to be unrelated to this restricted mechanism of activation7,8. For these reasons the purpose of our study was to elucidate the molecular mechanisms that lead to TAB1-induced, MAPKK-independent, p38α autoactivation during myocardial ischemia.
RESULTS
p38α is activated by autophosphorylation during myocardial ischemia
In the past we and others have shown p38 and TAB1 co-associate during myocardial ischemia10-13 and that dual phosphorylation of p38’s TGY activation motif is sensitive to p38 inhibitors that do not inhibit the immediately upstream MKKs10,23. The most likely explanation for these observations is TAB1-initiated p38 autophosphorylation9,15,23,24. However, the phosphorylated TGY epitope, that is monitored as a readout of autophosphorylation, is common to all p38 isoforms and it is therefore uncertain which specific isoform contributes to the reduction in signal intensity as a result of p38 kinase inhibition. Type I ATP competitive kinase inhibitors affect both p38α and p38β since they share a small Thr gatekeeper residue5,20. In p38γ and p38δ this residue is a larger Met which is responsible for their resistance to inhibition 5. Thus the autophosphorylation signal must arise from p38α and/or p38β.
Recently we have adopted a chemical genetic approach using mice in which either both p38α24 or both p38β25 alleles carried a T106M gatekeeper substitution allowing selective targeting of each of these two isoforms. Here we use these reagents to examine the relative contribution of p38α and p38β to p38 autoactivation and to determine whether transphosphorylation by upstream kinases can ultimately compensate for the block in autophosphorylation during early ischemia. Fig.1a demonstrates the pattern of p38 dual phosphorylation during global myocardial ischemia. In wild-type (WT) hearts ischemia initiated p38 dual phosphorylation that is sensitive to the p38α/β inhibitor, SB203580. In hearts expressing the p38α Thr106Met gatekeeper mutant that does not bind SB203580 (IRα) p38 remained active as determined by phosphorylation of the direct downstream substrate TAB1 and the indirect downstream substrate HSP27. In hearts in which p38β is resistant to SB203580 (IRβ) the dual autophosphorylation of p38 and phosphorylation of its downstream substrates was inhibited by SB203580. These patterns were similar at 5, 10 and 15 mins of ischemia. In combination with our previous observations24,25, the findings confirm (i) p38α is the dominant isoform activated during myocardial ischemia by a mechanism consistent with autoactivation; (ii) that transactivation mechanisms, that are not dependent on p38α catalytic activity, cannot overcome this blockade (see Fig. 1a, comparing IRα to WT at 10 and 15 mins) and (iii) that if the autoactivation of p38α can be prevented, myocardial total p38 activity will remain low. The question remains, however, whether this signature is consistent with TAB1-mediated p38 phosphorylation? We addressed this issue by the following experiments:
Figure 1.
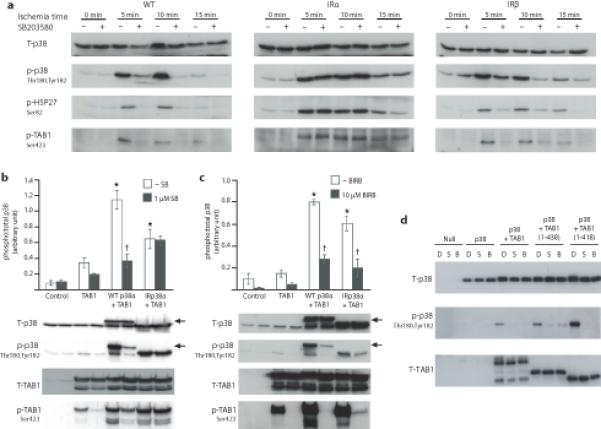
The dual phosphorylation of p38 during myocardial ischaemia is consistent with the pattern of TAB1-initiated p38 activation. (a). Activation of myocardial p38 after various durations of global ischemia in the absence (−) or presence (+) of the p38 inhibitor SB203580 (10 μM).. Each lane is prepared from protein harvested from the heart from an individual mouse. IRp38α and IRp38β samples bear a mutation in the gatekeeper residue of either both α or both β alleles (T106M) and are not inhibited by SB203580. (b and c) Western-blot analysis of p38MAPK and TAB1 phosphorylation after co transfection of HEK293 cells with TAB1 and p38α or IRp38α in the presence of SB203580 (b) or BIRB793 (c). Top, quantitation from three independent repetitions of cell transfection shown as average ± s.d. *= P< 0.05 vs Control. †= P< 0.05 vs WTp38α Western blots from one such experiment are shown in the bottom. The arrowheads indicate WTp38α with a higher apparent MW due to an HA tag. (d) Co-expression of mouse p38α and TAB1 in E.coli with and without exposure to SB203580 or BIRB796. E.coli were transformed with vectors encoding nothing (Null), p38α alone (p38), p38α/full-length TAB1 (p38+TAB1), p38α with TAB1(1-438) or p38α with TAB1(1-418). E.coli were grown with 0.01%DMSO alone (D) or with 10μM SB203580 (S) or 10μM BIRB796 (B) and induced with 1mM IPTG. The uncropped images from which the immunoblots in figure are derived appear in Supplementary Fig.6 appear in Supplementary Fig.6 together with the repetitions used to quantify panels b and c..
In Fig. 1b TAB1 and WTp38α or IRp38α were co-expressed in 293 cells. TAB1 enhanced p38α dual phosphorylation in an SB203580-sensitive manner, akin to the observation in WT hearts in Fig. 1a. Moreover, this pattern was lost when the IRp38α was coexpressed with TAB1; a pattern once again consistent with the observation in Fig. 1a. Finally this pattern was altered when cells were exposed to BIRB796, a Type II p38α/β inhibitor that does not depend on Thr106 for binding (see Fig. 1c). Once again this pattern is identical to that seen in hearts expressing IRα24. However, one complication interpreting co-expression data in eukaryotic cells is the fact that TAB1 also promotes the autoactivation of TAK1, a kinase that lies upstream of p3816. To exclude such a scenario, where TAB1-initiated transphosphorylation masquerades as autophosphorylation, we repeated these co-expression experiments in E. coli, a background that is devoid of almost all protein kinases (Fig. 1d). These experiments demonstrated that p38α phosphorylation was enhanced by TAB1 coexpression and diminished by pharmacological inhibitors of p38 catalytic activity. Moreover, the effect of TAB1 persisted following C-terminal truncations harbouring the TAK1 interacting domain26. The recapitulation in E. coli of TAB1-initiated p38α activation encouraged us to purify the individual proteins, map the interacting regions in both proteins and examine the biophysical characteristics of their interaction.
TAB1(371-416) interacts with p38α increasing ATP affinity
The outcome of the p38α-TAB1 co-expression experiments in E.coli (Fig. 1d) suggests a direct interaction between the two proteins and to test this hypothesis we employed Isothermal Titration Calorimetry (ITC). However, the inherent instability of full-length TAB1 when produced and purified from E.coli prevented its use in these experiments; instead we choose TAB1(1-438) as a positive control since it is known to encompass the entire region responsible for p38α autoactivation15,26,27 (and Fig. 1c) whilst still incorporating the portion that acts as a p38α substrate (Ser423 and Thr431)16.
ITC experiments confirmed a direct interaction between TAB1(1-438) and p38α, revealing in the experimental conditions used, a monophasic 1:1 binding isotherm and a binding affinity in the low micromolar range (Fig. 2a and Table 1). Based on an analysis of previously published characterizations15,27 we identified a 46-residue region of TAB1, TAB(371-416) which had similar ITC binding characteristics to TAB(1-438). Based on these data and the fact that the peptide is necessary and sufficient for TAB1-mediated activation of p38, we hypothesize that this short polypeptide contains the residues required for TAB1 recognition. Furthermore, the thermodynamics of binding observed in these experiments are compatible with some structural rearrangement upon TAB1-p38α interaction (see below).
Figure 2.
