

Abstract
Therapy-resistance and postoperative recurrence are causes of the poor prognosis in pancreatic cancer. Conventional therapies have a limited impact on the control of pancreatic cancer, resulting in the rapid re-growth of the tumor. The indispensable role of tumor-stromal interaction, which acts as a defender of cancer cells and enhances malignant potential, is being uncovered now. For example, specific signaling pathways for desmoplasia induction have been identified, such as sonic hedgehog (Shh) or connective tissue growth factor (CTGF), whose inhibition causes desmoplasia depletion and therapeutic advantages at least in in vivo mouse models of pancreatic cancer. Revolutions in drug delivery methods have led to the establishment of novel chemotherapeutic regimens, with better patient survival. Furthermore, mechanisms of immunosuppression in the pancreatic cancer-bearing host were clarified by the identification of myeloid-derived suppressor cells (MDSCs), which also promote disease progression. Strategies to target these components of the tumor stroma revealed certain anticancer effects in vitro and in vivo, suggesting the possibility of stroma-targeting therapy. Suppression of the stromal cell function increases the sensitivity of pancreatic cancer cells to therapeutic intervention. Further study will clarify the complex nature of the tumor microenvironment, the targeting of which has the potential to improve clinical outcome.
Keywords: desmoplasia, pancreatic stellate cells, cell survival, sonic hedgehog inhibitor, angiotensin receptor blocker, chemoresistance
Introduction
Radical surgical resection for pancreatic cancer is a curative therapy, but benefits only a small percentage (~20%) of pancreatic cancer patients. Even when such patients receive surgical resection, early recurrence and metastasis threaten their lives. Most pancreatic cancer patients show metastatic invasion to large blood vessels or distant organs, resulting in unresectable disease. The prognosis of inoperable patients is extremely poor, due to the lack of an effective therapy (Hidalgo, 2010; Michl and Gress, 2013). According to the Japan Pancreatic Cancer Registry, which summarized the clinical data of pancreatic cancer in Japan, the prognosis of pancreatic cancer overall has been improving for over 30 years, but the 5-year survival ratio is still lower than 20% (Egawa et al., 2012). The clinical problem of pancreatic cancer is its resistance to conventional therapies, such as chemotherapy or radiation. Even though these therapies reveal suppressive effects on tumor growth for a while, re-growth of the tumor frequently occurs. Furthermore, conventional chemotherapy, gemcitabine itself was shown to induce therapy-resistant populations of cancer cells in in vivo xenograft model in a previous study, suggesting specific mechanisms underlying the development of resistance (Jimeno et al., 2009). Therefore, an additional therapeutic strategy needs to be established to prevent the development of resistance.
Gemcitabine is a well-established therapeutic agent for unresectable pancreatic cancer, but complete remission of the disease rarely occurs (Burris et al., 1997). Gemcitabine alleviated disease-related symptoms in this study, but nearly 50% of the patients treated with gemcitabine showed only a partial response or static disease in imaging studies. Additional chemotherapeutic regimens using cytotoxic agents such as cisplatin, 5-fluorouracil (5-FU) or capecitabine in combination with gemcitabine were reported, but significant improvement in the patients' survival has not been achieved (Berlin et al., 2002; Heinemann et al., 2006; Herrmann et al., 2007). Targeted therapies were also tested alone or in combination with chemotherapy, such as vascular endothelial growth factor antibody and multikinase inhibitors. However, these agents have also failed to show improvement in patients' survival, so far (Kindler et al., 2010, 2012; O'Reilly et al., 2010). In addition, pancreatic cancer shows resistance against radiation therapy. A systematic review of the management of locally advanced pancreatic cancer demonstrated that radiation therapy alone does not have a survival benefit over that of chemoradiation therapy, suggesting the difficulties in controlling pancreatic cancer by radiation alone (Sultana et al., 2007). Recent studies indicated some beneficial effects of chemoradiation for patients with borderline resectable pancreatic cancer, but its effect on the patients with locally advanced disease remains controversial (Goodman and Hajj, 2013). Nevertheless, radiation has few benefits for metastatic pancreatic cancer.
These clinical features of pancreatic cancer have been considered to be the result of resistance in the cancer cells themselves, such as increased cell proliferation, enhanced survival signal, and blocked apoptotic pathways. Indeed, cumulative gene mutations provide these characteristics to cancer cells, which require more than 20 years for the establishment of metastatic disease (Yachida et al., 2010). However, host cells are also exposed to various signals from the pancreatic cancer cells at the same time. Recent research identified that cancer stromal cells play pivotal roles during the progression of pancreatic cancer, providing a cancer-promoting microenvironment. In pancreatic cancer tissue, cancer cells are surrounded by fibrotic stroma called desmoplasia, which sometimes occupy a larger area than cancer cells (Erkan et al., 2012). Pancreatic stellate cells (PSCs) play a central role in the formation of fibrotic tumor stroma (Apte et al., 2004; Bachem et al., 2005; Vonlaufen et al., 2008; Masamune and Shimosegawa, 2009, 2013). The interaction between cancer cells and stromal cells perpetuates inflammation within the pancreatic cancer tissue, which drives the formation and maintenance of desmoplasia. This tissue structure and extracellular matrix (ECM) proteins were reported to increase pancreatic cancer cell chemoresistance against gemcitabine and 5-FU (Erkan et al., 2007). Similarly, the ECM component hyaluronan, a megadalton glycosaminoglycan, was also reported to impair the vascular function and drug delivery in a genetically engineered mouse model of pancreatic cancer (Jacobetz et al., 2013). Another report described that the expression of Secreted protein acidic and rich in cysteine (SPARC) in the tumor stroma was inversely correlated with patients' survival. This study confirmed the invasion-promoting role of exogenous SPARC in pancreatic cancer cells, suggesting a tumor-promoting role of ECM proteins (Mantoni et al., 2008). In addition, pancreatic cancer-derived immunosuppression also contributes to the disease progression, which was confirmed by the existence of myeloid-derived suppressor cells (MDSCs) in pancreatic cancer tissue (Clark et al., 2007; Evans and Costello, 2012). These studies indicate that tumor-stromal interactions contribute to therapy-resistance in pancreatic cancer, which therefore could be an alternative therapeutic target.
