

Abstract
Four and a half LIM protein 1 (FHL1) belongs to the Lin-1, Isl-1 and Mec-3 (LIM)-only protein family and plays important roles in muscle growth and carcinogenesis. However, the biological function of FHL1 remains largely unknown. Here, we show that FHL1 physically and functionally interacted with oestrogen receptors (ERs), which are involved in breast cancer development and progression. FHL1 bound specifically to the activation function-1 domain of ER. Physical interaction of FHL1 and ER is required for FHL1 repression of oestrogen-responsive gene transcription. FHL1 affected recruitment of ER to an oestrogen-responsive promoter and ER binding to an oestrogen-responsive element. Overexpression of FHL1 in breast cancer cells decreased expression of oestrogen-responsive proteins, whereas knockdown of endogenous FHL1 with FHL1 small interfering RNA increased the expression of these proteins. Further analysis of 46 breast cancer samples showed that FHL1 expression negatively associated with oestrogen-responsive gene expression in breast cancer cells. FHL1 inhibited anchorage-dependent and -independent breast cancer cell growth. These results suggest that FHL1 may play an important role in ER signalling as well as breast cancer cell growth regulation.
Keywords: FHL1, ER, interaction, transcriptional activity, breast cancer
Introduction
Four and a half LIM protein 1 (FHL1) belongs to the LIM-only protein family, characterized by four complete LIM domains, preceded by an N-terminal half LIM domain [1]. LIM domains are cysteine-rich zinc finger motifs mediating protein–protein interactions with transcription factors, cell-signalling molecules and cytoskeleton-associated proteins. FHL1 plays important roles in skeletal and cardiac muscle growth [2, 3]. Recently, FHL1 has been shown to play roles in carcinogenesis. FHL1 expression is down-regulated in various types of malignancies including breast cancer, gastric cancer, lung cancer, prostate cancer, ovary cancer, colon cancer, thyroid cancer, brain tumour, renal cancer, liver cancer and melanoma [4]. FHL1 is a tumour suppressor gene that acts downstream of Src and Cas to specifically block mouse cancer cell growth and migration. Most recently, we showed that FHL1 physically and functionally interacts with Smad2, Smad3 and Smad4, important regulators of cancer development and progression, and suppresses human hepatoma cell growth [5]. However, the detailed mechanism by which FHL1 exerts its tumour suppressive role is still poorly understood.
Oestrogens play an important role in the development and progression of breast cancer [6]. The biological effects of oestrogens are mediated by two forms of oestrogen receptors (ERs), ERα and ERβ, which belong to a large superfamily of nuclear receptors [7, 8]. ERα and ERβ exhibit similar structural and functional features, with N-terminal oestrogen-independent activation function domain (AF1), C-terminal oestrogen-dependent activation function domain (AF2) and centrally located DNA binding domain (DBD) [9]. ERs, bound to oestrogen-responsive elements (EREs) as dimers, can recruit co-activators and co-repressors to regulate oestrogen-responsive gene expression and breast cancer cell growth [10–15]. However, the intracellular signalling pathways regulating ER activity are not fully elucidated.
In this study, we report that FHL1 physically and functionally interacts with ERα and ERβ. FHL1 expression inversely associates with ER target gene expression both in breast cancer cell lines and in breast cancer patients. FHL1 inhibits anchorage-dependent and-independent breast cancer cell growth.
Materials and methods
Plasmids
The reporter construct ERE-Luc (oestrogen-responsive element-containing luciferase reporter) and expression vectors for ERα and ERβ have been described previously [16]. The FLAG-tagged FHL1 expression plasmid was cloned into a pcDNA3 vector linked with FLAG at the amino terminus by PCR using mammary cDNA library (Clontech, Mountain View, CA, USA) as the template. Plasmids encoding glutathione S-transferase (GST)-fusion proteins were prepared by amplification of each sequence by standard PCR methods, and the resulting fragments were cloned in frame into pGEX-KG (Amersham Pharmacia Biotech, Little Chalfont, UK) using appropriate sites. Deletion mutants of FHL1 were constructed by inserting PCR-generated fragments from the corresponding FHL1 cDNA into the pcDNA3-FLAG vector. All of the constructs were confirmed by DNA sequencing.
Yeast two-hybrid assay
The bait plasmid was generated by inserting PCR-amplified cDNA fragment encoding the AF1 domain (amino acids 1–145) of ERβ into pGBKT7 (Clontech). The Matchmaker two-hybrid system (Clontech) was used to isolate proteins that interacted with the ERβ bait protein. The bait plasmid and a human mammary gland cDNA library (Clontech) were sequentially transformed into AH109 yeast cells as previously described [17].
GST pull-down assay
The GST alone and GST fusion proteins were expressed in bacteria and purified according to the manufacturer’s protocols (Amersham Pharmacia). ERα or ERβ was translated in vitro in the TNT system (Promega, Madison, WI, USA). 35S-labelled ERα or ERβ was incubated with GST or GST fusion proteins bound to glutathione-Sepharose beads, and the adsorbed proteins were analysed as previously described [18].
Co-immunoprecipitation
Cells were transfected with indicated plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were harvested and lysed in lysis buffer. Co-immunoprecipitation was performed with anti-FLAG (Sigma-Aldrich, St. Louis, MO, USA) or anti-ERα (Santa Cruz Biotechnology, Delaware Avenue, CA, USA) as previously described [19].
Luciferase assay
Cells were seeded in 24-well plates containing phenol red-free DMEM medium (Invitrogen) supplemented with 10% charcoal-stripped fetal bovine serum (FBS) (Hyclone, Logan, UT, USA). Transfections were performed with Lipofectamine 2000 (Invitrogen). After treatment with 1 nM 17β-estradiol (E2), 1 nM propyl pyrazoletriol (PPT), 1 nM diaryl-propionitrile (DPN), 100 nM 4-hydroxytamoxifen (4-OHT) or 100 nM ICI 182,780 for 24 hrs, the cells were harvested. Cell extracts were analysed for luciferase and β-galactosidase activities as described previously [18].
SiRNA experiments
The cDNA target sequences of siRNAs for FHL1 were AAGGAGGTGCACTATAAGAAC and AATCTGGCCAACAAGCGCTT T, and were cloned into pSilencer2.1-U6 neo (Ambion, Austin, TX, USA), respectively. Co-transfection of the two vector based siRNAs into breast cancer cells was performed with Lipofectamine 2000 (Invitrogen).