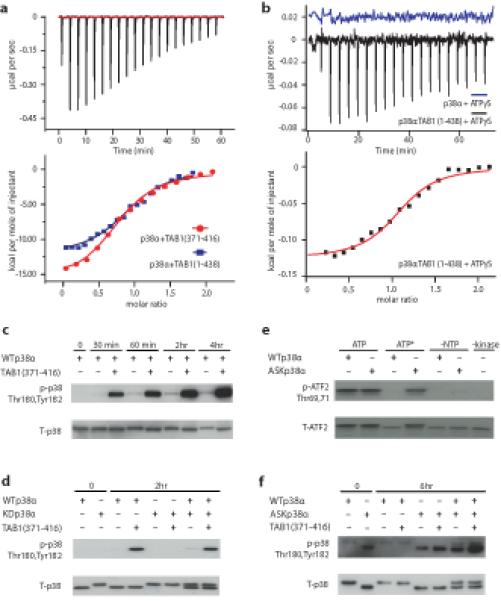
Thermodynamic characterization of TAB1:p38α complex formation on ATP affinity and autophosphorylation in cis. (a) ITC analysis of the interaction between p38α and TAB1. All but the first point were used for curve fitting and derived thermodynamic parameters are shown in Table 1. (b) ITC analysis of the interaction between p38α and ATPγS. A solution of ATPγS was titrated into p38α in the presence or absence of TAB1(1-438). Raw data of the titration of ATPγS into p38α alone revealed no detectable interaction (displaced by +0.02μcal/S for clarity). For a and b all but the first points were used for curve fitting and derived thermodynamic parameters are shown in Table 1. (c) Western blot analysis of p38 autophosphorylation during an in vitro kinase reaction in the absence (−) or presence (+) of TAB1(371-416). (d) Western blot analysis of in vitro kinase assay with wildtype (WTp38α), kinase dead, (KDp38α), or both in the absence or presence of TAB1(371-416) peptide. The HexaHis tag has been cleaved in the case of WTp38α but not KDp38α. (e) Comparison of nucleotide usage by WTp38α and ASKp38α. Activated dual phosphorylated WTp38α or ASKp38α were added to an in vitro kinase reaction containing either 1 mM ATP or an expanded form of ATP (ATP*), N6-(2-Phenylethyl)-ATP. Control reactions were without a ribonucleotide (-NTP) and without either kinase (-Kinase). (f) Western blot analysis of in vitro kinase assay with wildtype, (WTp38α), analogue sensitive kinase, (ASKp38α) or both forms of p38α in the absence or presence of TAB1(371-416) peptide. WTp38α had an intact HexaHis tag. These results confirm that TAB1 mediated autophosphorylation of p38α occurs in cis. The uncropped images from which the immunoblots are derived appear in Supplementary Fig.6.
Table 1. Thermodynamic parameters determined by Isothermal Titration Calorimetry of the association of p38α with TAB1, ATP, and SB203580 and of p38β with TAB1.
The values reported are averaged over three experiments, and the errors were found less than 5%. Non phospho p38α binding affinity to ATP is below the threshold that could be measured by ITC; the value reported here was obtained using time resolved FRET (Zhang et al., 2005). All the measurements were performed at 25°C except [p38α:TAB1(1-438)]complex / ATPγS which was run at 10°C.
| n | Kd (μM) | ΔG (kcal/mol) |
ΔH (kcal/mol) |
−TΔS (kcal/mol) |
|
|---|---|---|---|---|---|
| p38α/ TAB(1-438) | 0.8 | 1.5 | − 7.9 | − 13 | 5.1 |
| p38α/ TAB(371-416) | 0.8 | 2.5 | − 7.6 | − 16 | 8.4 |
| p38α/ ATP | nd | 13000 ± 5500 (TR-FRET) |
nd | nd | nd |
| [p38α:TAB1(1-438)]complex / ATPγS | 1.0 | 5.3 | − 7.1 | − 0.16 | − 6.9 |
| p38α/ SB203580 | 0.8 | 0.026 | − 10.3 | − 11.8 | 1.5 |
| [p38α:TAB1(1-438)]complex / SB203580 |
0.7 | 0.007 | − 11.1 | − 16.1 | 5.0 |
| [p38α:TAB(371-416)]complex / SB203580 |
0.9 | 0.004 | − 11.4 | − 16.1 | 4.7 |
| p38α/TAB1(371-416/A) | 0.9 | 2.3 | −7.6 | −2.4 | −5.2 |
| p38α/TAB1(371-416/B) | 1 | 1.2 | −8.0 | −1.6 | −6.4 |
| p38α/TAB1(371-416A/B) | nd | nd | nd | nd | nd |
| p38β/ TAB1(371-416) | 0.8 | 1.5 | −7.9 | −6.5 | −1.4 |
| p38β/TAB11(371-416/A) | 0.8 | 2.2 | −7.7 | −6.1 | −1.6 |
| p38β/TAB1(371-416/B) | nd | nd | nd | nd | nd |
nd, not detectable by ITC
Next, we examined whether TAB1, and in particular the shorter TAB1(371-416), could overcome the key conundrum underlying the Catch-22 paradox. As discussed above, during MAPK independent autophosphorylation ATP-binding must necessarily precede activation loop phosphorylation. We therefore asked does TAB1 binding switch p38α from its inactive low affinity ATP state to a high affinity ATP state competent of autophosphorylation? The titration of the non-hydrolysable (hence stable) ATPγS into p38α alone generated no measurable association by ITC (Fig. 2b), suggesting that due to low affinity no heat is detected. This is in agreement with previous results21. However, under identical conditions, a well interpolated sigmoid-shaped curve based on an independent and equivalent binding sites model centred on a 1:1 stoichiometry was observed following addition of ATPγS to a solution of p38α in the presence of TAB1(1-438), indicative of a p38α-ATP association with a low μM affinity (Fig. 2b and Table 1). An overall comparison of thermodynamic signatures of interactions (Fig. 2 and Table 1) endorse that the shorter TAB1 fragment contains the main determinants for association with unphosphorylated/inactive p38α and enables consequent marked enhancement of its ability to bind ATP reminiscent of that observed upon dual phosphorylation of the TGY motif21. However, to ensure that the effect observed in these experiments was indeed linked to the specific binding of ATP in the p38α nucleotide binding pocket, we performed analogous ITC experiments using SB203580, an ATP-competitive inhibitor that, contrary to ATP, is known to have equal affinity for the active and inactive conformation of p38α5,20. The interaction of SB203580 with p38α was found not to be appreciably affected by the presence of TAB1(1-438) or TAB1(371-416) (Supplementary Fig.1 and Table 1).
Taken together our ITC experiments point towards a model in which TAB1 binds to p38α increasing its affinity for ATP.
TAB1 accelerates p38α autophosphorylation in cis
To test the biochemical consequences of the binding of TAB1 to p38α and the binding of ATP to this complex we performed in vitro kinase assays with the dual phosphorylation of the TGY activation motif of p38α as the readout of autophosphorylation. The presence of the 46-mer TAB1(371-416) greatly accelerated the autophosphorylation of p38α (see Fig. 2c). It is unclear whether this event is intramolecular (autophosphorylation in cis) or due to the activation loop of one p38α molecule entering the substrate binding cleft of another (autophosphorylation in trans through activation loop exchange)28. To address this question we performed two complementary experiments. Fig. 2d shows the result of an in vitro kinase assay involving the recombinant purified p38α in wild type (WT) form and in kinase dead (KD) form (Lys53Met). The KDp38α was unable to undergo TAB1-initiated autophosphorylation. This deficit was apparent when KDp38α was present either in isolation or together with WTp38α. One possible explanation for the inability of KDp38α to autophosphorylate relates to the complex effect that such mutations, on the ATP-binding interface between N- and C- terminal lobes, have on the fold of a kinase29. For this reason the experiment was repeated using an analogue-sensitive (ASK) form of p38α engineered to accept an expanded form of ATP (see characterization in Fig. 2e)30. In this case WTp38α, rather than KDp38α, acted as the probe for the presence of autophosphorylation in trans. Despite WTp38α being capable of autophosphorylation, there was a minimal signal when an expanded form of ATP was present as the phosphate donor (Fig. 2f). In this case, the autophosphorylation was mainly limited to ASKp38α. Finally, to examine the possibility that nucleotide binding is a pre-requisite for autophosphorylation in trans we perfomed an additional experiment as outlined in supplementary figure 2. In combination these complementary experiments indicate that the binding of TAB1(371-416) to p38α greatly accelerates the autophosphorylation of its activation loop and that this event occurs in cis, as previously suggested15. We next attempted to characterise the rearrangements within p38α that enable autophosphorylation and the residues in TAB1 and p38α responsible for mutual recognition.
