

Abstract
Targeted therapy with inhibitors of epidermal growth factor receptor (EGFR) has produced a noticeable benefit to non-small cell lung cancer (NSCLC) patients whose tumors carry activating mutations (e.g. L858R) in EGFR. Unfortunately, these patients develop drug resistance after treatment, due to acquired secondary gatekeeper mutations in EGFR (e.g., T790M). Given the critical role of SHP2 in growth factor receptor signaling, we sought to determine whether targeting SHP2 could have therapeutic value for EGFR inhibitor resistant NSCLC. We show that SHP2 is required for EGF-stimulated ERK1/2 phosphorylation and proliferation in EGFR inhibitor resistant NSCLC cell line H1975, which harbors the EGFR T790M/L858R double-mutant. We demonstrate that treatment of H1975 cells with II-B08, a specific SHP2 inhibitor, phenocopies the observed growth inhibition and reduced ERK1/2 activation seen in cells treated with SHP2 siRNA. Importantly, we also find that II-B08 exhibits marked anti-tumor activity in H1975 xenograft mice. Finally, we observe that combined inhibition of SHP2 and PI3K impairs both the ERK1/2 and PI3K/AKT signaling axes and produces significantly greater effects on repressing H1975 cell growth than inhibition of either protein individually. Collectively, these results suggest that targeting SHP2 may represent an effective strategy for treatment of EGFR inhibitor resistant NSCLCs.
Keywords: SHP2, EGFR, lung cancer, drug resistance, phosphatase inhibitor
1. Introduction
Protein tyrosine phosphorylation-mediated signal transduction, governed by the coordinated and balanced activities of protein tyrosine kinases and protein tyrosine phosphatases (PTPs), plays a key role in controlling cell growth, differentiation, and apoptosis [1,2]. Disruption of the process can lead to numerous human diseases including cancer. Consequently, cellular pathways regulated by tyrosine phosphorylation offer a wealth of molecular targets for drug discovery, with more than two dozens of small molecule kinase inhibitors already in the clinic [3]. However, despite the fact that PTPs have been garnering attention as potential new targets, they still remain largely underexplored [4,5]. Indeed, the PTPs are generally viewed antagonistic of the kinase-mediated processes, although this is not always the case. Several PTPs are shown to function primarily as signal promoting agents [6]. Thus, inhibition of a signal-promoting PTP may represent a novel therapeutic strategy for anti-cancer agents.
The Src homology 2 (SH2) domain-containing PTP2 (SHP2), encoded by the PTPN11 gene, is required for Ras/ERK1/2 pathway activation downstream of receptor tyrosine kinases, cytokine receptors, and integrins, and is a positive regulator of cell growth and survival [7,8]. Considerable evidence support an oncogenic role for SHP2. Germline mutations in SHP2 cause Noonan syndromes [9], whereas somatic SHP2 mutations occur in several types of hematologic malignancies, most notably juvenile myelomonocytic leukemia and, in solid tumors such as lung cancer, colon cancer, melanoma, neuroblastoma, and hepatocellular carcinoma [10–13]. In view of the observed activating SHP2 mutants in various kinds of leukemia and solid tumor, SHP2 makes an exciting target for mechanism-based anti-cancer therapy. Moreover, given the obligatory requirement of SHP2 in multiple oncogenic receptor tyrosine kinase pathways, inhibition of SHP2 may also prove effective for cancers with coactivation of receptor tyrosine kinases, which respond poorly to kinase inhibitor monotherapy [14].