Gel shift assay
The ERE (5′-AGCTCTTTGATCAGGTCACTGTGACCTGACTTT-3′) or mutant ERE (EREM; 5′-AGCTCTTTGATCAGTACACTGTGACCTGACTTT-3′) probes were labelled with Biotin 3′-End DNA Labeling kit (Pierce) as instructed by the manufacturer. Gel-shift assays were performed with LightShift Chemi-luminescent EMSA kits (Pierce, Rockford, ID, USA). Briefly, binding reactions containing 10 μg of nuclear extracts and 1 nmol of oligonucleotide were performed for 30 min. in binding buffer (2.5% glycerol, 0.05% Nonidet P-40, 50 mM KCl, 5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM Tris, pH 7.6 and 50 ng of poly(dI-dC)). Protein–nucleic acid complexes were resolved using a non-denaturating polyacrylamide gel consisting of 6% acrylamide, and transferred to a 100% nitrocellulose membrane with 0.45 μM pore size (Amersham Biosciences, Bath, UK). The membrane was incubated in blocking solution followed by incubation with streptavidin-peroxidase. After extensive washing, signal was detected with chemiluminescence solution.
Cell growth assays
Anchorage-dependent cell proliferation was analysed by crystal violet assay as described previously [17]. For anchorage-independent growth assay, cells (2 × 104) were seeded on 6-cm plates, with a bottom layer of 0.6% low-melting-temperature agar in DMEM and a top layer of 0.35% agar in DMEM. Colonies with greater than 100 mm diameter were scored after 5 weeks of growth.
Chromatin Immunoprecipitation (ChIP)
Breast cancer cells were cultured in phenol red-free medium for at least 3 days and treated with either ethanol (vehicle) or 10 nM E2 for 1 hr. ChIP assays were performed as described previously with minor modification [20]. Briefly, cells were cross-linked with 1% formaldehyde, pelleted and resuspended in lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, at pH 8.1 and protease inhibitors). Cells were sonicated, followed by centrifugation to remove insoluble material. Supernatants were collected and incubated overnight at 4°C with anti-ERα antibody or Normal IgG (Santa Cruz Biotechnology). Protein G-Sepharose beads (Santa Cruz Biotechnology) were then added and incubated for 1 hr at 4°C. The beads were washed, and precipitated chromatin complexes were then eluted with 100 ml of elution buffer (1% SDS, 0.1 M NaHCO3). Cross-linking was reversed by an overnight incubation at 65°C. DNA was purified using Qiaquick PCR purification kit (Qiagen, Hamburg, Germany). The following primers were used for ChIP PCR analysis: pS2 promoter sense, 5′-GGCCATCTCTCACTATGAATCACT-3′; pS2 promoter antisense, 5′-GGCAGGCTCTGTTTGCTTAAA-3′; pS2 upstream sense, 5′-TGATTCTCCTGACTTAACCTCC-3′; pS2 upstream antisense, 5′-CACGCTGTAATCCCAACACTTTG-3′.
Immunohistochemistry
Breast cancer samples and adjacent non-cancerous tissues were obtained from the Chinese PLA General Hospital with the informed consent of patients and with approval for experiments from the Chinese PLA General Hospital and Beijing Institute of Biotechnology. Immunohistochemistry was performed as described previously [19]. Rabbit anti-FHL1 (Proteintech, Chicago, IL, USA) was used as primary antibody.
Statistical analysis
Statistical significance in the luciferase activity and cell growth assays among constructs was determined by two-tailed Student’s t-test. The association of FHL1 expression with single clinical factor was assessed by Mann-Whitney U-test, Fisher’s exact test or Pearson chi-square test. Statistical calculations were performed with SPSS 13.0. P-values of less than 0.05 were considered statistically significant.
Results
Interaction of FHL1 with ERα and ERβin vitro and in vivo
To identify potential co-regulators that interact with ERβ, we screened a human mammary cDNA library using amino acids 1–145 of ERβ containing N-terminal AF1 domain as bait in the yeast two-hybrid system. FHL1 was identified as an ERβ-interacting protein. The specificity of this interaction was confirmed by a direct two-hybrid binding assay (Fig. 1A). Transformation of yeast cells with FHL1 in pACT2 vector alone or together with the GAL4 DNA-binding domain (DBD) in pAS2–1 vector or together with an unrelated protein, lamin C, fused to the GAL4 DBD instead of ERβ(1–145), did not activate the his (growth) and lacZ (β-gal) reporter genes, suggesting the specific interaction of FHL1 with ERβ.
Fig 1.
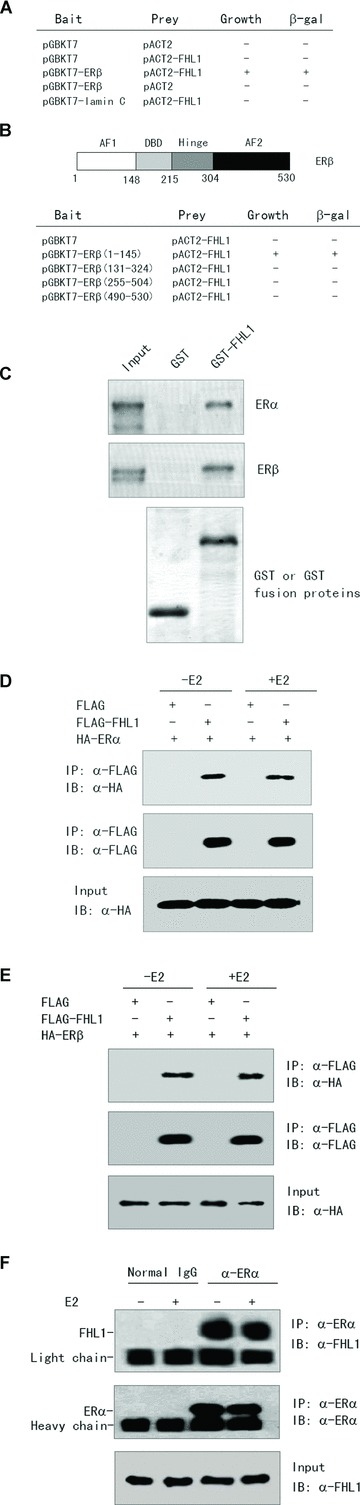
FHL1 interacts with ERα and ERβin vitro and in vivo. (A) AH109 yeast cells were transformed with different plasmids (bait and prey) and grown on SD/-Trp-Leu-His-Ade. +, grown within 96 hrs; −, no growth within 96 hrs. Positive colonies were tested for β-galactosidase (β-gal) activity. +, turned blue within 2 hrs; −, did not turn blue within 2 hrs. (B) Mapping of the FHL1 interaction region in ERβ. AH109 cells were transformed with indicated constructs and analysed as in (A). Also shown at top of the graph is a schematic diagram of the ERβ protein, illustrating the locations of various domains. (C) Glutathione-Sepharose beads bound with GST-FHL1 or with GST were incubated with 35S-labelled ERα or ERβ. After washing the beads, the bound proteins were subjected to SDS-PAGE followed by autoradiography. (D and E) FLAG-tagged FHL1 and HA-tagged ERα (D) or ERβ (E) were co-transfected into 293T cells. Cells were treated with or without 1 nM E2 for 2 hrs. Cell lysates were immunoprecipitated (IP) by anti-FLAG antibody, and the precipitates were immunoblotted with anti-HA antibody. (F) MCF7 cells, cultured in the absence of oestrogen for 3 days, were treated with 1 nM E2 for 2 hrs. Cell lysates were immunoprecipitated with either anti-ERα antibody or pre-immune control serum. The precipitates were analysed by immunoblot using anti-FHL1.