NMR analyses of p38α:TAB1(371-416) interaction
Nuclear Magnetic Resonance (NMR) was employed to map the interaction surfaces between p38α and TAB1. In particular, the chemical shift perturbations experienced by p38α amide groups in a 2H/15N-labelled sample were investigated upon titration of unlabelled TAB1(371-416) (Supplementary Fig. 3a). The chemical shift variation analysis identified several affected regions in p38α, in particular spread over the entire C-terminal lobe, which could be indicative of long-range conformational rearrangements upon peptide binding (see Supplementary material Table 1 and Fig. 3a).
Figure 3.
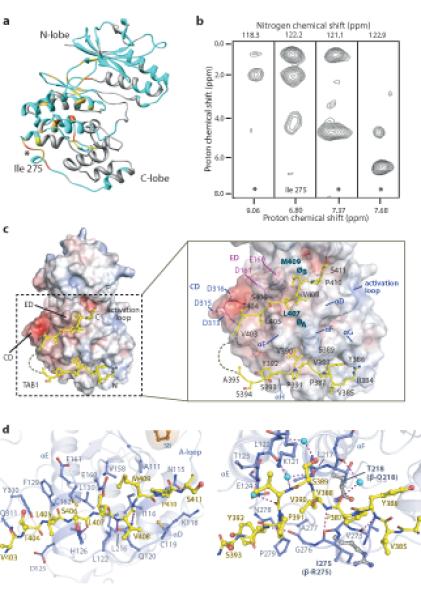
Structural features of the p38α:TAB1 interaction. (a) NMR chemical shift perturbations of p38α residues upon TAB1 binding are mapped onto the X-ray structure of p38α (PDB:1p38) (See also Supplementary Table S1). Unassigned residues are represented in gray, residues for which the assignment is available are shown in light blue, residues that show an average chemical shift perturbation (ΔδAV) larger than 0.05 are in orange, those with (ΔδAV) smaller than 0.05 are in yellow and the residues that disappear in the course of the NMR titration are gold. (b) Signals extracted from a 15N/13C filtered NOESY experiment acquired on a p38α:TAB1(371-416) complex. p38 was perdeutered and 15N ,13C labelled and TAB1 was unlabeled. * denoted unassigned residue as point of contact. (c) Structural overview of p38α:TAB1(384-412). TAB1 is shown in yellow on the surface of p38α represented as surface electrostatic potential between −10kT/e (red) and +10kT/e (blue). (d) The relationship between the C terminal portion of TAB1(403-412) and the upper canonical common docking domain and between the N terminal portion of TAB1(384-393) and the lower non canonical docking site of p38α. The key features of the docking domain are highlighted. Binding partners of p38α that utilise the upper site as well as analogous sites on other MAPKs are shown in supplementary figure S4b.
The chemical shift mapping alone is however unable to discriminate between regions responsible for direct protein-peptide contact and regions that undergo conformational rearrangement upon binding. To better define the direct points of contact between p38α and TAB1 peptide we performed 15N/13C filtered NOESY experiments on perdeuterated, 15N/13C labeled p38α and unlabelled TAB1 peptide. The combination of the labeling scheme with the appropriate NMR filter experiment (see methods) enabled the selective detection of NOE signals arising from protons bound to 12C-carbon and 14N-nitrogen (i.e. TAB1) to protons bound to 15N nitrogen (i.e. p38α amide protons). Each strip in Fig 3B showed a point of contact between TAB1 protons and p38α amides. Ile275, located on the C-lobe of p38α, was clearly identified to interact directly with TAB1, on the basis of unambiguous NOE signals from its amide proton to resonances at 0.8 and 1.9 ppm. Although 3 more NH resonances of p38α exhibit detectable NOE contacts with TAB1 protons, the responsible residues could not be unequivocally assigned prompting a complementary structural analysis.
Guided by the NMR experiments (Supplementary Fig. 3a and 3b), we ultimately used a peptide corresponding to TAB1(384-412) for crystallization trials. We confirmed that this shorter 29mer peptide recapitulated the effects of the 46mer [TAB1(371-416)] peptide to accelerate p38α autophosphorylation in vitro (see supplementary Fig. 3c) and in terms of the thermodynamic signature of its interaction with p38α by ITC (see supplementary Table 3 and Supplementary Fig. 3d).
Crystal structure of p38α:TAB1(384-412) complex
Complexes of TAB1(384-412) with p38α crystallized in two forms (see supplemental Table 2), and the electron density in all three structures revealed two distinct patches suggesting that TAB1 binds to the C-lobe of p38α in a bipartite manner (Fig. 3c and Supplementary Fig. 4). The C-terminal region of the TAB1 peptide bound within the groove defined by αD and αE helices and the reverse turn between β7 and β8 (canonical upper site – ED domain) (Fig. 3c and 3d), while its N-terminus bound a lower hydrophobic pocket created by αF and αH helices and the loop connecting helix α2L14 to helix αH (non-canonical lower site) (Fig. 3c and 3d). The intervening nine residues of the peptide, between these two interaction sites, were not visible in the crystal structures. The canonical upper site of the p38α:TAB1 complex overlaps with the binding surface used by other proteins that interact with MAPKs through canonical docking motifs; such as p38α:MKK3b, p38α:MEF2a, JNK1:JIP1 and ERK2:HePTP (see Supplementary Fig. 4b). The surface defined by the non-canonical lower site appears specific to the p38α:TAB1 interaction and was not identified in currently available structures of MAPKs in complex with binding partners.
TAB1(384-412) configures p38α for autophosphorylation
The rearrangements induced by TAB1 binding are highlighted through comparison with uncomplexed p38α31 and indicate the basis of the increased ATP binding affinity (Fig. 2b) and propensity to autophosphorylation in cis (Fig. 2c-f).
The accommodation of TAB1 at the boundaries of helices αD, αE and αF and sheets β7 and β8 was associated with displacements parallel to their long axes (see Fig. 4a). These changes, local to the TAB1 binding site, were accompanied by more marked distant rearrangements, perhaps reflecting networks of connectivity that are anchored to αF32. Most strikingly there is a closure and approximation of the N- and C- lobes of the kinase most usually associated with phosphorylation of the activation segment (see Fig. 4b). Furthermore, within the N- lobe there was an inward swing of αC bringing Glu71 within 2.7Å of Lys53, enabling the salt bridge prototypic of an active kinase (see Fig. 4b)32. It is expected that these changes within the nucleotide binding pocket will increase affinity towards ATP.
Figure 4.
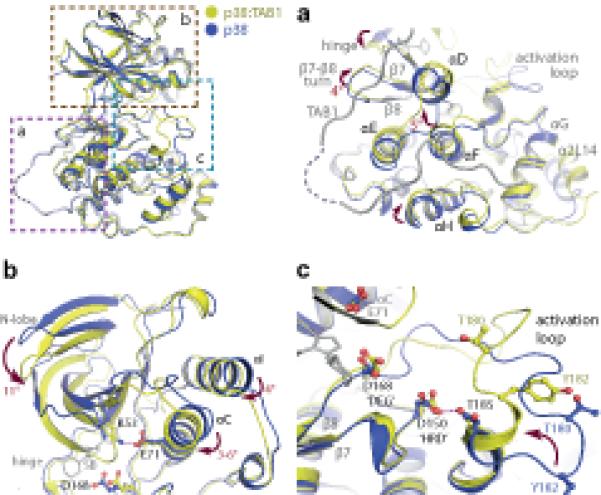
Overview of the key rearrangements within p38α on complex formation with TAB1(384-412). All comparisons are with PDB:1p38 (blue). (a) The accommodation of TAB1 peptide causes displacements in the C-terminal lobe of p38α. Amongst these is an approximate 5° swing of αF. This helix is thought to act as a key register aligning kinase regulatory and catalytic spines (see text). (b) Associated alterations in the N-terminal lobe. A downward swing approximates the 2 lobes of the kinase and is accompanied by a movement of αC towards the ATP binding pocket allowing a salt bridge to form between Lys53 and Glu71. (c) Reordering of the activation loop. The short α-helix at the C-terminus of the activation segment becomes extended. This swings Thr180 towards the key Asp residues (168 and 150) coordinating ATP binding. This loop is stabilised by an interaction between Asp150 and Thr185 that may mimic the function of the HRD motif in coordinating substrates.