Recently, attempts to treat pancreatic cancer by targeting tumor-stromal interactions have been reported. Various strategies have been examined such as targeting PSCs, inhibiting ECM deposition, suppressing angiogenesis and restoring the immune response in pancreatic cancer. Some of these strategies suggested the possibility of targeting the tumor stroma of pancreatic cancer as a novel therapeutic option. Since stromal cells maintain intact intracellular signals, these cells are assumed to show better responses to therapeutic intervention compared with cancer cells. In addition, the therapy-resistant evolution seen in cancer cells is a rare phenomenon in normal cells, based on the normal genomic regulation and lack of oncogenic mutations. The combination of novel strategies with conventional therapies should improve the clinical outcomes of pancreatic cancer. This review article summarizes the mechanisms of therapy-resistance in pancreatic cancer that are provided by tumor-stromal interactions. The current status and benefits of novel therapeutic strategies that modify drug delivery and target tumor-stromal interactions are discussed.
Critical mediators of tumor-stromal interaction
Desmoplasia consists of the deposition of ECM proteins and consistently activated stromal cells such as PSCs and fibroblasts. Among stromal cells, PSCs play a central role in ECM production and trigger continuous inflammation through cytokine production (Erkan et al., 2012). In addition to PSCs, cancer-associated fibroblasts suppress blood vessel formation leading to the sparse vasculature, making drug delivery more difficult (Olson and Hanahan, 2009). These stromal cells contribute to the establishment of desmoplasia that involve the activation of multiple signaling pathways and cell-to-cell interactions. However, the entire picture of these processes remains ambiguous. Recent research identified some of the critical pathways that induce desmoplasia in pancreatic cancer that could be pharmaceutically targeted.
For example, sonic hedgehog (Shh) is highly expressed in pancreatic cancer tissues and their precursor lesions, which suggests some contribution to the pancreatic cancer progression (Kayed et al., 2006). The hedgehog signal plays an important role in cell-fate determination during organ development by modulating multiple cellular functions. Recently, pancreatic cancer cell-derived Shh was found to induce desmoplasia in an orthotopic implantation model of pancreatic cancer in athymic nude mice (Bailey et al., 2008). Shh affected the differentiation of human PSCs and fibroblasts, demonstrating an indispensable role as a mediator of the desmoplastic reaction. Another study identified that connective tissue growth factor (CTGF) expression was elevated in pancreatic cancer tissue compared with normal pancreatic tissue. CTGF is able to bind various growth factors or integrins modifying their activity (Abreu et al., 2002; Heng et al., 2006). CTGF was found to stimulate the proliferation of PSCs, migration, and fibrogenesis (Gao and Brigstock, 2006). CTGF expression was associated with the elevated expression of the endogenous hypoxia marker carbonic anhydrase-IX in pancreatic cancer tissue, suggesting that pancreatic cancer cell-derived factors affect the tissue structure and microenvironment (Bennewith et al., 2009).
In turn, PSCs activate multiple signaling pathways in pancreatic cancer cells. Indirect co-culture of PSCs with human pancreatic cancer cell lines activated extracellular signal-regulated kinase (ERK) and Akt pathways in vitro, which are cell survival-related signaling pathways (Takikawa et al., 2013). Furthermore, PSCs promote cancer metastasis in an orthotopic implantation model and increase cellular migration of cancer cells (Vonlaufen et al., 2008).
Interaction between PSCs and pancreatic cancer cells also enhanced cancer-stem cell (CSC)-related phenotypes such as increased in vivo tumorigenicity, the in vitro ability to form spheroids and epithelial-mesenchymal transition (EMT) (Kikuta et al., 2010; Hamada et al., 2012). ECM proteins produced from PSCs were also found to play cancer-promoting role by previous studies. For instance, ECM protein SPARC promoted the EMT of cancer cells (Neuzillet et al., 2013). These cell-to-cell interactions form a feed-forward loop, which perpetuates the fibrogenic process within pancreatic cancer. Therefore, the inhibition of specific pathways indispensable for desmoplasia and broad suppression of the stromal function were evaluated for therapeutic application.