Non-small cell lung cancer (NSCLC) is the leading cause of death from cancer. Traditional chemotherapy is only modestly effective, slightly prolonging survival among patients with advanced disease, but at the cost of significant adverse effects [15]. The tyrosine kinase epidermal growth factor receptor (EGFR) is overexpressed or mutated in a large percentage of NSCLCs and many other epithelial cancers [16]. Recent advances with targeted therapies with EGFR inhibitors have provided a noticeable benefit to subsets of NSCLC patients whose tumors harbor specific mutations in the gene encoding EGFR [17–19]. The most frequent single activation-loop mutation (L858R) increases tumor sensitivity to EGFR blockade with EGFR inhibitors gefitinib and erlotinib [20]. Unfortunately, after treatment with these inhibitors, patients with EGFR/L858R mutation eventually develop resistance to the drugs [21,22]. The emergence of the secondary gatekeeper mutation (T790M) in EGFR/L858R reduces NSCLC responsiveness to gefitinib and erlotinib and has been reported as a key factor of drug resistance.
Given the critical role of SHP2 in receptor tyrosine kinase signaling and the difficulty of targeting EGFR due to secondary gatekeeper mutations (e.g., T790M) that prevent EGFR inhibition, we sought to determine whether small molecule SHP2 inhibitors could be used as potential treatment for EGFR inhibitor resistant NSCLC. To this end, we have identified an indole salicylic acid based SHP2 inhibitor II-B08, which blocks EGFR stimulated ERK1/2 activation and inhibits GM-CSF induced growth of bone marrow cells bearing gain-of-function SHP2 mutations (D61Y and E76K) commonly found in patients with Juvenile myelomonocytic leukemia [23]. We further demonstrated that inhibition of SHP2 with II-B08 suppresses constitutive growth of myeloid cells bearing oncogenic KIT/D814V as well as primary bone marrow derived acute myelogenous leukemia blasts. Importantly, treatment of a mouse model bearing KIT/D814V-induced mast cell leukemia with II-B08 prolonged mouse survival and reduced splenomegaly and hepatomegaly [24]. In this study we investigated the effect of inhibiting SHP2 activity in an NSCLC cell line H1975, which expresses the T790M/L858R double-mutated EGFR, rendering the cells resistant to EGFR inhibitor gefitinib and erlotinib [25,26]. Our results suggest that targeting SHP2 may represent an effective strategy for treatment of EGFR inhibitor resistant NSCLCs.
2. Methods
2.1. Cell culture and reagents
Human non small cell lung carcinoma cell line H1975 was obtained from the American Tissue Culture Collection and grown at 37 °C in humidified 5% CO2 in RPMI1640 (Invitrogen) supplemented with 10% fetal bovine serum. EGF was obtained from Sigma and prepared according to the manufacture’s instruction. EGF was used to treat cells for various times. For immunoblotting, we used the following primary antibodies: rabbit polyclonal antibodies against pAKT (Ser473), AKT, phosphorylated extracellular signal-regulated kinase 1 & 2 (pERK1/2), ERK1/2; and β-actin (Cell Signaling). The PI3K inhibitor LY294002 was purchased from Calbiochem. SHP2 inhibitor II-B08 [23] was prepared using a new strategy optimized for large-scale synthesis provided as Supplementary data.
2.2. RNA interference studies
Small interfering RNA (siRNA) specific for SHP2 (5′-PCACGCAUGACGCCAUAUUCTT-3′) and scrambled siRNA (5′-PGCACGACCGCCUUAUAACUTT-3′) were synthesized by Dharmacon Research, Inc. SiRNAs were transfected into H1975 cells by using Lipofectamine 2000 reagent from Invitrogen Life Technologies using the protocol recommended by the manufacturer.
2.3. Cell proliferation assay
Cell proliferation was measured by MTT assay according to manufacturer’s specifications (Promega). Briefly, H1975 cells seeded in 96-well plates were treated with up to 100 μM II-B08 for 3 days. Subsequently, fresh media and MTT reagent were added to the wells, and the plates were incubated for 2–4 h at 37 °C to maximize signal-to-background. Wells containing only media were used for background correction. Each experiment was performed at least three times with each condition plated in three replicate wells.