Of particular interest is a long-range effect observed in the activation loop (Leu171-Val183) where major conformational rearrangement occured (see Fig. 4c). The loop contains the phosphorylation sites Thr180 and Tyr182 of the TGY motif, which in apo p38α adopts an extended conformation with the side chains of both threonine and tyrosine residues pointing away from the catalytic site. Binding to TAB1 caused residues Tyr182 to Thr185 to extend the short α helical segment that was stabilized by an interaction between Thr185 and Asp150 of the HRD motif. This structural alteration displaced the TGY motif by ~10Å and brought it towards the catalytic site with the side chain of Thr180 in a favourable position for ATP γ-phosphate transfer (see Fig. 4c). The formation of the extended helical segment within the activation loop was present in all crystal forms and seems unique to the p38:TAB1 complex. It is, therefore, tempting to speculate that it represents a transition intermediate in the cis-autophosphorylation reaction.
Disrupting the p38α:TAB1 interaction
To assess the individual role of the two recognition sites in the p38:TAB1 complex, we prepared the following TAB1 variants, in which residues were mutated in pairs as follows; V390A Y392A (designated TAB1(371-416A) to disrupt lower, non-canonical site); V408G M409A (designated TAB1(371-416B) to disrupt upper, canonical site); V390A Y392A V408G M409A (designated TAB1(371-416A-B) to disrupt both sites). We used ITC to examine the binding behaviour of these TAB1 mutants to p38α. Fig. 5a shows that no binding was detected with TAB1(371-416A-B), whereas the individual mutation pairs exhibited changes in the thermodynamic signature of binding but with a similar dissociation constant to that of wild-type TAB1 (Table 1). However, whilst the wild-type TAB1 interaction was enthalpically driven (with a negative entropic contribution), with TAB1(371-416A) and TAB1(371-416B) the interaction was entropically driven (with a smaller enthalpic contribution), reflecting differences in the interaction and/or its consequences.
Figure 5.
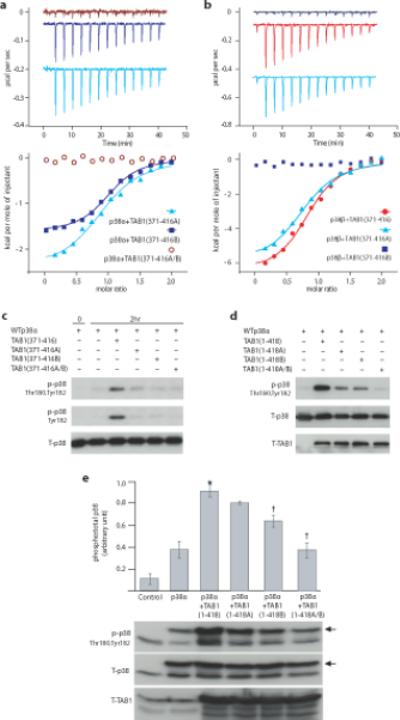
Verification of the residues within TAB1 responsible for p38α autoactivation and their discriminatory effect on p38β. (a) ITC analysis of the interaction between p38α and TAB1(371-416) mutants; TAB1(371-416A), TAB1(371-416B) or TAB1(371-416A-B) (see text). Raw data of the heat produced by p38α titrated into a solution of either TAB1(371-416A) (offset ~ −0.045μcal/sec), TAB1(371-416B) (offset ~ −0.2μcal/sec) or TAB1(371-416A-B) (see Table 1 and text). (b) ITC analysis of the interaction between p38β and wild-type TAB1(371-416) and mutants. Raw data of the heat produced by p38β titrated into a solution of either wild type TAB1(371-416) (offset ~−0. 09μcal/sec), TAB1(371-416A) (offset ~−0. 45μcal/sec) or TAB1(371-416B). (c) The residues crucial for the TAB1 mediated p38 autoactivation in vitro are investigated. Western blot analysis of in vitro kinase reaction with WTp38α in the absence (−) or presence (+) of peptides corresponding to TAB1(371-416), TAB1(371-416A), TAB1(371-416B) or TAB1(371-416A-B). The pattern differs markedly from that seen with p38β (see supplementary fig. 5a). (d) The residues crucial for the TAB1 mediated p38 autoactivation are confirmed in E.coli. Co-expression of p38α and TAB1 in E.coli transformed using the following pET duet vector constructs; p38α alone, p38α/TAB1(1-418), p38α/TAB1(1-418A), p38α/TAB1(1-418B) and p38α/TAB1(1-418A-B). (e) The residues crucial for the TAB1 mediated p38 autoactivation are confirmed in a human cell line. Co transfection into HEK293 cells of p38α with TAB1 (1-418), TAB1 (1-418A), TAB1 (1-418B) or TAB1 (1-418A-B). The arrowheads indicates ectopic WTp38α with an HA tag. Blot is aligned with quantitative data derived from three separate transfection which appear in Supplementary Fig.6. Bars represent mean ± s.d. * = P< 0.05 vs p38α. †= P< 0.05 vs p38α+TAB1 (1-418). The uncropped images from which the immunoblots are derived appear in Supplementary Fig.6.
p38β lacks the non-canonical site for TAB1interaction
p38α and p38β are highly homologous and equally sensitive to pharmacological inhibition. Nonetheless, global ablation of p38α in mice results in early embryonal lethality whilst there is barely a discernible phenotype when p38β is targeted3. In p38β the upper canonical TAB1 binding site is conserved whilst the lower pocket is disrupted by bulky Ile275Arg and Thr218Gln substitutions (see Fig 3d, right panel). The ITC data of the interaction between p38β and TAB1 peptide confirm that the beta isoform binds using only the upper canonical site (see Fig. 5b). The thermodynamic signature of the binding between p38β and non-mutated TAB1 is reminiscent of that observed between p38α and the individually mutated forms TAB1 (see Table 1 and Fig. 5a), the functional consequence is that p38β is not similarly autoactivated by TAB1 (see Supplementary Fig. 5a). Furthermore, we have examined the importance of Ile275, highlighted as a point of contact in Fig 3b, by its substitution in isolation within the p38α backbone. The autophosphorylation of p38α (Ile275Gly) by TAB1(371-416) is slower than seen with wild type p38α (see Supplementary Fig. 5b) and this is also reflected by a different thermodynamic signature of interaction (see Supplementary table 2 and Supplementary Fig. 3d).
Effect of TAB1 mutations on myocardial p38 and function
Having confirmed there was a deficit in the binding of TAB1(371-416A-B) to p38α, we next determined if this was also accompanied by the loss of its ability to induce p38α autophosphorylation in vitro and in vivo. Fig. 5c shows the marked difference between TAB1(371-416) and TAB1(371-416A-B) 46-mer peptides in their ability to accelerate p38α autophosphorylation in vitro, confirming the importance of the A and B residue pairs to the functional interaction.
To examine the importance of these residue pairs in the context of the TAB1 protein backbone we ectopically co-expressed p38α with TAB1(1-418), TAB1(1-418A), TAB1(1-418B) or TAB1(1-418A-B) in both prokaryotic (see Fig. 5d) and eukaryotic (see Fig. 5e) systems. The data show that each individual pair of mutations (A or B) is sufficient to diminish TAB1-induced autophosphorylation of p38α and that when present in combination (A and B) TGY motif phosphorylation is reduced to that seen when p38α is expressed in isolation.