Effect of inhibiting desmoplasia-promoting pathways
Recently, the novel chemotherapeutic agent nab-paclitaxel became available for the treatment of pancreatic cancer (Von Hoff et al., 2011). This albumin-bound paclitaxel-based formula enables the hydrophobic paclitaxel to be administered at higher doses of the drug without solvent, and increases the maximum tolerated dose (Ibrahim et al., 2002). In addition, preoperative nab-paclitaxel administration decreased collagen deposition and cancer-associated fibroblasts in resected specimens, suggesting that nab-paclitaxel has an additional effect, tumor stroma disruption (Alvarez et al., 2013). Accordingly, the combination of nab-paclitaxel with gemcitabine increased the intratumoral gemcitabine concentration, leading to enhanced antitumor activity. The effect of nab-paclitaxel on the tumor stroma was also confirmed in the genetically engineered mouse model of pancreatic cancer, with impaired collagen maturation (Neesse et al., 2013b). This new regimen improved the overall survival and progression-free survival of patients with metastatic pancreatic cancer (Heinemann et al., 2013). These lines of evidence are an excellent example of the clinical benefits that can be provided by the evolution of drug delivery methods. Of note, the additional effect of nab-paclitaxel on the tumor stroma, which synergistically potentiates gemcitabine's antitumor effect, suggests the possibility of tumor-stromal interaction-targeting therapy.
Several studies reported more specific strategies directed at desmoplasia. As mentioned earlier, critical signals required for the inductions of desmoplasia have been identified. The Shh pathway plays an important role during embryonic development, and is aberrantly expressed in pancreatic cancer tissue. The Shh ligand binds to its receptor. Patched allows cell membrane-associated signal activator Smoothened to mediate the downstream signal. Small-molecule inhibitors of the Shh pathway were identified such as cyclopamine, a naturally occurring teratogenic molecule (Stanton and Peng, 2010). Additional Shh inhibitors have been identified thereafter, and their effects on desmoplasia were extensively studied. Conditional expression of mutant K-ras (constitutively active mutation G12D) and mutant p53 (inactivating mutation R172H) in mice pancreas recapitulates pancreatic cancer development, which shows a progression pattern similar to that of human pancreatic cancer such as liver metastasis and desmoplasia (Hingorani et al., 2005). Pancreatic tumors in this KPC mouse were resistant to gemcitabine, as confirmed by the sustained tumor growth under gemcitabine administration (Olive et al., 2009). Isolated pancreatic cancer cells from KPC mice were sensitive to gemcitabine, and the accumulation of active metabolites of gemcitabine in isolated cancer cells was not impaired, suggesting that the tumor stroma is responsible for this resistance. Oral administration of IPI-926, a derivative of cyclopamine successfully depleted desmoplasia in the tumors of KPC mice (Olive et al., 2009). IPI-926 caused a transient increase in the vascularity of the tumors, which increased the intratumoral gemcitabine concentration. Gemcitabine or IPI-926 alone did not show survival benefits, but combination therapy with IPI-926 and gemcitabine prolonged survival. Based on these results, clinical trial using IPI-926 in combination with gemcitabine was carried out. Unfortunately, the phase II clinical trial of IPI-926 halted due to the significantly shorter survival in patients on the IPI-926 arm. This result might be due to the heterogeneity of human pancreatic cancer, which could not be recapitulated by genetically engineered mouse model. In addition, depletion of desmoplasia by a single agent might be insufficient to eliminate cancer cells completely.
An alternative pathway has also been examined as a therapeutic target. Conditional expression of mutant K-ras (constitutively active mutation G12D) with TGF-β receptor type II knockout in pancreas also developed pancreatic cancer in mice, accompanied by desmoplasia. Elevated expression of CTGF was detected in these pancreatic tumors, and its induction was mediated by Cxc chemokine signal. Cxc receptor inhibitor SB225002 administration alone could decrease the CTGF expression in pancreatic tumors and prolonged the survival of the cancer-bearing mice (Ijichi et al., 2011). In another study using a monoclonal antibody against CTGF (FG-3019), an enhancement of the chemotherapy response by combination therapy with gemcitabine in KPC mice was described. In this study, cytidine deaminase inhibitor administration did not show therapeutic advantages despite the elevated gemcitabine concentration within the pancreatic tumors, whereas FG-3019 monoclonal antibody could enhance the effect of gemcitabine (Neesse et al., 2013a). Since cytidine deaminase inhibitor did not alter the tumor microenvironment, the poorer response to gemcitabine and the cancer cell survival might be largely due to environmental factors derived from the desmoplasia.
These targeted therapies against tumor stroma have certain effect, but several problems need to be solved for the establishment of effective regimen. Resistance against single targeted therapy is a common phenomenon, such as the resistance against epidermal growth factor receptor tyrosine kinase inhibitor in non-small cell lung cancer (Sun et al., 2013). To overcome this resistance mechanism, downstream target of epidermal growth factor pathway, MEK inhibition was tested that induced apoptosis in epidermal growth factor receptor tyrosine kinase inhibitor-resistant lung cancer cells (Song et al., 2013). Taken together, targeted therapy against tumor stroma will also suffer from the resistance, which needs to be prevented by a combination approach.
