

Abstract
Abnormal activation of mitochondrial translocator protein (TSPO) contributes to arrhythmogenesis during cardiac metabolic compromise; however, its role in the antiarrhythmic activities of chronic hypoxia adaptation remains unclear. Our results demonstrated that 80% of normoxic rats developed ischaemic VF, whereas this condition was seldom observed in rats with 14 days of chronic intermittent hypobaric hypoxia (CIHH). TSPO stimulation or inhibition affected the arrhythmias incidence in normoxic rats, but did not change the CIHH-mediated antiarrhythmic effects. Abrupt and excessive elevation of TSPO activity was positively linked to ischaemic VF, and CIHH preserved TSPO activity during ischaemia. The preservation of TSPO activity by CIHH also contributed to the maintenance of intracellular Ca homeostasis. These results suggest that the blunt sensitivity of TSPO to ischaemic stress may be responsible for the antiarrhythmic effects by CIHH.
Keywords: mitochondria, ventricular fibrillation, hypoxia
Introduction
Ventricular fibrillation (VF) is a fatal arrhythmia, and coronary artery disease (CAD) is the most common cause for VF [1]. Fewer CAD incidents have been best documented in sojourners at high altitude, which evokes the investigation of chronic and/or intermittent hypoxia-mediated responses of human and animal hearts [2]. There is impressive evidence that chronic or intermittent hypoxia protects the heart against ischaemia/reperfusion injury and arrhythmias [3, 4]. However, the beneficial cardiac effects depend on the training protocols used, such as hypoxic duration and degree, and the mechanisms remain multiplex.
Mitochondrial dysfunction is increasingly recognized as an important mechanism for cardiac diseases [5]. Mitochondrial translocator protein (TSPO, also known as peripheral benzodiazepine receptor) locates at the contact site of the mitochondrial inner and outer membrane and is ubiquitously expressed [6]. It has been implicated for the role of TSPO in mitochondrial functions, especially in the regulation of cholesterol transport into mitochondria [6, 7]. At cellular levels, the TSPO expression was correlated with the resistance of the cell to oxidative stress [8]. In brain, kidney and heart, TSPO levels were also found to be increased in a variety of acute stresses, indicating an important role of TSPO in stress response [6]. A recent report and our previous study illustrated that its activation during myocardial ischaemia contributed to arrhythmogensis [9, 10].
In this study, it has been suggested that chronic intermittent hypobaric hypoxia (CIHH) training might exert an influence over myocardial TSPO, thereby preventing the heart from fatal arrhythmias.
Materials and methods
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and the policy of Animal Care and Use Committee of the Tongji University.
Chronic intermittent hypobaric hypoxia model
Adult male Sprague-Dawley (SD) rats (aged 10 weeks, weighing 220–250 g) were separated into two groups. One group (CIHH rats) was exposed to a simulated altitude of 4500–5000 m (barometric pressure 360–380 mmHg) in a well-ventilated, temperature-controlled hypobaric chamber for 14 days for 4 hrs each day (14:00 p.m. ∼18:00 p.m.). Upon completing 4 hrs of CIHH, rats were kept in normoxic environments. The control group (normoxic rats) was kept in the same room, but not in the hypobaric chamber, with the same 12–12 hrs light–dark cycle. Free access to a standard rat diet and water was allowed throughout the experimental period.
In vitro global ischaemic VF model
Animals were anaesthetized with pentobarbital sodium (60 mg/kg, i.p.) and heparinized with sodium heparin (250 IU, i.p.). Hearts were quickly excised and placed in ice-cold normoxic Krebs–Henseleit (KH) solution containing NaCl 118 mM/l, KCl 4.7 mM/l, MgSO4 1.2 mM/l, CaCl2 1.25 mM/l, KH2PO4 1.2 mM/l, NaHCO3 25 mM/l, glucose 11 mM/l (pH 7.4 adjusted with gas 95% O2 and 5% CO2). After washing out the blood in the chambers of the heart, the heart was connected to the Langendorff setup and perfused with standard KH solution. Low-flow ischaemia was induced by reducing the perfusion pressure from 80 to 15 mmHg or by constant low-flow leading to a reduction of coronary flow of ≍ 90%[10, 11]. VF was induced by one basic cycle length pacing (8 Hz, cycle length 67 msec, duration 30 sec) at the end of 24 min low-flow via a pair of platinum pacemaker wires implanted in the right ventricular free wall and connected to a pulse generator (AD instruments, Australia). Electrocardiographic electrodes were attached to the aorta and the apex of the heart, and connected to the chart recorder.
Experimental groups
Hearts were randomly assigned to one of six groups: (1) and (2): Control (normoxic) and CIHH rat hearts, respectively, were subjected to an ischaemic VF inducing protocol. (3)–(6): Control and CIHH rat hearts were treated with FGIN-1–27 (N,N-dihexyl-2-(4-fluorophenyl)indole-3-acetamide, 4.6 μM, TSPO agonist; Sigma-Aldrich, St. Louis, MO, USA) and 4’-Cl-DZP (4-chlorodiazepam, 16 μM, TSPO antagonist; Sigma-Aldrich), respectively.