Finally to determine the effect of the TAB1-mediated p38α interaction on the heart we exposed isolated adult rat cardiac myocytes and perfused isolated mouse hearts to TAB1(371-416), TAB1(371-416A-B) or a scrambled form of the peptide TAB1(371-416SCR) each fused to a cell penetration peptide derived from the HIV1 transactivator of transcription (TAT) (see Fig. 6a). Using fluorescein-labelled TAT-TAB1(371-416) we confirmed that the peptide penetrates cardiac myocytes in culture and perfused isolated hearts (Fig. 6b). In isolated cardiac myocytes the peptide is widely distributed with greatest accumulation in nuclei and at intercalated discs (Fig. 6b). TAB1(371-416) but not TAB1(371-416A-B) caused p38 activation (Fig. 6c) and at later time points cell death (see rounded appearance of cells with homogenous distribution of fluorescein label). Similarly in isolated perfused hearts TAT-TAB1(371-416), but not the other peptides, caused an elevation in the end-diastolic pressure and a marked reduction in developed pressure (see Fig. 6e). These changes were accompanied by a reduction in coronary flow (see Fig. 6f) and with p38 dual phosphorylation (see Fig. 6g). Furthermore these effects were reversed by pharmacological inhibition of p38 (see Fig. 6h). In combination, these data indicate that artificially driving the p38α:TAB1 association in myocardium has detrimental effects, suggesting that specifically inhibiting the association documented during myocardial infarction10,12,13 would be cardioprotective.
Figure 6.
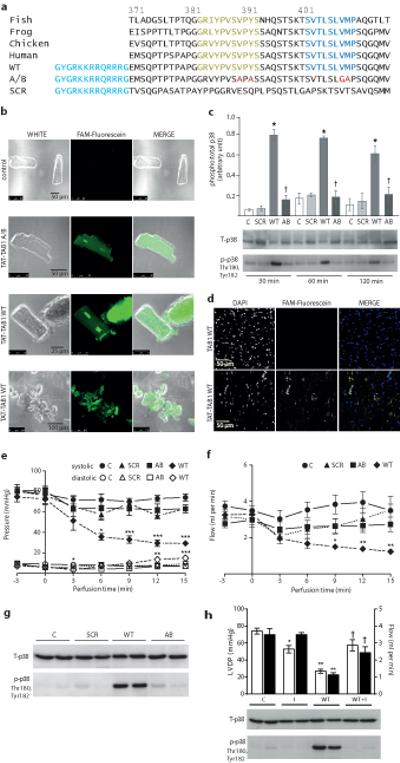
The effects of cell permeable forms of TAB1(371-416) on the myocardium. (a) The sequences of TAB1 cell permeable peptides and sequence conservation across vertebrates. Fish, Danio rerio, (GenBank: AAH58295.1), starting at 362. Frog, Xenopus Laevus, (GenBank: AAC14009.1) starting at 368. Chicken, Gallus gallus, (Accession: NP_001006240.1) starting at 371. Human, Homo sapiens, (GenBank: AAC12660.1) starting at 371 with omission of Ala-Ala insert at 381-382. A cell penetration domain derived from HIV TAT is shown in red. The residues interacting directly with p38α in the crystal structure (see Fig. 3) are highlighted. The A-B peptide harbours mutations preventing interaction with p38α (see Fig. 5) highlighted in blue. SCR is scrambled control. (b) Confocal images of cultured adult rat ventricular myocytes (ARVMs) incubated with WT TAB1 peptide conjugated to fluorescein. (c) Immunoblot analysis of p38 in ARVMs exposed to cell permeable forms of TAB1(371-416). The quantitative data are derived from three separate transfections which appear in Supplementary Fig.6 (d) Confocal images of mouse hearts perfused for 15 minutes with WT TAB1 peptide conjugated to fluorescein. (e) Contractile performance of isolated mouse hearts during intracoronary perfusion of TAB1 peptides. Perfusion with peptides begins at time 0min. (f) Coronary flow corresponding to e. For panels e and f, n=5 mouse hearts per group. Error bars represent mean±s.e.m. * p<0.05 vs. Control; **p<0.01 vs. Control and ***p<0.005 vs. Control. Comparisons by 2 way ANOVA with repeated measures and post-hoc comparison by Bonferroni. (g) Immunoblot analysis of p38 activation in mouse hearts after 15mins exposure to cell permeable forms of TAB1(371-416) (h) Inhibition of p38α with BIRB796 attenuates the effects of perfusion with TAB1(371-416) (WT) or vehicle DMSO (0.1% v/v) (C). The upper panel is of left ventricular developed pressure (LVDP) [open bars] and coronary blood flow (Flow) [shaded bars] with or without BIRB796 (I). Numbers per group C=5, C+I=4, WT=7 and WT+I=5. Bars are mean±s.e.m. *P<0.05 vs. C; **P<0.01 vs. C; † P<0.05 vs. WT. Comparisons by one-way ANOVA with post-hoc comparison by Bonferroni within LVDP or Flow. The uncropped images from which the immunoblots are derived appear in Supplementary Fig.6.
DISCUSSION
This study describes the molecular mechanism of TAB1-mediated p38α autophosphorylation. The salient findings are (i) that TAB1 interacts directly with p38α, (ii) this interaction increases the affinity of p38α towards ATP, (iii) as a consequence p38α autophosphorylates its activation loop in cis, (iv) the ability to induce p38α autophosphorylation resides in residues 371-416 of TAB1, (v) TAB1 makes direct contact with p38α in a bipartite manner with occupancy of both sites required for optimal autophosphorylation, (vi) the TAB1-induced autoactivation mechanism is specific to p38α, sparing p38β (vii) the autoactivation of p38α through the TAB1 interaction can be artificially driven in isolated cardiac myocytes and perfused hearts, reducing viability and function.
The autophosphorylation of Thr180 and Tyr182 within the activation loop is conceptually difficult to accept: p38α is a serine/threonine kinase and should therefore have difficulty phosphorylating Tyr182. Nonetheless, there are at least four other examples in the literature of wild type Ser/Thr kinases autophosphorylating on a Tyr, specifically DYRK, GSK3β, fus3 and PLK419,22. Fus3 is the only other kinase for which the molecular mechanism of auto activation is understood at an atomic level33. Fus3 is a highly conserved p38 yeast homologue which is similarly activated by dual phosphorylation of its TGY activation motif by an upstream MAPKK. However, Bhattacharyya et al., have shown that it can also be activated and autophosphorylated following binding to full length Ste5, a scaffold protein, and such activity resides in a 28-residue region (encompassing 288-316) (see Fig. 4b33, and Supplementary Fig. 4b). Co-crystallisation of Fus3 with this peptide revealed an allosteric mechanism of activation where Ste5 establishes contact points in both the N- and C- terminal lobes of Fus3 resulting in their reorientation to achieve kinase activity33. Nevertheless, despite many similarities, the Fus3:Ste5 and p38α:TAB1 systems use different molecular mechanisms to achieve autophosphorylation. Firstly, our structural data show that the TAB1 peptide establishes contact points only with the C-lobe of p38α, we also identify a novel binding site on the kinase surface that at present is TAB1-specific. Moreover, in the Fus3-Ste5 system the autophosphorylation is only of the Tyr whereas in the p38α:TAB1 system it is dual-autophosphorylation.
The atomic details of the interaction of TAB1 with p38α presented here unambiguously show that TAB1 is capable of overcoming the ‘Catch 22’ paradox, since its binding to p38α is accompanied by a dramatic increase in affinity of p38α towards ATP. At the molecular level, this is linked to distant conformational rearrangements of the ATP binding site and of the activation loop.