2.4. Human NSCLC tumor xenograft experiments
Five-week old male NOD/SCID mice were housed under pathogen-free conditions. Xenograft experiments were performed in compliance with the relevant laws and guidelines set forth by the Institutional Laboratory Animal Care and Use Committee of Indiana University. H1975 cells (3 × 106) were injected into one flank of each mouse. Tumor size was measured with calipers every other day. Tumor volume (V) was determined by the equation V = (L × W2) × 0.5, in which L is the length and W is the width of the tumor. When xenografts reached volumes of ~80 mm3, the mice were randomly assigned to either control group (n = 5), or II-B08 treated group (n = 5). These groups received the following treatments daily by IP injection: solvent control (10% DMSO/PBS), or II-B08 (100 mg/kg). The experiment was terminated and the mice were sacrificed when the control tumors reached ~1,000 mm3 (on day 18). The tumors were removed, weighted and photographed.
2.5. Statistical analysis
Results are mean values ± standard error. Statistical analyses were performed using a Student t-test.
3. Results
3.1. Effect of SHP2 siRNA on H1975 cells proliferation and ERK1/2 activation
To determine whether SHP2 is required for H1975 cell proliferation, we knocked down SHP2 expression by siRNA interference. After transfection with SHP2 siRNA for 72 hours, SHP2 protein level in the SHP2 siRNA knockdown cells decreased by more than 80% compared with that in cells treated with a scrambled siRNA (Figure 1A). Decreased proliferation was observed for H1975 cells transfected with SHP2 siRNA compared to H1975 cells transfected with the scrambled siRNA (Figure 1B). Since SHP2 is required to transduce signals from receptor tyrosine kinases to downstream ERK1/2 pathway, we determined whether downregulation of SHP2 could block ERK1/2 activation. As shown in Figure 1C, knockdown of SHP2 significantly obliterated the EGF-induced ERK1/2 activation, whereas no change in phospho-AKT was observed as a result of SHP2 reduction.
Figure 1. Effects of SHP2 knockdown in H1975 cells.
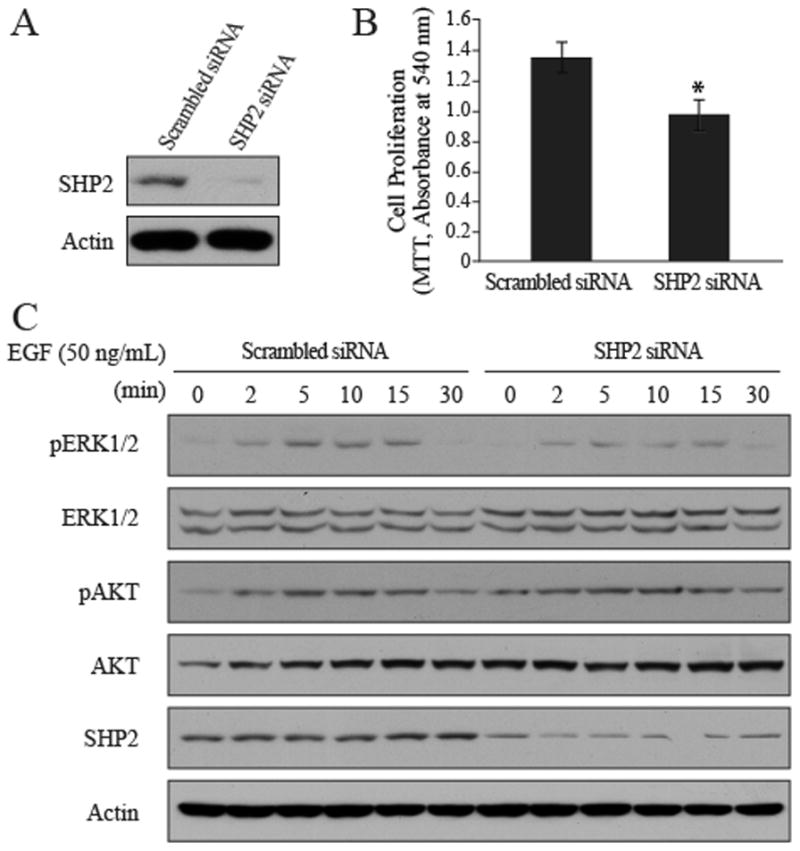
A. SHP2 siRNA decreased SHP2 protein level. B. SHP2 knockdown decreased H1975 cell proliferation. H1975 cells were transfected with SHP2 siRNA or scrambled siRNA for 72 hours in RPMI1640 with 0.5% FBS. Cell proliferation was measured by MTT after 72-hour transfection, and the results were expressed as mean ± SD (n=4), *P<0.05 relative to the scrambled siRNA. C. Effects of SHP2 knockdown on ERK1/2 and AKT phosphorylation. H1975 cells were transfected with SHP2 siRNA or scrambled siRNA for 72 hours in RPMI1640 with 10% FBS, and then serum starved in RPMI 1640 without FBS for another 6 hours. Cells were then treated with EGF (50 ng/mL) for various times, rinsed with cold PBS once and harvested for Western blotting.