Targeting PSCs' function
Broad inhibition of the stromal function is an additional therapeutic strategy to attenuate tumor-stromal interactions. A wide variety of agents inhibit ECM production or the constitutive activation of PSCs. These agents include plant-derived polyphenol (ellagic acid, curcumin, and green tea polyphenol), nicotinamide adenine dinucleotide phosphate oxidase inhibitor (diphenylene iodonium and apocynin) and angiotensin II type 1 receptor blocker (ARB) (Masamune et al., 2005a,b, 2006, 2008; Sakurai et al., 2011). Treatment with these agents resulted in decreased ECM protein production such as collagen from PSCs. The attenuation of cytokine production from PSCs, inhibition of pro-inflammatory signals and reduced proliferation of PSCs were also observed by these treatments. Since these agents could be administered orally, tumor stroma-repressive effects have been well examined in vivo (Masamune et al., 2008; Sakurai et al., 2011).
Among these agents, ARB is an antihypertensive drug with established feasibility and safety in clinical use. An anti-inflammatory effect of ARB in the pancreas was found in one of the clinically-available ARBs, candesartan. The administration of candesartan in a rat model of chronic pancreatitis attenuated pancreatic inflammation and fibrosis (Yamada et al., 2003). Candesartan decreased the alpha-smooth muscle actin-positive cells in pancreas, suggesting suppressed activation of PSCs. A retrospective study of patients with pancreatic cancer who received angiotensin I-converting enzyme inhibitors (ACEIs) and ARBs demonstrated the contribution of both drugs to a better prognosis (Nakai et al., 2010). The overall survival was 15.1 months in the ACEI/ARB group and 8.9 months in the non-ACEI/ARB group (Nakai et al., 2010). Another report described the use of olmesartan to treat subcutaneous-tumor bearing immunodeficient mice. The growth of subcutaneous tumors derived from the co-injection of human pancreatic cancer cell line AsPC-1 with human PSC cell line was significantly suppressed by olmesartan (Masamune et al., 2013). Furthermore, olmesartan treatment attenuated the cell viability of PSCs and suppressed collagen production in vitro. Accordingly, decreased expression of alpha-smooth muscle actin and collagen deposition in subcutaneous tumors was confirmed, indicating an alteration of the microenvironment in the cancer tissue. In this study, delayed administration (2 weeks after the subcutaneous implantation of cancer cells with PSCs) of olmesartan also attenuated the growth of subcutaneous tumors, suggesting the contribution of PSCs to sustained tumor growth as well as successful tumor implantation. In addition to the inhibitory effect on PSCs, losartan, another ARB, reduced stromal collagen and hyaluronan that led to the reduction of solid stress and increased blood perfusion. This study suggested that ARB has a potential to remodel tumor microenvironment (Chauhan et al., 2013).
Together with the inhibition of specific signaling pathways such as Shh or CTGF, these inhibitors of PSC functions could become candidates for therapeutic applications that target tumor-stromal interaction. PSC functions that promote pancreatic cancer and possible therapeutic interventions are summarized in Figure 1.
Figure 1.
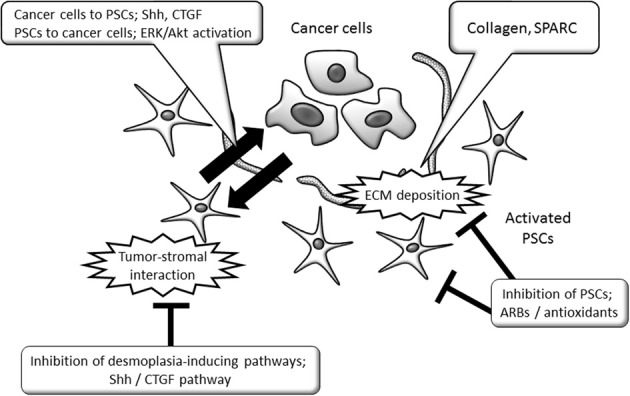
Schematic view of tumor-promoting PSC functions and PSC-targeting strategies in pancreatic cancer. The tumor-promoting interaction between cancer cells and PSCs could be therapeutic targets, by the inhibition of specific signaling pathways or PSC's functions. ARB, angiotensin II type 1 receptor blocker; CTGF, connective tissue growth factor; ECM, extracellular matrix; ERK, extracellular signal-regulated kinase; PSCs, Pancreatic stellate cells; Shh, sonic hedgehog; SPARC, Secreted protein acidic and rich in cysteine.
Modification of immune reaction against cancer cells
Interactions between pancreatic cancer cells and host immune cells play critical roles during the progression of pancreatic cancer. Recently, progress has been made in identifying the detailed mechanisms of immune suppression in pancreatic cancer. Infiltration of inflammatory cells is observed, in addition to desmoplasia, in pancreatic cancer tissue. These cells consist of immature myeloid cells that have immunosuppressive functions, known as MDSCs (Clark et al., 2007; Scarlett, 2013). These cells produce arginase, nitric oxide, and reactive oxygen species that suppress cytotoxic T-cell functions (Ostrand-Rosenberg and Sinha, 2009). Recently, several approaches have been devised to target MDSCs. The elimination of MDSCs by effector T cells targeting CD11b+Gr1+ MDSCs efficiently inhibited tumor growth (Zhang et al., 2008). Treatment with interleukin-12 was reported to attenuate the immunosuppressive effect of MDSCs by enhancing differentiation and decreasing nitric oxide synthase expression (Steding et al., 2011). Furthermore, IL-12 changed the function of MDSCs to enhance the effect of CD8+ T-cells, which could lead to tumor regression in the mouse model (Kerkar et al., 2011). Since MDSCs have immature nature, induction of differentiation using several reagents such as all-trans-retinoic acid or vitamin D3 was also tested (Lathers et al., 2004; Mirza et al., 2006).