The interaction of p38α and TAB1 is circumstance specific, interestingly all the circumstances so far reported in the literature are harmful to the cell and this is particularly well-illustrated by myocardial ischemia/infarction10-13 and cardiac amyloidosis14. p38α and TAB1 are ubiquitously expressed and the residues involved in TAB1:p38α recognition are conserved across species suggesting that the interaction may have broader relevance. However, tool compounds are needed to examine other contexts in which this interaction is of importance and to explore the utility of disrupting it in the context of myocardial infarction. Ultimately, such compounds may enable circumstance-specific p38α inhibition that circumvents the toxicity and/or lack of efficacy seen in recent clinical trials7,8.
ONLINE METHODS
SUPPLEMENTARY EXPERIMENTAL PROCEDURES
Expression and purification of p38α, 2H/15N p38α and 2H/13C/15N p38α
DNA encoding mouse p38α was derived from a pET14b vector kindly donated by Yibin Wang 34 and sub-cloned into a pETDuet-1 vector. The full coding sequence included an N-terminal HexaHistidine tag followed by a TEV cleavage site and mouse p38α sequence. The vector was transformed in E. coli strain Rosetta II cells (Novagen). The protein expression and purification procedure followed is described in 35. For 2H/15N p38α, 1 L culture was grown in minimal media containing 0.8 g/L15N –ammonium chloride and 95% D2O. For 2H/13C/15N p38α, 1 L culture was grown using Silantes E-coli OD2 growth media (13C,98%;2H,98%;15N,98%). For the labelled proteins, purification was performed as above.
Expression and purification of TAB1(1-438)
Chemically synthesised DNA encoding mouse TAB1 was sub-cloned into a pETDuet-1 vector. The full coding sequence included an N-terminal HexaHistidine tag followed by a TEV cleavage site and mouse TAB1(1-438). The vector was transformed in E. coli strain Rosetta II cells. Recombinant TAB1(1-438) was expressed and purified following the same protocols used for p38α.
TAB1(371-416)
The peptides corresponding to TAB1(371-416) and TAB1(384-412) were purchased from Activobiotech.
Peptide for NMR study was selectively 15N, 13C labelled on residues shown below: E(15N,13C)MSQPTPTP(15N,13C)APGGR(15N,13C)VYP(15N,13C)VSVPYSS(15N,13C)AQS TSKTS(15N,13C)VT(15N,13C)LS(15N,13C)L(15N,13C)V(15N,13C)MPSQGQ(15N,13C)MV and purchased from Thermofisher.
The TAT-TAB1 peptide were purchased from Activobiotech and the sequences are: wt TAT-TAB1 GYGRKKRRQRRRGEMSQPTPTPAPGGRVYPVSVPYSSAQSTSKTSVTLSLVMPSQGQMV TAT-TAB1(A/B) GYGRKKRRQRRRGEMSQPTPTPAPGGRVYPVSAPASSAQSTSKTSVTLSLGAPSQGQMV TAT-TAB1(SCR) GYGRKKRRQRRRGTVSQGPASATPAYPPGGRVESQPLPSQSTLGAPSKTSVTSAVQSMM The fluorescently labelled peptides were conjugated to FAM.
Cloning and co-expression of p38α, p38αLys53Met(KD), p38αIle275Gly, p38α:TAB1(1-502), p38α:TAB1(1-438), p38α:TAB1(1-418), p38α:TAB1(1-418A; Val390Ala/Tyr392Ala), p38-TAB1(1-418B;Val408Gly/Met409Ala) and p38-TAB1(1-418A/B; Val390Ala/Tyr392Ala/Val408Gly/Met409Ala)
pETDuet-1 vector was used for bacterial co-expression of p38α and the different TAB1 mutants. In all the bacterial vectors p38α was sub-cloned in the first multiple cloning site whereas TAB1 was sub-cloned in the second multiple cloning site. All the TAB1 mutants were obtained using the same strategy: overlapping C and N terminus fragments were produced by PCR with complementary internal primers harbouring the desired mutations. The PCR products from this reaction were then combined to form the template for a second PCR reaction with external primers.
In vitro kinase assays and immunoblot analysis
Depending on the experiment WTp38α, KDp38α, ASp38α, p38αIle275Gly or an equimolar mixture of two (3 μM) were incubated with wild type TAB1(371-416), TAB1(384-412) or mutated TAB1(371-416A/B) (15 μM) in buffer containing 100 mM NaCl, 20 mM Tris, pH 7.5, 2 mM DTT, 2 mM MgCl2 and 600 μM ATP. The reactions were monitored for at least 2 h.
Samples were resolved on a 10% reducing sodium dodecyl sulphate-polyacrylamide gel and blotted onto a polyvinylidene difluoride membrane. Membranes were incubated in Ponceau S solution (Sigma) for 5 mins. After proteins had been visualized, membranes were rinsed in distilled water and destained in 0.1 M NaOH. Membranes were blocked for 1 h in 1.33% low-fat milk and 0.33% bovine serum albumin in Tris-buffered saline, pH 7.4, 0.1% Tween-20. Primary antibodies were incubated overnight at 4 °C with agitation. Antibodies used included anti dual-phosphorylated p38 (Thr180, Tyr182) (#M8177, Sigma, #9211, Cell Signalling Technology) 1:4,000, Total p38 (#9212, Cell Signalling Technology) 1:15,000, anti-phospho-p38 Tyr182 (#sc-7975-R, Santa Cruz Biotechnology) 1:2000. Following washing and incubation with the appropriate HRP-conjugated secondary antibody (GE Healthcare, #NA 943ML for rabbit primary and #NA931ML for mouse primary, both at 1:2000, and ThermoScientific, #31480 for sheep primary at 1:1000), antibody-antigen complexes were visualized by enhanced chemiluminescence (Pierce). Original images of blots used in this study can be found in Supplementary Fig. 6.
ITC titrations
All ITC experiments were carried out on an iTC200microcalorimeter from Microcal (GE Healthcare). SB203508 and ATPγS were purchased from Tocris Bioscience at the highest purity available.
Heat produced by titrant dilution was verified to be negligible by a control experiment, titrating into buffer alone, under the same conditions. Integrated heat data obtained for the titrations corrected for heats of dilution were fitted using a nonlinear least-squares minimisation algorithm to a theoretical titration curve, using the MicroCal-Origin 7.0 software package. ΔH° (reaction enthalpy change in kcal·mol−1), Kb (equilibrium binding constant in M−1), and n (molar ratio between the two species in the syringe and calorimetric cell respectively) were the fitting parameters. The reaction entropy was calculated using the relationships ΔG = −RT·lnKb (R 1.987 cal·mol−1*K−1, T 298 K) and ΔG = ΔH-TΔS.
NMR spectroscopy
Spectra of free p38α and of p38α:TAB1(371-416) complex were acquired in buffer containing 50 mM NaCl, 20 mM Tris (pH 7.5) and 2 mM DTT, while those of TAB1(371-416) were acquired in buffer containing 20 mM Tris (pH 7.5) and 2 mM DTT. Sample concentrations of free p38α and free TAB1(371-416) were 0.5 mM. Samples of p38α:TAB1(371-416) in which p38α was [2H/15N/13C]-labeled and TAB1 unlabeled were prepared by mixing the components in a molar ratio of 1:2 at low micromolar concentration and concentrating the complex to a final concentration of 0.5 mM. The same procedure, with the inverted stoichiometric ratio, was used to prepare the NMR sample for the complex of unlabeled p38α and selectively [15N/13C]-labeled TAB1 (371-416).
Spectra were acquired on Bruker Avance spectrometers operating at 700 and 500 MHz (1H frequency) and a Varian Inova spectrometer operating at 800 MHz, all equipped with cryoprobes. The spectra were processed using the manufacturers’ software or using NMRpipe 36, NMRViewJ 37, and XEASY 38. The spectra were typically apodised in each dimension by applying a Gaussian or sine –bell window function and zero-filled to double the size of the data prior to Fourier transformation.