3.2. SHP2 inhibitor II-B08 decreases H1975 cell proliferation and EGF-mediated ERK1/2 activation
We then investigated whether inhibition of SHP2 by small molecule inhibitor II-B08 [23] could also reduce H1975 cell proliferation. To this end, H1975 cells were treated with various concentrations of II-B08 for 3 days, and the cell number was measured. Similar to the siRNA interference experiment, II-B08 elicited a dose-dependent anti-proliferative response in H1975, with an IC50 value of 16 ± 3 μM (Figure 2A). To delineate the biochemical mechanism by which II-B08 exerts its growth inhibitory activity, we analyzed the effect of II-B08 on EGF-mediated ERK1/2 activation, which plays a critical role in cell proliferation. SHP2 phosphatase activity is known to be required for growth factor induced activation of the Ras-ERK1/2 cascade [7]. We found that treatment H1975 cells with 20 μM II-B08 significantly abrogated the EGF-stimulated ERK1/2 activation over a time period of 5 min to 30 min (Figure 2B). Again, II-B08 exhibited no effect on the EGF-induced AKT activation. To make certain that the cellular activity displayed by II-B08 was not due to off-target effects, we also evaluated II-B05, a structurally related but inactive analog of II-B08 [23]. As expected, II-B05 at 20 μM had no appreciable effect on the levels of ERK1/2 and AKT phosphorylation (Figure 2B).
Figure 2. SHP2 inhibitor II-B08 reduces ERK1/2 activation, cell proliferation and tumor growth.

A. II-B08 dose dependently inhibited H1975 proliferation in 0.5% FBS with an IC50 of 16 ± 3 μM. Cell proliferation was measured by MTT. B, II-B08 inhibits EGF-induced ERK1/2 activation. H1975 cells were cultured in 6-well plates (3×105 per well), serum-starved for 6 hours, and then treated with 20 μM II-B08 or II-B05 for 1 hr, followed by treatment with EGF (50 ng/mL) for various times. C. Suppression of tumor growth by SHP2 inhibitor II-B08. H1975 xenografts in NOD-SCID mice were treated with II-B08 (100 mg/kg, ip injection daily) or vehicle control (DMSO). Tumor size was measured every other day. Each data point represents the mean ± SD for 5 mice.
3.3. II-B08 blocks SHP2-dependent signaling
To provide additional evidence that the effect of II-B08 on intracellular signaling is SHP2 dependent, we evaluated the effect of II-B08 on EGF-mediated ERK1/2 activation after the level of SHP2 was downregulated by siRNA. As can be seen in Figure 3A, II-B08 could attenuate EGF-induced ERK1/2 activation in H1975 cells treated with scrambled siRNA, but the ability of II-B08 to inhibit EGF-mediated ERK1/2 activation was blunted when SHP2 was knocked down with siRNA. Again, as a negative control, II-B05 had no effect on ERK activation. To further establish that SHP2 inhibitors specifically block SHP2-dependent signaling, we analyzed the effect of II-B08 on ERK1/2 activation after stimulation of the cells by phorbol 12-myristate 13-acetate (PMA). The PMA-induced ERK1/2 activation has been shown to be independent of SHP2 [27] and instead depends on activation of protein kinase C and Raf [28] in a Ras-independent manner [29]. Thus, we predicted that SHP2 inhibitors would not inhibit PMA-induced ERK1/2 phosphorylation. As predicted, we found that II-B08 did not inhibit PMA-induced ERK1/2 phosphorylation (Figure 3B). Together, these experiments establish that II-B08 most likely affects only SHP2-mediated signaling events.