To examine whether FHL1 specifically interacts with amino acids 1–145 of ERβ, different ERβ mutants were made for yeast two-hybrid experiments. FHL1 did not interact with ERβ(131–324) containing the DBD and hinge region, ERβ(255–504) containing part of hinge region and entire AF2 domain, and ERβ(490–530) (Fig. 1B).
Since ERβ shares similarity with ERα, the possibility that FHL1 interacts with ERα was investigated using GST pull-down assay. As shown in Fig. 1C, GST-FHL1, but not GST, bound to in vitro translated ERα and ERβ proteins, with comparable binding affinity.
To investigate FHL1 and ER protein interaction in mammalian cells, co-immunoprecipitation assays were performed with human embryonic kidney 293T cells. FLAG-tagged FHL1 co-immunoprecipitated HA-tagged ERα and ERβ in 17b-estradiol (E2)-independent manner (Fig. 1D, E). The physiological interaction of FHL1 and ERα was confirmed by co-immunoprecipitation assays with human breast cancer MCF7 cells. Endogenous ERα co-precipitated with FHL1 in E2-independent manner (Fig. 1F). Taken together, these data suggest that FHL1 interacts with ERα and ERβin vitro and in vivo.
FHL1 modulates the transcriptional activity of ERα and ERbb
To determine whether the FHL1-ERα/ERβ interaction affects oestrogen-responsive gene transcription, ERα− and ERβ+ MCF7 cells were co-transfected with the oestrogen-responsive reporter, ERE-Luc, and FLAG-tagged FHL1 or FHL1 small interfering RNAs (siRNAs). Both in the presence and absence of E2, FHL1 overexpression decreased ERE-Luc reporter activity (Fig. 2A). With ERα-specific agonist, PPT, or ERβ-specific agonist, DPN, FHL1 overexpression also inhibited ERE-Luc reporter activity, suggesting that FHL1 regulates both ERα and ERβ transcriptional activity. FHL1 also inhibited ERE-Luc reporter activity in human breast cancer ZR75–1 cells (Fig. 2B). Transient co-expression of FHL1 and ER did not affect the protein levels of ERα and ERβ in MCF7 and ZR75–1 cells (data not shown), so the decrease of the transactivation of ERα and ERβ by FHL1 was not due to a modulation of the protein levels of ERα and ERβ. Consistent with the results of FHL1 overexpression, siRNA knockdown of endogenous FHL1 increased ERE-Luc reporter activity (Fig. 2C).
Fig 2.
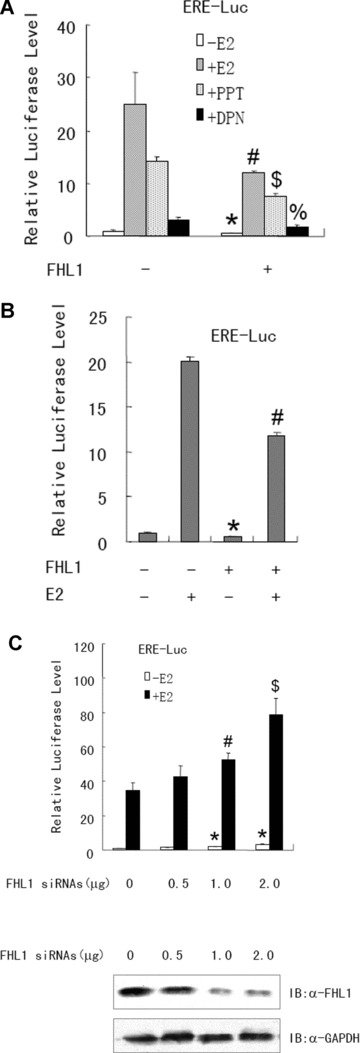
FHL1 regulates oestrogen-responsive reporter gene transcription. (A) MCF7 cells were co-transfected with 0.2 μg of ERE-Luc and 1.0 mg of the expression vector for FHL1. Cells were treated with or without 1 nM E2, 1 nM PPT and 1 nM DPN, and analysed for luciferase activity. Values are mean ± S.D. of triplicate measurements and have been repeated three times with similar results. *P < 0.05 versus empty vector without E2. #P < 0.05 versus empty vector with E2. $P < 0.01 versus empty vector with PPT. %P < 0.05 versus empty vector with DPN. (B) ZR75–1 cells were co-transfected with 0.2 mg of ERE-Luc and 1.0 mg of the expression vector for FHL1. Cells were then treated with 1 nM E2 for 24 hrs before the luciferase assay. Values are mean ± S.D. of triplicate measurements and have been repeated three times with similar results. *P < 0.05 versus empty vector without E2. #P < 0.01 versus empty vector with E2. (C) ZR75–1 cells were co-transfected with 0.2 mg of ERE-Luc and increasing amounts of FHL1 siRNAs as indicated. Cells were treated and analysed as in (A). The representative immunoblot with anti-FHL1 indicates specific knockdown of endogenous FHL1 by 0.5, 1.0, 2.0 mg of FHL1 siRNAs in the presence of E2 (lower panel). *P < 0.01 versus control siRNA without E2. #P < 0.05 versus control siRNA with E2. $P < 0.01 versus control siRNA with E2.