15N-TROSY39 and 13C-HSQC 40,41 spectra were acquired in a standard manner. Chemical shift variations were determined by comparing the 15N-TROSY spectrum of free p38α with that of p38α:TAB1(371-416). The weighted average chemical shift variation (ΔδAV) was calculated as follows ([(ΔHN)2 + (ΔN)2/5]/2) 1/2 where ΔHN and ΔN are the chemical shift variations in the two dimensions. (42,43. Chemical shift variations were classified as being weak (ΔδA ≤ 0.5), strong (ΔδAV > 0.5), undefined (where signals are lost upon interaction) or unclear (unassigned peaks and where spectral overlapped prevented the unambiguous analysis). The classes were mapped onto the structure of p38α (PDB ID: 1P38) 31.
Crystallization and structure determination
Complex of p38α-TAB1 at ~11 mg/ml pre-incubated with 1 mM SB220025 was crystallized by sitting drop vapour diffusion at 4 °C. The complex crystals grew in three different conditions yielding three different forms; i) p38α:TAB1 Monoclinic - 25% w/v medium-molecular weight PEG Smears (PEG 2000, PEG 3350, PEG 4000, PEG 5000 MME), 0.1M MES pH 6.5, ii) p38α:TAB1 Tetragonal - 20% w/v PEG 3350, 0.2 M Na/K tartrate, 0.1 M bis-tris propane pH 6.5, 10% ethylene glycol, and iii) p38α:TAB1:SO4 Tetragonal - 25% w/v medium-molecular weight PEG Smears, 0.2 M Ammonium sulphate, 0.01 M CdCl2, 0.1 M HEPES pH 7.5. Crystals were cryo-protected with mother liquor supplemented with 20% ethylene glycol and flash-cooled in liquid nitrogen. Diffraction data were collected at the Diamond Light Source beamline I03 for the former two crystals, and I04-1 for the latter crystal and processed and scaled with MOSFLM and SCALA from the CCP4 suite 44. The structure was solved by molecular replacement using PHASER 45 and a p38α monomer (PDB id: 3que 46) as a search model. Iterative cycles of manual building alternated with structure refinement were performed using COOT 47 and REFMAC 48, respectively. Data collection and refinement statistics are summarized in Table 2
Table 2.
Data collection and refinement statistics
| p38α:TAB1 Monoclinic |
p38α:TAB1 Tetragonal |
p38α:TAB1:SO4 Tetragonal |
|
|---|---|---|---|
| Data collection | |||
| Space group | P 21 | P 41 | P 41 |
| Cell dimensions | |||
| a, b, c (Å) | 43.3, 73.6, 59.2 | 86.5, 86.5, 226.9 | 87.1, 87.1, 228.3 |
| α, β, γ (°) | 90.0, 91.2, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 59.18-1.95 (2.06-1.95) * | 80.86-2.05 (2.16-2.05) * | 43.55-2.32 (2.45-2.32) * |
| R merge | 0.061 (0.758) | 0.084 (0.422) | 0.077 (0.707) |
| I / σI | 12.3 (2.0) | 8.5 (2.1) | 10.4 (2.0) |
| Completeness (%) | 99.4 (98.7) | 99.5 (97.1) | 98.7 (99.6) |
| Redundancy | 4.1 (3.9) | 3.2 (2.5) | 3.3 (3.4) |
| Refinement | |||
| Resolution (Å) | 46.11-1.95 | 86.55-2.05 | 42.78-2.32 |
| No. reflections | 27,022 (3,898) | 103494 (14756) | 72211 (10608) |
| Rwork / Rfree | 0.198/ 0.250 | 0.148/ 0.186 | 0.192/ 0.236 |
| No. atoms | |||
| Protein p38α | 2779 | 11262 | 11254 |
| Protein TAB1 | 155 | 595 | 598 |
| Others | 196 | 1045 | 626 |
| B factors | |||
| Protein p38α | 45 | 32 | 47 |
| Protein TAB1 | 71 | 43 | 61 |
| Others | 43 | 35 | 44 |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.016 | 0.015 | 0.013 |
| Bond angles (°) | 1.6 | 1.6 | 1.5 |
Values in parentheses are for highest-resolution shell.
The Protein Data Bank accession codes are 4LOO, 4LOP, 4LOQ, respectively
Adult rat ventricular myocyte (ARVM) culture
ARVMs were isolated as previously described 49, and washed with M199 complete medium (M199 medium with added 100 I.U./ml penicillin, 100 I.U./ml streptomycin, 2mM L-carnitine, 5mM creatine and 5mM taurine). The cell suspension was allowed to settle by gravity centrifuged at 100 g for 2 min in order to pellet the myocytes, which were then resuspended in M199 complete medium and placed in laminin-coated 6-well plates prior to incubation in 5% CO2/room air at 37 °C. After 1 h, the medium was aspirated, leaving only adherent cells, and fresh pre-warmed M199 complete medium added with specified TAT-TAB1 peptides.
Retrograde perfusion of isolated mouse hearts
All experiments were approved under the Animals Scientific Procedures Act, UK Home Office. After intraperitoneal pentobarbital (300mg/kg) and heparin (150 units) the hearts were rapidly isolated from outbred male c57BL/6 mice (24-27g, Harlan, UK), and placed in ice cold modified Krebs-Henseleit buffer (KHB) (118.5 mM NaCl, 25.0 mM NaHCO3, 4.75 mM KCl, 1.18 mM KH2PO4, 1.19 mM MgSO4, 11.0 mM D-glucose, and 1.4 mM CaCl2). The excised hearts were mounted on a Langendorff apparatus and retrogradely perfused at a constant pressure of 80 mm Hg with KHB equilibrated with 95% O2 and 5% CO2 at 37°C. A fluid-filled balloon inserted into the left ventricle monitored contractile function and was set to an end-diastolic pressure of 2-8 mmHg. Atrial pacing was at 600 bpm. For inclusion isolated mouse hearts after stabalization had to have a left ventricular developed pressure of at least 60mmHg with coronary flow between 1.5 to 3.5ml/min whilst being pacing at ~600bpm. For more detailed methods see 10,49.
The maximum concentration of TAT-TAB1(371-416A/B) that did not cause discernible contractile dysfunction (15μM) was chosen as the mid-range peptide concentration. Preliminary experiments were carried out using three different concentrations of the peptide(s) to test for toxicity in the isolated heart. WT, SCR and MUT peptide and buffer controls were perfused through an isolated heart for 15 minutes following a 30 min baseline perfusion (n=2 for each condition). These studies demonstrated a clear effect of WT peptide on the function of the heart in a concentration-dependent manner. Preliminary data revealed control mean LVDP=67 mmHg and WT peptide perfusion mean=43 mmHg (with an n=2 data set) and a combined Standard Deviation (SD) of 12 mmHg , resulting in a recommended sample size of >4<6 for the concentration of peptide used in the final study given 95% confidence and 80% probability (www.biomath.info/power/ttest.html). For the main study, peptides were randomized into falcon tubes in KH buffer and stored in a −80 freezer labeled 1-28 in a random order assigned by another member of staff, not involved in the perfusion protocol. Samples were taken from the freezer in order (1-28) and used in the perfusion study. Hearts were exposed to peptide at 15μM which were recirculated for 15 minutes in a total volume of 20mL KHB.
Outbred c57/bl6 mice from a commercial supplier were used in this study. All mice were 8-10 weeks of age and weighed 28-31g. Blinding of mice was not required since the selection of mouse was unlikely to impact on the outcome of the experiments
Statistical Analysis
Results are expressed as mean±s.e.m for hemodynamic performance of individual isolated mouse heart and mean±s.d. for quantification of immunoblots from individual cell transfection experiments. Data sets were analysed by one-way, (figs 1b/c, 5e, 6c and 6h) or two-way, (figs 6e/f) analysis of variance followed by Bonferonni comparison. A value of P < 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by project grants from the UK Medical Research Council (MRC) (G0802033 to M.S.M., G1001138 to M.S.M. and M.R.C. and J007501 to M.S.M.) and UK Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s & St Thomas’ NHS Foundation Trust. All the ITC and part of the NMR experiments were performed using the facilities of the Centre for Biomolecular Spectroscopy, King’s College London, established with a Capital Award from the Wellcome Trust to M.R.C. (085944/Z/08/Z). S.K. is grateful for support from the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. A.C. is supported by the European Union FP7 Grant No. 278568 “PRIMES”. We thank the scientists at the Diamond Light Source for help with data collection. We are grateful to the MRC Biomedical NMR Centre, Mill Hill, and its staff for a generous allocation of NMR time and for expert technical assistance. We also thank N. Drinkwater and B. Sutton for help at an early stage of the work.