Figure 3. II-B08 blocks SHP2 dependent signaling in H1975 cells.
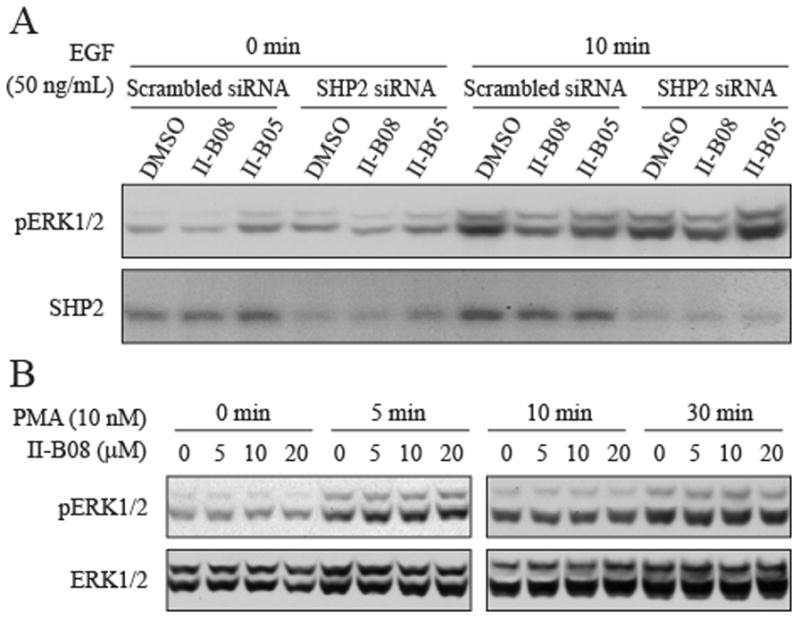
A. Inhibition of II-B08 on ERK1/2 phosphorylation was attenuated in H1975 cells transfected with SHP2 siRNA. B, II-B08 has no effect on PMA-stimulated ERK1/2 phosphorylation.
3.4. SHP2 inhibitor II-B08 suppresses H1975 xenograft tumor growth
Given the growth inhibitory activity and specificity for II-B08 in cell-based assays, we next evaluated the in vivo efficacy of II-B08 in a xenograft mouse model of NSCLC. H1975 cells (3×106) were injected into the right flank of NOD/SCID mice. All mice were monitored for tumor growth at the site of inoculation by palpation. Once tumors become palpable, tumor size was measured twice weekly using calipers. When tumor sizes reach ~80 mm3, the mice were randomized and treated with either vehicle control or II-B08 (100 mg/kg, ip once a day). We found that II-B08 treatment inhibited H1975 tumor growth by 25% compared with the vehicle control (Figure 2C).
3.5. SHP2 inhibitor II-B08 synergizes with PI3K inhibitor in suppressing H1975 cell growth
EGF can activate both the Ras-ERK1/2 and PI3K-AKT pathways, and both of which are important for EGF stimulated cell proliferation and survival. However, as shown above II-B08 treatment has no effect on AKT phosphorylation. Consequently, we hypothesized that targeting SHP2 in combination with PI3K may improve therapeutic efficacy over SHP2 inhibition alone. To test this hypothesis, we examined the effects of SHP2 and PI3K inhibition, individually and in combination, on EGF-mediated ERK1/2 and AKT activation, and cell proliferation. As shown in Figure 4A, inhibition of SHP2 by II-B08 attenuated EGF-induced ERK1/2 phosphorylation, whereas inhibition of PI3K with LY294002 abrogated EGF-induced AKT phosphorylation. Importantly, combined inhibition of SHP2 and PI3K blocked both ERK1/2 and AKT activation (Figure 4A) and produced significantly greater effects on H1975 cell proliferation than inhibition of either protein individually (Figure 4B). Thus, SHP2 inhibitor II-B08 can synergize with PI3K inhibitor LY294002 in repressing EGF-stimulated H1975 NSCLC proliferation.