Interaction of FHL1 and ER is required for repression of oestrogen-responsive transcription
To investigate whether the interaction of FHL1 and ERα regulates oestrogen-responsive transcription, we used deletion analysis to map the interaction domains of ERα in co-immunoprecipitation experiments. Similar to the ERβ domain mapping results in the yeast two-hybrid, the ERα(1–185) fragment containing the AF1 domain bound specifically to FHL1. In contrast, the ERα(180–282) fragment containing the DBD domain, and the ERα(282–595) containing the AF2 domain, did not bind FHL1 (Fig. 3A). Next we examined which FHL1 protein region mediates interaction with ERα. Deletion of the C-terminal fourth LIM domain of FHL1 (FHL1[1–228]) had no effect on the binding to ERα and deletion of the N-terminal half LIM domain (FHL1[56–280]) reduced but did not abolish the ability of the FHL1 protein to associate with ERα (Fig. 3B). Larger deletions (FHL1[1–169] and FHL1[168–280]) abolished the interaction. These results suggest that the LIM domains 1, 2 and 3 of FHL1 are necessary for the interaction with ERα. Importantly, unlike FLAG-tagged wild-type FHL1 and FHL1(1–228) that bind ERα, FLAG-tagged FHL1(1–169), which failed to bind ERα, did not repress ERE-Luc reporter activity but increased the activity (Fig. 3C). Notably, FLAG-tagged FHL1, FHL1(1–228) and FHL1(1–169) were expressed at comparable levels (Fig. 3C).
Fig 3.
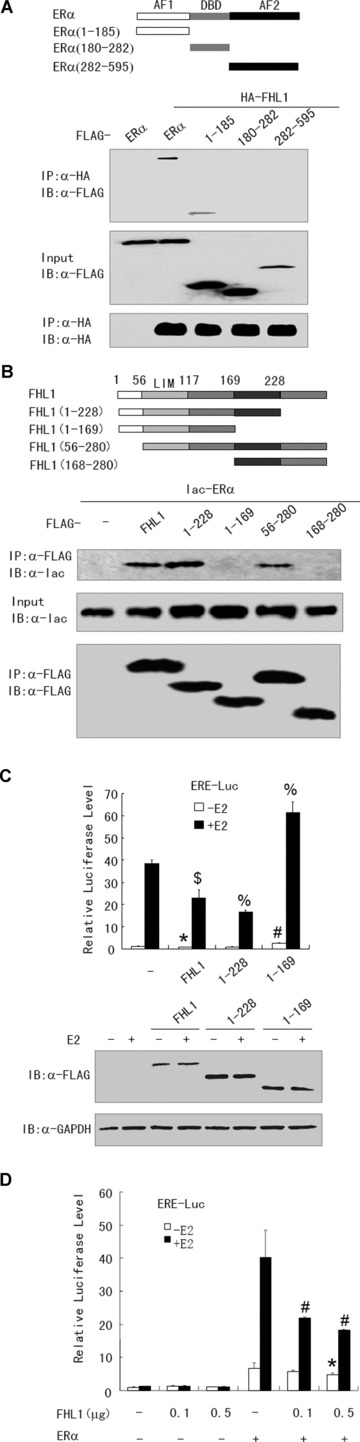
Interaction of FHL1 and ER is required for repression of oestrogen-responsive reporter activity. (A) Mapping of the FHL1 interaction region in ERα. HA-tagged FHL1 and FLAG-tagged full-length ERα or its deletion mutants were co-transfected into 293T cells in the presence of E2. Cell lysates were immunoprecipitated by anti-HA and the precipitates were then immunoblotted with anti-FLAG antibody. Also shown are schematic diagrams of the constructs used in this study. (B) Mapping of the ERα interaction region of FHL1. 293T cells were co-transfected with lac-tagged ERα and FLAG-tagged full-length FHL1 or its mutants in the presence of E2. Lysates from the transfected cells were immunoprecipitated with anti-FLAG antibody, and the immunoprecipitates were assayed with anti-lac antibody. Also shown are schematic diagrams of the constructs used in this study. (C) Luciferase assay with ZR75–1 cells co-transfected with 0.2 mg of ERE-Luc and 1.0 mg of FLAG-tagged FHL1, FHL1(1–228), or FHL1(1–169) as indicated (upper panel). Values are mean ± S.D. of triplicate measurements and have been repeated three times with similar results. Expression of FLAG-tagged FHL1, FHL1(1–228) and FHL1-(1–169) was detected by immunoblotting with anti-FLAG (lower panel). *P < 0.05 versus empty vector without E2. #P < 0.01 versus empty vector without E2. $P < 0.05 versus empty vector with E2. %P < 0.01 versus empty vector with E2. (D) Luciferase assay with SKBR3 cells co-transfected with ERE-Luc and increasing amounts of FHL1, in the absence and presence of ERα. Values are mean ± S.D. of triplicate measurements and have been repeated three times with similar results. *P < 0.05 versus ERα without E2. #P < 0.05 versus ERα with E2.
To further determine whether ERα is required for FHL1-mediated repression of ERE-Luc reporter activity, human breast cancer SKBR3 cells, which lack endogenous ERα were used in the ERE-Luc assay. Co-transfection of FHL1 and the ERE-Luc reporter into SKBR3 cells did not decrease ERE-Luc reporter transcription, whereas co-transfection of these genes with human ERα expression vector led to repression of the ERE-Luc (Fig. 3D). These data strongly suggest that FHL1 acts through ERα to decrease ERE-Luc reporter transcription.
FHL1 decreases the expression of endogenous oestrogen-responsive genes
To corroborate the results of the luciferase reporter assay, the effect of FHL1 on the expression of endogenous oestrogen-responsive genes was determined. The E2-deprived ZR75–1 cells transfected with either empty vector, FLAG-tagged wild-type FHL1 or FHL1(1–169) were treated with E2 and then harvested for immunoblotting. As expected, E2 stimulated the expression of pS2 and cathepsin D [21], two well-studied oestrogen-responsive genes (Fig. 4A). Importantly, overexpression of FHL1 decreased the expression of pS2 and cathepsin D with or without E2. Consistent with the results of the luciferase reporter assay, FLAG-tagged FHL1(1–169), which fails to bind ERα, did not inhibit the expression of pS2 and cathepsin D but increased that of pS2 and cathepsin D. Moreover, suppression of the endogenous expression of FHL1 by the specific FHL1 siRNAs markedly increased the expression of pS2 and cathepsin D in ZR75–1 cells (Fig. 4B). Similar results were obtained in MCF7 cells (Fig. 4C and D).
Fig 4.