Reinforcement of the host immune reaction by vaccination or dendritic cell therapy is an additional approach for modification of the immune reaction. Currently, survivin2B is targeted as a possible antigen in pancreatic cancer treatment, and immunological responses in patients have been reported (Kameshima et al., 2013). Another clinical trial targeting vascular endothelial growth factor receptor 2 by oral DNA vaccine is now in progress (Niethammer et al., 2012). As an immunotherapy, the combination of dendritic cell injection with gemcitabine administration was administered in mice that received subcutaneous implantation of pancreatic cancer cells. This treatment significantly delayed the growth of subcutaneous tumors by decreasing MDSCs within them (Ghansah et al., 2013). Previous attempts to develop cancer vaccines did not achieve favorable results, but novel targets and technical improvements might yield success. Since these therapeutic strategies targeting immune systems largely remain experimental, further examination and validation of their efficacy are required.
Conclusion
Tumor-stromal interactions contribute to the specific microenvironment of pancreatic cancer and hamper effective cancer cell elimination. The relationship between tumor-stromal interaction, therapy-resistance and future perspectives for stroma-targeting therapy is summarized in Figure 2. The novel anticancer agent nab-paclitaxel provided an additional therapeutic option in pancreatic cancer treatment, with proper inhibitory effects on the tumor stroma. Present strategies for depleting the pancreatic cancer stroma itself have revealed promising effects, though their clinical application has not yet been established. Specific inducers of desmoplasia and broad inhibition of PSC functions might be combined for effective therapy. Further study will enable novel therapeutic options targeting the critical mechanisms that maintain pancreatic cancer.
Figure 2.
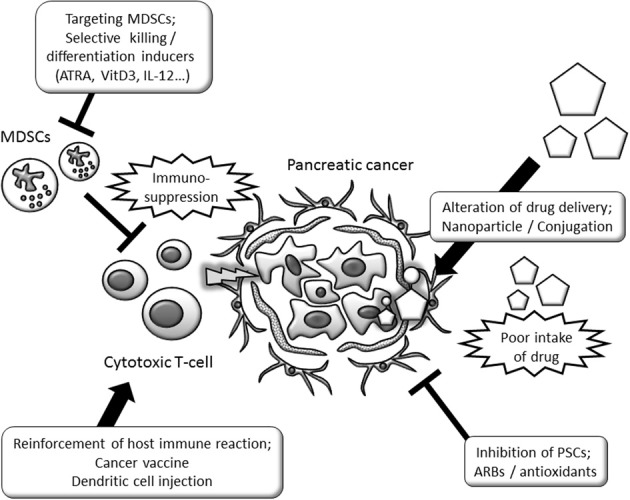
Schematic view of therapy-resistance related tumor-stromal interaction and possibility as a tumor stroma-targeting therapy. Stroma-targeting strategies include the modification of drug delivery, inhibition of PSC functions and restoration of immune functions. ARB, angiotensin II type 1 receptor blocker; ATRA, all-trans retinoic acid; IL, interleukin; MDSCs, myeloid-derived suppressor cells; PSCs, Pancreatic stellate cells; Shh, sonic hedgehog; SPARC, Secreted protein acidic and rich in cysteine.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by Grant-in-Aid from the Japan Society for the Promotion of Science (23591008, 24790674, and 23390194) and by the Research Committee of Intractable Pancreatic Diseases (Principal investigator: Tooru Shimosegawa) provided by the Ministry of Health, Labor and Welfare of Japan.
Glossary
Abbreviations
- ACEI
angiotensin I-converting enzyme inhibitor
- ARB
angiotensin II type 1 receptor blocker
- CSC
cancer-stem cell
- CTGF
connective tissue growth factor
- ECM
extracellular matrix
- ERK
extracellular signal-regulated kinase
- 5-FU
5-fluorouracil
- MDSCs
myeloid-derived suppressor cells
- PSCs
Pancreatic stellate cells
- Shh
sonic hedgehog
- SPARC
Secreted protein acidic and rich in cysteine.