Figure 4. SHP2 inhibitor II-B08 synergizes PI3K inhibitor LY294002 in repressing H1975 cell proliferation.

A. H1975 cells were treated with II-B08 (30 μM), LY294002 (20 μM), or both for 1 hour, then stimulated with 50 ng/mL EGF for 5 and 15 min. Cell lysates were collected for Western blotting. B. H1975 cells were treated with II-B08 (30 μM), LY294002 (20 μM), or both for 72 hour, followed by cell viability by the MTT assay. Data were expressed as mean ± SD (n=4), *P<0.05 relative to DMSO control, **P<0.01 relative to II-B08 or LY294002 single treatment.
4. Discussion
Cellular pathways regulated by tyrosine phosphorylation offer a rich source of drug targets for developing novel therapeutics. For example, deregulated tyrosyl phosphorylation, evoked by gain-of-function mutations and/or over-expression of tyrosine kinases, contributes to the pathogenesis of many cancers. The potential of such target-based therapeutic approaches has been well demonstrated by the successful treatment of a subpopulation of NSCLC patients carrying specific activating EGFR mutations with small molecule EGFR inhibitors [17–19]. Unfortunately, patients inevitably acquire secondary mutations in EGFR upon drug treatment, which interfere with binding of the inhibitors to the receptor, rendering the tumors drug-resistant [21,22]. Thus, it is vital to identify novel targets for NSCLC already resistant to EGFR inhibitors. Given the critical role of SHP2 in growth factor receptor (including EGFR) signaling, inhibitors of SHP2 are expected to have therapeutic value. In addition, as observed with tyrosine kinases, deregulation and gain-of-function mutations in SHP2 also contributes to the pathogenesis of a number of leukemias and cancers [7,8]. The goal of this study is to evaluate the potential utility of SHP2 inhibition in an EGFR inhibitor gefitinib and erlotinib resistant NSCLC cell line H1975, which carries the T790M/L858R double-mutated EGFR [25,26].
We showed that SHP2 knockdown inhibits EGF-stimulated ERK1/2 phosphorylation and proliferation in H1975 cells. We established that the small molecule SHP2 inhibitor II-B08 displays highly efficacious cellular activity and can specifically block the SHP2-dependent signaling. We then demonstrated that inhibition of SHP2 with II-B08 in H1975 cells phenocopies the observed growth inhibition and reduced ERK1/2 activation seen in cells treated with SHP2 siRNA. Importantly, we provided evidence that SHP2 inhibitor II-B08 exhibits significant anti-tumor activity in H1975 xenograft mice. Finally, we observed that combined inhibition of SHP2 and PI3K impairs activity of both the ERK1/2 and PI3K/AKT signaling axes and produces significantly greater effects on repressing H1975 cell growth than inhibition of either protein individually. This observation is similar to our previous finding that SHP2 inhibitor II-B08 synergizes with PI3K inhibitor (LY294002) in suppressing KIT/D814V induced mast cell leukemia [24]. Collectively, these results suggest that targeting SHP2 may represent an effective strategy for treatment of EGFR inhibitor resistant NSCLCs. Given that SHP2 is essential for the signaling capacity of multiple growth factor receptors and that activating mutations in SHP2 have been found in multiple types of leukemias and solid tumors, further development of SHP2 inhibitors should find broad applications in neoplastic disorders.