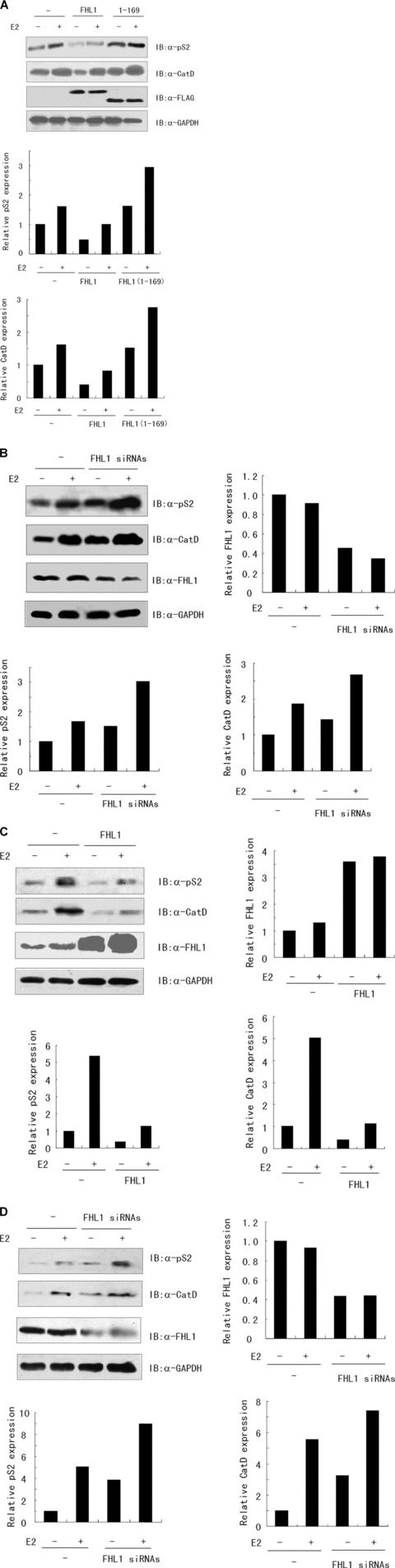
FHL1 regulates oestrogen-responsive gene expression. (A) ZR75–1 cells transfected with expression vector of FLAG-tagged FHL1 or FHL1(1–169) were treated with or without 1 nM E2 for 24 hrs. Whole cell lysate was used for western blot analysis of the expression of the FLAG-tagged proteins and oestrogen-responsive target genes, pS2 and cathepsin D. The densitometric quantitation of pS2 and cathepsin D bands normalized to GAPDH from a representative experiment is presented as a histogram. (B) ZR75–1 cells transfected with FHL1 siRNAs or control vector were treated as in (A). Whole cell lysate was used for western blot analysis with the indicated antibodies. The densitometric quantitation of FHL1, pS2 and cathepsin D bands normalized to GAPDH from a representative experiment is presented as a histogram. (C and D) MCF7 cells transfected with expression vector of FHL1 (C) or FHL1 siRNAs (D) were treated and analysed as in (B).
FHL1 regulates binding of ERα to oestrogen-responsive promoter
To investigate molecular mechanism by which FHL1 modulates ER transcriptional activity, the effect of FHL1 on ERα binding to ERE sequence was determined by gel shift assay. As expected, the biotin-labelled ERE, but not mutant ERE (EREM), bound to proteins from ER+ MCF7 nuclear extracts in the absence or presence of E2 (Fig. 5A and data not shown). The binding was specifically inhibited by a 100-fold molar excess of a cold ERE oligonucleotide. The addition of human anti-ERα antibody to the reaction caused a supershift, suggesting that ERα protein from MCF7 nuclear extracts specifically binds to ERE sequence. Importantly, overexpression of FHL1 abolished the binding of ERα to ERE (Fig. 5A). Opposite effects were observed with FHL1 siRNAs. Consistent with the results of the reporter gene transcription assays, the FHL1(1–228) mutant, which is as active as wild-type FHL1 in repression of ER transcriptional activity, also abolished ERα binding to ERE. Conversely, the FHL1(1–169) mutant, which increases ER transcriptional activity, enhanced the binding of ERα to ERE.
Fig 5.
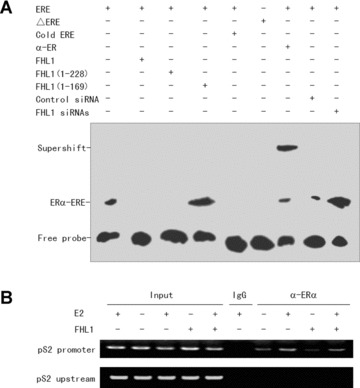
FHL1 affects binding of ERα to ERE sequence. (A) Gel shift assay was performed with biotin-labelled ERE probe and nuclear proteins extracted from MCF-7 cells transfected with FLAG-tagged FHL1, FHL1(1–169) or FHL1(1–228), or FHL1 siRNAs. For competition experiments, a 100-fold molar excess of unlabelled ERE was incubated with the labelled probe. The biotin-labelled mutant EREM probe was used as a negative control. Supershifts were performed with specific anti-ERα antibody. (B) Soluble chromatin was prepared from FLAG-tagged FHL1 expressed MCF7 cells treated with 10 nM E2 and subjected to immunoprecipitation with normal serum (IgG) or anti-ERα. Immunoprecipitated DNA was PCR-amplified with primers that annealed to the proximal region of the pS2 promoter or to the region approximately 2 kb upstream of the promoter.
To test whether FHL1 affects recruitment of ERα to an oestrogen-responsive promoter in vivo, chromatin immunoprecipitation (ChIP) experiments were performed for the pS2 promoter. As shown in Fig. 5B, ERα displayed a clear E2-stimulated recruitment to the pS2 promoter, but not a region approximately 2 kb upstream of the pS2 promoter. Consistent with the results of the gel shift assay, FHL1 overexpression decreased recruitment of ERα to the pS2 promoter (Fig. 5B). Taken together, these data suggest that FHL1 modulation of ER transcription activity may be through repression of ERα binding to the ERE sequence within oestrogen-responsive genes.
FHL1 inhibits breast cancer cell growth
To test the effect of FHL1 on breast cancer cell growth, the anchorage-dependent growth rate of ZR75–1 cells transfected with FHL1 or empty vector, or with FHL1 siRNAs or control siRNA, was examined. In the presence or absence of E2, ZR75–1 cells transfected with FHL1 grew more slowly than those with empty vector (Fig. 6A), whereas ZR75–1 cells transfected with FHL1 siRNAs grew faster than those with control siRNA (Fig. 6B). Similar results were observed in MCF7 cells (data not shown). In addition, in agreement with the results of the transcriptional experiments, the anti-oestrogens 4-OHT and ICI 182,780 further promoted the effect of FHL1 on breast cancer cell growth (Fig. 6C).
Fig 6.