References
- Abreu J. G., Ketpura N. I., Reversade B., De Robertis E. M. (2002). Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 4, 599–604 10.1038/ncb826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez R., Musteanu M., Garcia-Garcia E., Lopez-Casas P. P., Megias D., Guerra C., et al. (2013). Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 109, 926–933 10.1038/bjc.2013.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte M. V., Park S., Phillips P. A., Santucci N., Goldstein D., Kumar R. K., et al. (2004). Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 29, 179–187 10.1097/00006676-200410000-00002 [DOI] [PubMed] [Google Scholar]
- Bachem M. G., Schunemann M., Ramadani M., Siech M., Beger H., Buck A., et al. (2005). Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921 10.1053/j.gastro.2004.12.036 [DOI] [PubMed] [Google Scholar]
- Bailey J. M., Swanson B. J., Hamada T., Eggers J. P., Singh P. K., Caffery T., et al. (2008). Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 14, 5995–6004 10.1158/1078-0432.CCR-08-0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennewith K. L., Huang X., Ham C. M., Graves E. E., Erler J. T., Kambham N., et al. (2009). The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 69, 775–784 10.1158/0008-5472.CAN-08-0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin J. D., Catalano P., Thomas J. P., Kugler J. W., Haller D. G., Benson A. B., 3rd. (2002). Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J. Clin. Oncol. 20, 3270–3275 10.1200/JCO.2002.11.149 [DOI] [PubMed] [Google Scholar]
- Burris H. A., 3rd., Moore M. J., Andersen J., Green M. R., Rothenberg M. L., Modiano M. R., et al. (1997). Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15, 2403–2413 [DOI] [PubMed] [Google Scholar]
- Chauhan V. P., Martin J. D., Liu H., Lacorre D. A., Jain S. R., Kozin S. V., et al. (2013). Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 4, 2516 10.1038/ncomms3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C. E., Hingorani S. R., Mick R., Combs C., Tuveson D. A., Vonderheide R. H. (2007). Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 67, 9518–9527 10.1158/0008-5472.CAN-07-0175 [DOI] [PubMed] [Google Scholar]
- Egawa S., Toma H., Ohigashi H., Okusaka T., Nakao A., Hatori T., et al. (2012). Japan pancreatic cancer registry; 30th year anniversary: Japan pancreas society. Pancreas 41, 985–992 10.1097/MPA.0b013e318258055c [DOI] [PubMed] [Google Scholar]
- Erkan M., Adler G., Apte M. V., Bachem M. G., Buchholz M., Detlefsen S., et al. (2012). StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 61, 172–178 10.1136/gutjnl-2011-301220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan M., Kleeff J., Gorbachevski A., Reiser C., Mitkus T., Esposito I., et al. (2007). Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 132, 1447–1464 10.1053/j.gastro.2007.01.031 [DOI] [PubMed] [Google Scholar]
- Evans A., Costello E. (2012). The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 3:270 10.3389/fphys.2012.00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R., Brigstock D. R. (2006). A novel integrin alpha5beta1 binding domain in module 4 of connective tissue growth factor (CCN2/CTGF) promotes adhesion and migration of activated pancreatic stellate cells. Gut 55, 856–862 10.1136/gut.2005.079178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghansah T., Vohra N., Kinney K., Weber A., Kodumudi K., Springett G., et al. (2013). Dendritic cell immunotherapy combined with gemcitabine chemotherapy enhances survival in a murine model of pancreatic carcinoma. Cancer Immunol. Immunother. 62, 1083–1091 10.1007/s00262-013-1407-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman K. A., Hajj C. (2013). Role of radiation therapy in the management of pancreatic cancer. J. Surg. Oncol. 107, 86–96 10.1002/jso.23137 [DOI] [PubMed] [Google Scholar]
- Hamada S., Masamune A., Takikawa T., Suzuki N., Kikuta K., Hirota M., et al. (2012). Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 421, 349–354 10.1016/j.bbrc.2012.04.014 [DOI] [PubMed] [Google Scholar]
- Heinemann V., Quietzsch D., Gieseler F., Gonnermann M., Schonekas H., Rost A., et al. (2006). Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J. Clin. Oncol. 24, 3946–3952 10.1200/JCO.2005.05.1490 [DOI] [PubMed] [Google Scholar]
- Heinemann V., Reni M., Ychou M., Richel D. J., Macarulla T., Ducreux M. (2013). Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer Treat. Rev. [Epub ahead of print]. 10.1016/j.ctrv.2013.04.004 [DOI] [PubMed] [Google Scholar]
- Heng E. C., Huang Y., Black S. A., Jr., Trackman P. C. (2006). CCN2, connective tissue growth factor, stimulates collagen deposition by gingival fibroblasts via module 3 and alpha6- and beta1 integrins. J. Cell. Biochem. 98, 409–420 10.1002/jcb.20810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann R., Bodoky G., Ruhstaller T., Glimelius B., Bajetta E., Schuller J., et al. (2007). Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss group for clinical cancer research and the Central European cooperative oncology group. J. Clin. Oncol. 25, 2212–2217 10.1200/JCO.2006.09.0886 [DOI] [PubMed] [Google Scholar]
- Hidalgo M. (2010). Pancreatic cancer. N. Engl. J. Med. 362, 1605–1617 10.1056/NEJMra0901557 [DOI] [PubMed] [Google Scholar]