Footnotes
ACCESSION CODES The Protein Data Bank accession codes for p38α-TAB1 are 4LOO, 4LOP, 4LOQ,
REFERENCES
- 1.Cuenda A, Rousseau S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim.Biophys.Acta. 2007 doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Mudgett JS, et al. Essential role for p38alpha mitogen-activated protein kinase in placental angiogenesis. Proc.Natl.Acad.Sci.U.S.A. 2000;97:10454–10459. doi: 10.1073/pnas.180316397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beardmore VA, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–10464. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JC, et al. A protein-kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 5.Gum RJ, et al. Acquisition of sensitivity of stress-activated protein kinases to the p38 inhibitor, SB 203580, by alteration of one or more amino acids within the ATP binding pocket. J Biol Chem. 1998;273:15605–15610. doi: 10.1074/jbc.273.25.15605. [DOI] [PubMed] [Google Scholar]
- 6.Dominguez C, Powers DA, Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr.Opin.Drug Discov.Devel. 2005;8:421–430. [PubMed] [Google Scholar]
- 7.Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009;60:317–320. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- 8.Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann.Rheum.Dis. 2010;69(Suppl 1):i77–i82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin DE, Felice De Nicola G, Marber MS. New Therapeutic Targets in Cardiology: p38 Alpha Mitogen-Activated Protein Kinase for Ischemic Heart Disease. Circulation. 2012;126:357–368. doi: 10.1161/CIRCULATIONAHA.111.071886. [DOI] [PubMed] [Google Scholar]
- 10.Tanno M, et al. Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ.Res. 2003;93:254–261. doi: 10.1161/01.RES.0000083490.43943.85. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, III, Young LH. AMP-Activated Protein Kinase Activates p38 Mitogen-Activated Protein Kinase by Increasing Its Recruitment to TAB1 in the Ischemic Heart. Circ.Res. 2005;97:872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 12.Ota A, Zhang J, Ping P, Han J, Wang Y. Specific Regulation of Noncanonical p38{alpha} Activation by Hsp90-Cdc37 Chaperone Complex in Cardiomyocyte. Circ Res. 2010;106:1404–1412. doi: 10.1161/CIRCRESAHA.109.213769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiedler B, et al. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. J Biol.Chem. 2006;281:32831–32840. doi: 10.1074/jbc.M603416200. [DOI] [PubMed] [Google Scholar]
- 14.Shi J, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc.Natl.Acad.Sci.U.S.A. 2010;107:4188–4193. doi: 10.1073/pnas.0912263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge B, et al. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- 16.Cheung PC, Campbell DG, Nebreda AR, Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 2003;22:5793–5805. doi: 10.1093/emboj/cdg552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diskin R, Lebendiker M, Engelberg D, Livnah O. Structures of p38alpha active mutants reveal conformational changes in L16 loop that induce autophosphorylation and activation. J Mol Biol. 2007;365:66–76. doi: 10.1016/j.jmb.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 18.Salvador JM, et al. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat.Immunol. 2005;6:390–395. doi: 10.1038/ni1177. [DOI] [PubMed] [Google Scholar]
- 19.Lochhead PA. Protein kinase activation loop autophosphorylation in cis: overcoming a Catch-22 situation. Sci.Signal. 2009;2:e4. doi: 10.1126/scisignal.254pe4. [DOI] [PubMed] [Google Scholar]
- 20.Wilson KP, et al. Crystal structure of p38 mitogen-activated protein kinase. J Biol.Chem. 1996;271:27696–27700. doi: 10.1074/jbc.271.44.27696. [DOI] [PubMed] [Google Scholar]
- 21.Zhang WX, et al. Time-resolved Forster resonance energy transfer assays for the binding of nucleotide and protein substrates to p38alpha protein kinase. Analytical biochemistry. 2005;343:76–83. doi: 10.1016/j.ab.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Shrestha A, Hamilton G, O’Neill E, Knapp S, Elkins JM. Analysis of conditions affecting auto-phosphorylation of human kinases during expression in bacteria. Protein Expr Purif. 2012;81:136–143. doi: 10.1016/j.pep.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacquet S, et al. The role of RIP2 in p38 MAPK activation in the stressed heart. J.Biol.Chem. 2008;283:11964–11971. doi: 10.1074/jbc.M707750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumphune S, et al. A chemical genetic approach reveals that p38alpha MAPK activation by diphosphorylation aggravates myocardial infarction and is prevented by the direct binding of SB203580. J Biol.Chem. 2010;285:2968–2975. doi: 10.1074/jbc.M109.079228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sicard P, et al. The activation of p38alpha, and not p38beta, mitogen-activated protein kinase is required for ischemic preconditioning. J Mol Cell Cardiol. 2010;48:1324–1328. doi: 10.1016/j.yjmcc.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ge B, et al. TAB1beta (transforming growth factor-beta-activated protein kinase 1-binding protein 1beta ), a novel splicing variant of TAB1 that interacts with p38alpha but not TAK1. J Biol.Chem. 2003;278:2286–2293. doi: 10.1074/jbc.M210918200. [DOI] [PubMed] [Google Scholar]
- 27.Zhou H, et al. Determinants that control the specific interactions between TAB1 and p38alpha. Mol Cell Biol. 2006;26:3824–3834. doi: 10.1128/MCB.26.10.3824-3834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rothweiler U, et al. p38alpha MAP kinase dimers with swapped activation segments and a novel catalytic loop conformation. J Mol Biol. 2011;411:474–485. doi: 10.1016/j.jmb.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 29.Cameron AJ, Escribano C, Saurin AT, Kostelecky B, Parker PJ. PKC maturation is promoted by nucleotide pocket occupation independently of intrinsic kinase activity. Nat.Struct.Mol Biol. 2009 doi: 10.1038/nsmb.1606. [DOI] [PubMed] [Google Scholar]
- 30.Blethrow J, Zhang C, Shokat KM, Weiss EL. Design and use of analog-sensitive protein kinases. Curr.Protoc.Mol Biol. 2004 doi: 10.1002/0471142727.mb1811s66. Chapter 18, Unit. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, et al. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc Natl Acad Sci U S A. 1997;94:2327–2332. doi: 10.1073/pnas.94.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc.Natl.Acad.Sci.U.S.A. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattacharyya RP, et al. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, et al. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen activated protein kinase family. J Biol Chem. 1998;273:4, 2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
REFERENCES
- 35.Bukhtiyarova M, et al. Improved expression, purification, and crystallization of p38alpha MAP kinase. Protein Expr Purif. 2004;37:154–161. doi: 10.1016/j.pep.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 36.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 38.Bartels C, Xia TH, Billeter M, Guntert P, Wuthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 39.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer AG, 3rd, Fairbrother WJ, Cavanagh J, Wright PE, Rance M. Improved resolution in three-dimensional constant-time triple resonance NMR spectroscopy of proteins. Journal of biomolecular NMR. 1992;2:103–108. doi: 10.1007/BF02192804. [DOI] [PubMed] [Google Scholar]
- 41.Schleucher J, et al. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. Journal of biomolecular NMR. 1994;4:301–306. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- 42.Foster MP, McElroy CA, Amero CD. Solution NMR of large molecules and assemblies. Biochemistry. 2007;46:331–340. doi: 10.1021/bi0621314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martino L, et al. Analysis of the interaction with the hepatitis C virus mRNA reveals an alternative mode of RNA recognition by the human La protein. Nucleic Acids Res. 2012;40:1381–1394. doi: 10.1093/nar/gkr890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 46.Koeberle SC, et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat Chem Biol. 2012;8:141–143. doi: 10.1038/nchembio.761. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 49.Bellahcene M, et al. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol. 2006;48:545–555. doi: 10.1016/j.jacc.2006.02.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.