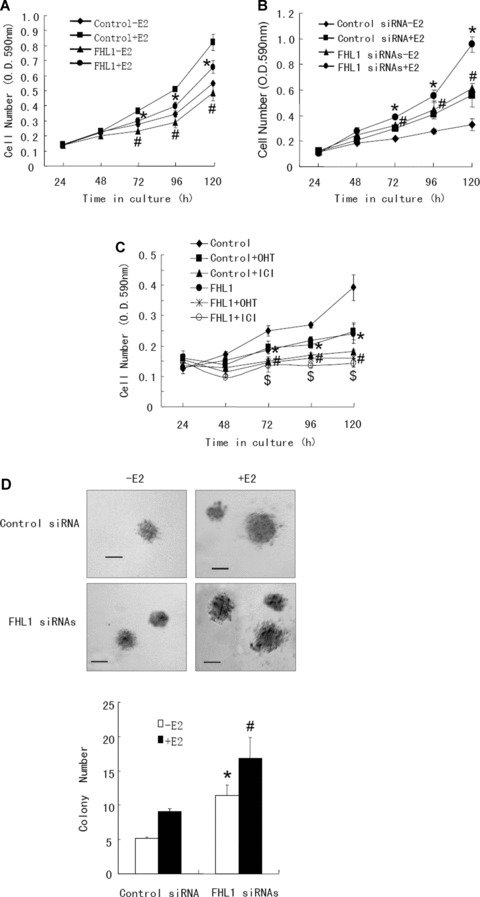
FHL1 inhibits breast cancer cell growth. (A and B) ZR75–1 cells transfected with expression vector of FHL1 (A) or FHL1 siRNAs (B) were treated with or without 1 nM E2 and harvested at the indicated times. Cell number was determined by crystal violet assay. Values shown are mean ± S.D. of triplicate measurements and have been repeated three times with similar results. #P < 0.05 versus empty vector or control siRNA without E2. *P < 0.05 versus empty vector or control siRNA with E2. (C) ZR75–1 cells transfected with expression vector for FHL1 were treated with 100 nM 4-OHT or 100 nM ICI 182,780 in regular medium at the indicated times. Cell number was measured by crystal violet assay. *P < 0.05 versus empty vector without 4-OHT or ICI 182,780. #P < 0.05 versus empty vector with 4-OHT. $P < 0.05 versus empty vector with ICI 182,780. (D) ZR75–1 cells transfected with FHL1 siRNAs or control siRNA were plated in soft agar and assayed for colony number after 5 weeks. Representative images show colonies in soft agar (upper panel). Scale bar: 100 μm. Values shown are mean ± S.D. of triplicate measurements (lower panel) and have been repeated three times with similar results. *P < 0.01 versus control siRNA without E2. #P < 0.01 versus control siRNA with E2.
We then detected the effect of FHL1 on anchorage-independent breast cancer cell growth. As shown in Fig. 6D, reduction of endogenous FHL1 with FHL1 siRNAs increased the anchorage-independent growth of ZR75–1 cells. Similar results were seen in MCF7 cells (data not shown). Taken together, these results indicate that FHL1 represses breast cancer cell growth.
FHL1 expression in breast cancer patients and its correlation with clinical factors
FHL1 has been shown to be down-regulated in 15 breast cancer patients [22]. We further detected the expression of FHL1 by immunohistochemistry in 46 pairs of human breast tumours and matched non-tumour breast tissues. Similar to the previous report, 91.3% (42/46) of non-tumour breasts expressed FHL1, while only 30.4% (14/46) of cancerous tissues stained positive for FHL1 (Fig. 7). Focusing on paired tumour and normal tissues, in 67.4% (31/46) of patients, the expression levels of FHL1 in tumours were lower than those in adjacent normal tissues; in 26.1% (12/46) of patients, normal tissue and breast cancer had similar staining patterns; and in only 6.5% (3/46) of patients, the staining scores in cancers were higher than that in normal tissues. The specificity of the staining was confirmed by immunohistochemistry using phosphate-buffered saline substituted for anti-FHL1 antibody or by immunofluorescence analysis of FHL1 overexpression or knockdown cells with anti-FHL1 (Fig. 7).
Fig 7.
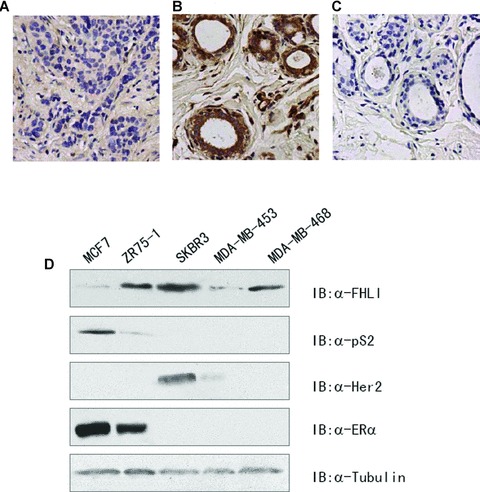
FHL1 expression in human breast cancer specimens and cell lines. (A–C) A case of breast carcinoma showed negative staining of cancerous cells (A) compared with positive staining of adjacent non-cancerous cells (B) using anti-FHL1. The same normal tissue was immunostained in the absence of the primary antibody (C). (A–C), original magnification, ×200. (D) Protein extracts from five breast cancer cell lines were analysed by Western blot analysis with the indicated antibodies. Tubulin was used as a loading control.
FHL1 expression did not correlate with age, tumour size, lymph node status, ER expression and progesterone receptor (PR) expression (P . 0.05) (Table 1), but positively correlated with the expression of epidermal growth factor 2 (Her2/n), a prognostic biomarker in breast cancer (P < 0.05). Interestingly, consistent with the results of the in vitro cultured breast cancer cells, FHL1 expression in breast cancer patients negatively associated with the expression of the oestrogen-responsive target gene pS2 (P < 0.05) (Table 1).
Table 1.
Correlations between FHL1 expression and clinical factors
| Clinical characteristics | FHL1−n= 32 | FHL1+n= 14 | P |
|---|---|---|---|
| Age | |||
| Mean ± S.D. | 53.0 ± 10.6 | 50.4 ± 15.8 | *P= 0.403 |
| Median | 53.5 | 50.5 | |
| Tumour size | |||
| <20 mm | 12 | 2 | †P= 0.169 |
| ≥20 mm | 20 | 12 | |
| Lymph node status | |||
| Node negative | 16 | 6 | ‡P= 0.655 |
| Node positive | 16 | 8 | |
| ER | |||
| Negative | 6 | 6 | †P= 0.143 |
| Positive | 26 | 8 | |
| PR | |||
| Negative | 4 | 4 | †P= 0.222 |
| Positive | 28 | 10 | |
| Her2/ν | |||
| Negative | 24 | 6 | †P= 0.048 |
| Positive | 8 | 8 | |
| pS2 | |||
| Negative | 4 | 6 | †P= 0.047 |
| Positive | 28 | 8 | |
Mann-Whitney U-test; †Fisher’s exact test; ‡Pearson chi-square test.