- Hingorani S. R., Wang L., Multani A. S., Combs C., Deramaudt T. B., Hruban R. H., et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 10.1016/j.ccr.2005.04.023 [DOI] [PubMed] [Google Scholar]
- Ibrahim N. K., Desai N., Legha S., Soon-Shiong P., Theriault R. L., Rivera E., et al. (2002). Phase I and pharmacokinetic study of ABI-007, a Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel. Clin. Cancer Res. 8, 1038–1044 [PubMed] [Google Scholar]
- Ijichi H., Chytil A., Gorska A. E., Aakre M. E., Bierie B., Tada M., et al. (2011). Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Invest. 121, 4106–4117 10.1172/JCI42754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobetz M. A., Chan D. S., Neesse A., Bapiro T. E., Cook N., Frese K. K., et al. (2013). Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120 10.1136/gutjnl-2012-302529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimeno A., Feldmann G., Suarez-Gauthier A., Rasheed Z., Solomon A., Zou G. M., et al. (2009). A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol. Cancer Ther. 8, 310–314 10.1158/1535-7163.MCT-08-0924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameshima H., Tsuruma T., Kutomi G., Shima H., Iwayama Y., Kimura Y., et al. (2013). Immunotherapeutic benefit of alpha-interferon (IFNalpha) in survivin2B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci. 104, 124–129 10.1111/cas.12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed H., Kleeff J., Osman T., Keleg S., Buchler M. W., Friess H. (2006). Hedgehog signaling in the normal and diseased pancreas. Pancreas 32, 119–129 10.1097/01.mpa.0000202937.55460.0c [DOI] [PubMed] [Google Scholar]
- Kerkar S. P., Goldszmid R. S., Muranski P., Chinnasamy D., Yu Z., Reger R. N., et al. (2011). IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J. Clin. Invest. 121, 4746–4757 10.1172/JCI58814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuta K., Masamune A., Watanabe T., Ariga H., Itoh H., Hamada S., et al. (2010). Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 403, 380–384 10.1016/j.bbrc.2010.11.040 [DOI] [PubMed] [Google Scholar]
- Kindler H. L., Niedzwiecki D., Hollis D., Sutherland S., Schrag D., Hurwitz H., et al. (2010). Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 28, 3617–3622 10.1200/JCO.2010.28.1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler H. L., Wroblewski K., Wallace J. A., Hall M. J., Locker G., Nattam S., et al. (2012). Gemcitabine plus sorafenib in patients with advanced pancreatic cancer: a phase II trial of the University of Chicago Phase II Consortium. Invest. New Drugs 30, 382–386 10.1007/s10637-010-9526-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathers D. M., Clark J. I., Achille N. J., Young M. R. (2004). Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol. Immunother. 53, 422–430 10.1007/s00262-003-0459-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantoni T. S., Schendel R. R., Rodel F., Niedobitek G., Al-Assar O., Masamune A., et al. (2008). Stromal SPARC expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol. Ther. 7, 1806–1815 10.4161/cbt.7.11.6846 [DOI] [PubMed] [Google Scholar]
- Masamune A., Hamada S., Kikuta K., Takikawa T., Miura S., Nakano E., et al. (2013). The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand. J. Gastroenterol. 48, 602–609 10.3109/00365521.2013.777776 [DOI] [PubMed] [Google Scholar]
- Masamune A., Kikuta K., Satoh M., Suzuki N., Shimosegawa T. (2005a). Green tea polyphenol epigallocatechin-3-gallate blocks PDGF-induced proliferation and migration of rat pancreatic stellate cells. World J. Gastroenterol. 11, 3368–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamune A., Satoh M., Kikuta K., Suzuki N., Satoh K., Shimosegawa T. (2005b). Ellagic acid blocks activation of pancreatic stellate cells. Biochem. Pharmacol. 70, 869–878 10.1016/j.bcp.2005.06.008 [DOI] [PubMed] [Google Scholar]
- Masamune A., Shimosegawa T. (2009). Signal transduction in pancreatic stellate cells. J. Gastroenterol. 44, 249–260 10.1007/s00535-009-0013-2 [DOI] [PubMed] [Google Scholar]
- Masamune A., Shimosegawa T. (2013). Pancreatic stellate cells–multi-functional cells in the pancreas. Pancreatology 13, 102–105 10.1016/j.pan.2012.12.058 [DOI] [PubMed] [Google Scholar]
- Masamune A., Suzuki N., Kikuta K., Satoh M., Satoh K., Shimosegawa T. (2006). Curcumin blocks activation of pancreatic stellate cells. J. Cell. Biochem. 97, 1080–1093 10.1002/jcb.20698 [DOI] [PubMed] [Google Scholar]
- Masamune A., Watanabe T., Kikuta K., Satoh K., Shimosegawa T. (2008). NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G99–G108 10.1152/ajpgi.00272.2007 [DOI] [PubMed] [Google Scholar]
- Michl P., Gress T. M. (2013). Current concepts and novel targets in advanced pancreatic cancer. Gut 62, 317–326 10.1136/gutjnl-2012-303588 [DOI] [PubMed] [Google Scholar]