Supplementary Material
Highlights.
SHP2 is required for EGFR inhibitor resistant NSCLC H1975 cell proliferation.
SHP2 inhibitor blocks EGF-stimulated ERK1/2 activation and proliferation.
SHP2 inhibitor exhibits marked anti-tumor activity in H1975 xenograft mice.
SHP2 inhibitor synergizes with PI3K inhibitor in suppressing cell growth.
Targeting SHP2 represents a novel strategy for EGFR inhibitor resistant NSCLCs.
Acknowledgments
This work was supported in part by the National Institutes of Health Grant RO1CA152194.
Abbreviations
- SHP2
Src homology 2 (SH2)-domain containing protein tyrosine phosphatase-2
- ERK
extracellular signal-regulated protein kinase
- PTP
Protein Tyrosine Phosphatase
- EGFR
Epidermal Growth Factor Receptor
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://xxx.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P, Alessi DR. Kinase Drug Discovery - What’s Next in the Field? ACS Chem Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang ZY. Protein Tyrosine Phosphatases: Prospects for Therapeutics. Curr Opin Chem Biol. 2001;5:416–423. doi: 10.1016/s1367-5931(00)00223-4. [DOI] [PubMed] [Google Scholar]
- 5.Julien SG, Dubé N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 6.Jiang ZX, Zhang ZY. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer and Metastasis Reviews. 2008;27:263–272. doi: 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 8.Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci. 2009;100:1786–1793. doi: 10.1111/j.1349-7006.2009.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tartaglia M, Gelb BD. Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2005;6:45–68. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Arico M, Masera G, Basso G, Sorcini M, Gelb BD, Biondi A. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104:307–313. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 12.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, Richards WG, Du J, Girard L, Minna JD, Loh ML, Fisher DE, Velculescu VE, Vogelstein B, Meyerson M, Sellers WR, Neel BG. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 13.Miyamoto D, Miyamoto M, Takahashi A, Yomogita Y, Higashi H, Kondo S, Hatakeyama M. Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene. 2008;27:3508–3515. doi: 10.1038/sj.onc.1211019. [DOI] [PubMed] [Google Scholar]
- 14.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 15.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. Eastern Cooperative Oncology Group, Comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N Engl J Med. 2002;346:92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 16.Arteaga CL. ErbB-targeted therapeutic approaches in human cancer. Exp Cell Res. 2003;284:122–130. doi: 10.1016/s0014-4827(02)00104-0. [DOI] [PubMed] [Google Scholar]
- 17.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 18.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 19.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 22.Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, Paz-Ares L, Bover I, Garcia-Campelo R, Moreno MA, Catot S, Rolfo C, Reguart N, Palmero R, Sánchez JM, Bastus R, Mayo C, Bertran-Alamillo J, Molina MA, Sanchez JJ, Taron M. Spanish Lung Cancer Group, Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–967. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, Dong Y, Nabinger SC, Wu L, Gunawan AM, Wang L, Chan RJ, Zhang ZY. Salicylic acid-based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) J Med Chem. 2010;53:2482–2493. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mali RS, Ma P, Zeng LF, Martin H, Ramdas B, He Y, Sims E, Nabinger S, Ghosh J, Sharma N, Munugalavadla V, Chatterjee A, Li S, Sandusky G, Craig AW, Bunting KD, Feng GS, Chan RJ, Zhang ZY, Kapur R. Role of SHP2 phosphatase in KIT induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood. 2012;120:2669–2678. doi: 10.1182/blood-2011-08-375873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 26.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc Natl Acad Sci USA. 1995;92:664–668. doi: 10.1073/pnas.92.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marquardt B, Frith D, Stabel S. Signalling from TPA to MAP kinase requires protein kinase C, raf and MEK: Reconstitution of the signalling pathway in vitro. Oncogene. 1994;9:3213–3218. [PubMed] [Google Scholar]
- 29.Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent of Raf. J Biol Chem. 1996;271:23512–23519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.