We next examined the expression of FHL1, pS2 and Her2 proteins in two ERα+ (MCF7 and ZR75–1) and three ERα– (SKBR3, MDA-MB-453 and MDA-MB-468) breast cancer cell lines (Fig. 7D). FHL1 was highly expressed in SKBR3, MDA-MB-468 and ZR75–1 cells, and low levels seemed to exist in MCF7 and MDA-MB-453 cells. pS2 proteins were detected in the ERα+ but not the ERα– breast cancer cell lines. As previously reported [23], Her2 was expressed in SKBR3 and MDA-MB-453 cells. Interestingly, FHL1 protein levels correlated with pS2 levels in the two ERα+ breast cancer cell lines, and SKBR3 cells, which expressed the highest level of FHL1, also expressed the highest level of Her2.
Discussion
Our present study establishes a new role for FHL1 as a negative regulator of ER-mediated transcription as well as breast cancer cell growth. FHL1 physically and functionally interacts with ER, and represses ER transcriptional activity as well as ER target protein expression in breast cancer cell lines and/or breast cancer patients, suggesting that FHL1 is an important co-repressor for ER. Most recently, FHL1 gene was found to be mutated in X-linked dominant scapuloperoneal myopathy and reducing body myopathy [24, 25]. It will be interesting to investigate whether FHL1 gene is mutated in breast cancer patients. FHL1 is down-regulated in the majority of breast cancer patients. Overexpression of FHL1 reduced both anchorage-dependent and anchorage-independent breast cancer cell growth. Therefore, FHL1 may be a useful target for breast cancer gene therapy.
ERα and ERβ have been reported to regulate distinct biological functions. Oestrogen via ERα induces cell proliferation and suppresses apoptosis, whereas ERβ opposes the proliferative effect of ERα in normal murine mammary epithelial cells [26]. When ERβ is co-transfected with ERα, it inhibits ERα transcriptional activity, indicating that ERβ is a negative regulator of ERα[27]. The facts that FHL1 inhibits both ERα and ERβ transcriptional activities and breast cancer cell growth suggest that the relative ratio of ERβ or FHL1 to ERα may be important factors responsible for breast cancer development and progression. Conceivably, cells with higher ERα levels and lower FHL1 or ERβ levels might readily develop breast cancer and have more malignant phenotype. Although many ERα-induced target genes [21], such as pS2 and cathepsin D, have been identified and well characterized, little is known for ERβ-induced downstream genes. Stossi et al. reported that, of 85 up-regulated genes, 52 were commonly regulated by ERα and ERβ in U2OS human osteosarcoma cells [28]. Thus, we also cannot exclude the possibility that FHL1 may modulate genes regulated commonly through ERα or ERβ. The detailed function of FHL1 on ERβ remains to be investigated.
To date, a number of co-repressors for ERα have been identified. The majority of reported ERα co-repressors, such as nuclear receptor corepressor (NCoR) [29], silencing mediator for retinoid and thyroid hormone receptors (SMRT) [30], receptor-interacting protein (RIP)140 [31] and breast cancer susceptibility gene (BRCA)1 [32, 33], bind to the AF2 domain of ERα. These co-repressors can decrease ERα transcriptional activity in a ligand-dependent manner. However, it has become clear that the ligand-independent AF-1 domain of ER is also important in regulation of oestrogen signalling. For instance, repressor of tamoxifen transcriptional activity (RTA) has been shown to interact with the AF-1 region of ERα and inhibit ERα transcriptional activity [34]. Like RTA, FHL1 binds to the AF-1 domain in the presence and absence of oestrogen, and FHL1 regulates ER transcriptional activity in a ligand-independent manner.
It has been reported that FHL family members play important roles in regulation of gene transcription. For instance, FHL2, FHL3 and activator of CREM in testis (ACT) bind and regulate the activity of multiple transcription factors, including androgen receptor (AR) [35], activator protein-1 (AP-1) [36], cyclic-AMP response element binding protein (CREB) [37], promyelocytic leukaemia zinc finger protein (PLZF) [38], serum response factor (SRF) [39] and Forkhead box class O protein 1 (FOXO1) [40]. Notably, FHL2 has been shown to interact with the AF1 domain of ERα, but have no effect on ERα-dependent transcriptional activity in African green monkey kidney COS-1 cells [41]. Like FHL2, FHL1 binds to the AF1 domain of ER, possibly due to 47.9% identical sequence between FHL1 and FHL2. However, our study showed that FHL1 can regulate ER transcriptional activity in breast cancer cells. It will be of interest to investigate whether FHL2 and other FHL family members can modulate oestrogen signalling in breast cancer cells.
Different co-repressors regulate steroid receptor activity through a variety of mechanisms, including chromatin remodelling, histone deacetylation, modulation of basal transcriptional apparatus, competition with coactivators, interference with DNA binding and ERα homodimerization, alteration of ERα stability, sequestration of ERα in the cytoplasm and effects on RNA processing. Most fully characterized are NCoR and SMRT, which function by recruiting different HDAC protein complexes. The fact that FHL1 affects ERα binding to ERE in vitro and in vivo and such function correlates with FHL1 repression of ERα transcriptional activity suggests that FHL1 regulates ERα transcriptional activity through interference with DNA binding. Since most co-repressors modulate the activity of nuclear receptor through more than one mechanism, it is possible that FHL1 has other ways to inhibit ERα transcriptional activity.
Acknowledgments
This work was supported by the Major State Basic Research Development Program (2007CB914603 and 2006CB943501), 863 Program (2007AA02Z113 and 2006BAI23B01), the National Key Technologies R&D Program for New Drugs (2009ZX09301–002), the National Natural Science Foundation (30530320, 30625035 and 30500091) and the Medicine and Health Research Foundation of PLA (06J021).