- Mirza N., Fishman M., Fricke I., Dunn M., Neuger A. M., Frost T. J., et al. (2006). All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 66, 9299–9307 10.1158/0008-5472.CAN-06-1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y., Isayama H., Ijichi H., Sasaki T., Sasahira N., Hirano K., et al. (2010). Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br. J. Cancer 103, 1644–1648 10.1038/sj.bjc.6605955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neesse A., Frese K. K., Bapiro T. E., Nakagawa T., Sternlicht M. D., Seeley T. W., et al. (2013a). CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 12325–12330 10.1073/pnas.1300415110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neesse A., Frese K. K., Chan D. S., Bapiro T. E., Howat W. J., Richards F. M., et al. (2013b). SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut. [Epub ahead of print]. 10.1136/gutjnl-2013-305559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzillet C., Tijeras-Raballand A., Cros J., Faivre S., Hammel P., Raymond E. (2013). Stromal expression of SPARC in pancreatic adenocarcinoma. Cancer Metastasis Rev. [sEpub ahead of print]. 10.1007/s10555-013-9439-3 [DOI] [PubMed] [Google Scholar]
- Niethammer A. G., Lubenau H., Mikus G., Knebel P., Hohmann N., Leowardi C., et al. (2012). Double-blind, placebo-controlled first in human study to investigate an oral vaccine aimed to elicit an immune reaction against the VEGF-Receptor 2 in patients with stage IV and locally advanced pancreatic cancer. BMC Cancer 12:361 10.1186/1471-2407-12-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., McIntyre D., Honess D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 10.1126/science.1171362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson P., Hanahan D. (2009). Cancer. Breaching the cancer fortress. Science 324, 1400–1401 10.1126/science.1175940 [DOI] [PubMed] [Google Scholar]
- O'Reilly E. M., Niedzwiecki D., Hall M., Hollis D., Bekaii-Saab T., Pluard T., et al. (2010). A Cancer and Leukemia Group B phase II study of sunitinib malate in patients with previously treated metastatic pancreatic adenocarcinoma (CALGB 80603). Oncologist 15, 1310–1319 10.1634/theoncologist.2010-0152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S., Sinha P. (2009). Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182, 4499–4506 10.4049/jimmunol.0802740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T., Kudo M., Fukuta N., Nakatani T., Kimura M., Park A. M., et al. (2011). Involvement of angiotensin II and reactive oxygen species in pancreatic fibrosis. Pancreatology 11(Suppl. 2), 7–13 10.1159/000323478 [DOI] [PubMed] [Google Scholar]
- Scarlett C. J. (2013). Contribution of bone marrow derived cells to the pancreatic tumor microenvironment. Front. Physiol. 4:56 10.3389/fphys.2013.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J. Y., Kim C. S., Lee J. H., Jang S. J., Lee S. W., Hwang J. J., et al. (2013). Dual inhibition of MEK1/2 and EGFR synergistically induces caspase-3-dependent apoptosis in EGFR inhibitor-resistant lung cancer cells via BIM upregulation. Invest. New Drugs. [Epub ahead of print]. 10.1007/s10637-013-0030-0 [DOI] [PubMed] [Google Scholar]
- Stanton B. Z., Peng L. F. (2010). Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol. Biosyst. 6, 44–54 10.1039/b910196a [DOI] [PubMed] [Google Scholar]
- Steding C. E., Wu S. T., Zhang Y., Jeng M. H., Elzey B. D., Kao C. (2011). The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology 133, 221–238 10.1111/j.1365-2567.2011.03429.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana A., Tudur Smith C., Cunningham D., Starling N., Tait D., Neoptolemos J. P., et al. (2007). Systematic review, including meta-analyses, on the management of locally advanced pancreatic cancer using radiation/combined modality therapy. Br. J. Cancer 96, 1183–1190 10.1038/sj.bjc.6603719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. M., Ahn M. J., Choi Y. L., Ahn J. S., Park K. (2013). Clinical implications of T790M mutation in patients with acquired resistance to EGFR tyrosine kinase inhibitors. Lung Cancer. [Epub ahead of print]. 10.1016/j.lungcan.2013.08.023 [DOI] [PubMed] [Google Scholar]
- Takikawa T., Masamune A., Hamada S., Nakano E., Yoshida N., Shimosegawa T. (2013). miR-210 regulates the interaction between pancreatic cancer cells and stellate cells. Biochem. Biophys. Res. Commun. 437, 433–439 10.1016/j.bbrc.2013.06.097 [DOI] [PubMed] [Google Scholar]
- Von Hoff D. D., Ramanathan R. K., Borad M. J., Laheru D. A., Smith L. S., Wood T. E., et al. (2011). Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J. Clin. Oncol. 29, 4548–4554 10.1200/JCO.2011.36.5742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonlaufen A., Joshi S., Qu C., Phillips P. A., Xu Z., Parker N. R., et al. (2008). Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093 10.1158/0008-5472.CAN-07-2477 [DOI] [PubMed] [Google Scholar]
- Yachida S., Jones S., Bozic I., Antal T., Leary R., Fu B., et al. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 10.1038/nature09515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T., Kuno A., Masuda K., Ogawa K., Sogawa M., Nakamura S., et al. (2003). Candesartan, an angiotensin II receptor antagonist, suppresses pancreatic inflammation and fibrosis in rats. J. Pharmacol. Exp. Ther. 307, 17–23 10.1124/jpet.103.053322 [DOI] [PubMed] [Google Scholar]
- Zhang B., Zhang Y., Bowerman N. A., Schietinger A., Fu Y. X., Kranz D. M., et al. (2008). Equilibrium between host and cancer caused by effector T cells killing tumor stroma. Cancer Res. 68, 1563–1571 10.1158/0008-5472.CAN-07-5324 [DOI] [PubMed] [Google Scholar]