References
- 1.Johannessen M, Moller S, Hansen T, et al. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell Mol Life Sci. 2006;63:268–84. doi: 10.1007/s00018-005-5438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu PH, Ruiz-Lozano P, Zhou Q, et al. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech Dev. 2000;95:259–65. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 3.McGrath MJ, Mitchell CA, Coghill ID, et al. Skeletal muscle LIM protein 1 (SLIM1/FHL1) induces α5b 1-integrin-dependent myocyte elongation. Am J Physiol Cell Physiol. 2003;285:1513–26. doi: 10.1152/ajpcell.00207.2003. [DOI] [PubMed] [Google Scholar]
- 4.Shen Y, Jia Z, Nagele RG, et al. SRC uses Cas to suppress Fhl1 in order to promote nonanchored growth and migration of tumor cells. Cancer Res. 2006;66:1543–52. doi: 10.1158/0008-5472.CAN-05-3152. [DOI] [PubMed] [Google Scholar]
- 5.Ding LH, Wang ZY, Yan JH, et al. Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-β-like signaling pathway. J Clin Invest. 2009;119:349–61. doi: 10.1172/JCI35930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferguson AT, Davidson NE. Regulation of estrogen receptor a function in breast cancer. Crit Rev Oncog. 1997;8:29–46. doi: 10.1615/critrevoncog.v8.i1.20. [DOI] [PubMed] [Google Scholar]
- 7.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 8.McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–44. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- 9.Gustafsson JA. New insights in oestrogen receptor (ER) research-the ERβ. Eur J Cancer. 2000;36:S16. doi: 10.1016/s0959-8049(00)00206-9. [DOI] [PubMed] [Google Scholar]
- 10.Cowley SM, Hoare S, Mosselman S, et al. Estrogen receptors α and β form heterodimers on DNA. J Biol Chem. 1997;272:19858–62. doi: 10.1074/jbc.272.32.19858. [DOI] [PubMed] [Google Scholar]
- 11.Kuiper GG, Carlsson B, Grandien K, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. 1997;138:863–70. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 12.Pace P, Taylor J, Suntharalingam S, et al. Human estrogen receptor β binds DNA in a manner similar to and dimerizes with estrogen receptor α. J Biol Chem. 1997;272:25832–8. doi: 10.1074/jbc.272.41.25832. [DOI] [PubMed] [Google Scholar]
- 13.Kuiper GG, Lemmen JG, Carlsson B, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology. 1998;139:4252–63. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 14.Tremblay A, Tremblay GB, Labrie F, et al. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol Cell. 1999;3:513–9. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9:140–7. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 16.Ding LH, Yan JH, Zhu JH, et al. Ligand-independent activation of estrogen receptor α by XBP-1. Nucleic Acids Res. 2003;31:5266–74. doi: 10.1093/nar/gkg731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XH, Yang ZH, Zhang H, et al. The estrogen receptor-interacting protein HPIP increases estrogen-responsive gene expression through activation of MAPK and AKT. Biochim Biophys Acta. 2008;1783:1220–8. doi: 10.1016/j.bbamcr.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 18.Han J, Ding L, Yuan B, et al. Hepatitis B virus X protein and the estrogen receptor variant lacking exon 5 inhibit estrogen receptor signaling in hepatoma cells. Nucleic Acids Res. 2006;34:3095–106. doi: 10.1093/nar/gkl389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Xie XY, Zhu XD, et al. Stimulatory cross-talk between NFAT3 and estrogen receptor in breast cancer cells. J Biol Chem. 2005;280:43188–97. doi: 10.1074/jbc.M506598200. [DOI] [PubMed] [Google Scholar]
- 20.Aiyar SE, Sun JL, Blair AL, et al. Attenuation of estrogen receptor α-mediated transcription through estrogen-stimulated recruitment of a negative elongation factor. Genes Dev. 2004;18:2134–46. doi: 10.1101/gad.1214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–19. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Jia Z, Shen Y, et al. Coordinate suppression of Sdpr and Fhl1 expression in tumors of the breast, kidney, and prostate. Cancer Sci. 2008;99:1326–33. doi: 10.1111/j.1349-7006.2008.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meric F, Lee WP, Sahin A, et al. Expression profile of tyronsine kinases in breast cancer. Clin Cancer Res. 2002;8:361–67. [PubMed] [Google Scholar]
- 24.Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–13. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–12. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helguero LA, Faulds MH, Gustafsson JA, et al. Estrogen receptors α (ERα) and β (ERβ) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24:6605–16. doi: 10.1038/sj.onc.1208807. [DOI] [PubMed] [Google Scholar]
- 27.Omoto Y, Eguchi H, Yamamoto-Yamaguchi Y, et al. Estrogen receptor (ER) β1 and ERbcx/β2 inhibit ERα function differently in breast cancer cell line MCF7. Oncogene. 2003;22:5011–20. doi: 10.1038/sj.onc.1206787. [DOI] [PubMed] [Google Scholar]
- 28.Stossi F, Barnett DH, Frasor J, et al. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) α or ERβ in human osteosarcoma cells: distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–86. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- 29.Lavinsky RM, Jepsen K, Heinzel T, et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci USA. 1998;95:2920–5. doi: 10.1073/pnas.95.6.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–49. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 31.Cavailles V, Dauvois S, Lopez G, et al. Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J. 1995;14:3741–51. doi: 10.1002/j.1460-2075.1995.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan S, Wang J, Yuan R, et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284:1354–6. doi: 10.1126/science.284.5418.1354. [DOI] [PubMed] [Google Scholar]
- 33.Fan S, Ma YX, Wang C, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20:77–87. doi: 10.1038/sj.onc.1204073. [DOI] [PubMed] [Google Scholar]
- 34.Norris JD, Fan D, Sherk A, et al. A negative coregulator for the human ER. Mol Endocrinol. 2002;16:459–68. doi: 10.1210/mend.16.3.0787. [DOI] [PubMed] [Google Scholar]
- 35.Muller JM, Isele U, Metzger E, et al. FHL2, a novel tissue-specific coactivator of the androgen receptor. EMBO J. 2000;19:359–69. doi: 10.1093/emboj/19.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morlon A, Sassone-Corsi P. The LIM-only protein FHL2 is a serum-inducible transcriptional coactivator of AP-1. Proc Natl Acad Sci USA. 2003;100:3977–82. doi: 10.1073/pnas.0735923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fimia GM, De CD, Sassone-Corsi P. A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol. 2000;20:8613–22. doi: 10.1128/mcb.20.22.8613-8622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLoughlin P, Ehler E, Carlile G, et al. The LIM-only protein DRAL/FHL2 interacts with and is a corepressor for the promyelocytic leukemia zinc finger protein. J Biol Chem. 2002;277:37045–53. doi: 10.1074/jbc.M203336200. [DOI] [PubMed] [Google Scholar]
- 39.Philippar U, Schratt G, Dieterich C, et al. The SRF target gene Fhl2 antagonizes RhoA/MAL-dependent activation of SRF. Mol Cell. 2004;16:867–80. doi: 10.1016/j.molcel.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Hou H, Haller EM, et al. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–32. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobayashi S, Shibata H, Yokota K, et al. FHL2, UBC9, and PIAS1 are novel estrogen receptor α-interacting proteins. Endocr Res. 2004;30:617–21. doi: 10.1081/erc-200043789. [DOI] [PubMed] [Google Scholar]