In these experiments, none of the concentrations used altered intrinsic electrophysiological properties, including ventricular action potential duration during normal perfusion (data not shown).
Mitochondria isolation and TSPO activity assay
Mitochondria were isolated from the hearts that underwent the VF-inducing protocol. Briefly, hearts were removed into an ice-cold isolation buffer containing sucrose, 50 mM; mannitol, 200 mM; KH2PO4, 5 mM; EGTA, 1 mM and Mops, 5 mM (pH 7.3 adjusted with KOH), with 0.2% bovine serum albumin (BSA). Ventricles were cut into 1 mm3 pieces. Two 10 sec-long homogenization cycles were performed with a PT 10/35 Polytron (Brinkman, Westbury, NY, USA), followed by a 10 min centrifugation at 750 ×g (Sorvall II centrifuge equipped with a GSA rotor). The supernatant was stored on ice, and the pellet resuspended in isolation buffer, and homogenized using a Potter–Elvehjem tissue grinder with a Teflon pestle to release the remaining mitochondria. The homogenate was then centrifuged at 750 ×g, and the supernatant combined with that obtained from the previous step and further centrifuged at 7000 ×g for 15 min. The obtained pellet was suspended in 40 ml of isolation buffer containing no EGTA or BSA, and sedimented at 7000 ×g for 15 min. Finally, the mitochondrial fraction, resuspended in EGTA- and BSA-free isolation buffer at 20–30 mg protein/ml, was kept on ice prior to experiments. Mitochondrial protein concentration was determined using the BCA protein assay method. TSPO-binding experiments were performed as previously described, with minor variations [12].
Single ventricular cardiomyocyte isolation and measurement of [Ca2+]i
Single cells were enzymatically isolated from adult (250–300 g) SD rat left ventricles. Each rat was anaesthetized with sodium pentobarbital. The heart was rapidly removed, mounted on a Langendorff apparatus, and perfused at 37°C with the following sequence of solutions: Ca2+-free (control, no added calcium) Tyrode solution for 5 min to remove the blood, low-Ca2+ (0.1 mM) solution containing 1 mg/ml type 2 collagenase (Worthington, Lakewood, NJ, USA) and 0.1 mg ml21 type XIV protease (Sigma-Aldrich) for 20 min, and enzyme-free low-Ca2+ solution for 5 min. The left ventricle was then minced and shaken for 10 min in the low-Ca2+ solution. Myocytes were stored at room temperature in the control solution with 0.5 mM Ca2+. All experiments were performed within 2–8 hrs after isolation.
Enzymatically isolated ventricular cardiomyocytes of normoxic and CIHH rats were loaded with a calcium indicator (Fura-2 AM, 1 μsM or Fluo-3 AM, 10 μM; Fluka, USA) for about 30 min at room temperature and then washed three times with standard Tyrode’s solution. Cells were alternately excited at 340/380 nm and the fluorescence emission was collected through a 510 ± 10 nm bandpass filter (T.I.L.L. Photonics GmbH, Germany). The F340/F380 fluorescence ratio values were used for the evaluation of [Ca21]i.
Calcium kinetics, such as transient amplitude and decay rate, were evaluated by F488/F520 fluorescence ratio values (Bio-radiance 2000, Nikon, Japan). Data was analyzed by Image-pro plus 6.0 software (Media Cybernetics, MD, USA).
Quantification of mRNA expression
RNA was extracted and reverse transcribed as previously described [13, 14]. A real-time polymerase chain reaction (PCR) procedure with SYBR Green, which was validated with respect to reproducibility and linearity within the measuring range, was performed in quadruplicate with the Stratagene 3500 (Stratagene, USA). To correct for potential variances among samples in mRNA extraction or in the efficiency of reverse transcription, the mRNA content of each gene was normalized to the expression of the stably expressed reference gene, β-actin, in the same sample. cDNA sequences were obtained from the public GenBank sequence database of the National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov), and primers were designed with the Primer software (http://fokker.wi.mit.edu/primer3/input.htm). Sequences for all PCR primers are shown as follows: forward 5’-TGT, CAC, CAA, CTG, GGA, CGA, TA-3’ and reverse 5’-AGG, AAG, GAA, GGC, TGG, AAG, AG-3’ for β-actin; forward 5’-CTT, TGG, TGG, ACC, TCA, TGC, TT-3’, and reverse 5’-TTC, CTG, ATG, GCT, AGG, TGT, CC-3’ for TSPO; forward 5’-TGC, TGG, AAC, TTG, TGA, TCG, AG-3’, and reverse 5’-ATC, CGC, TGC, ACA, CTC, TTT, CT-3’ for Ca-ATPase (SERCA2a); forward 5’-CTG, CCA, GAT, GCT, GGT, GTC, TA-3’, and reverse 5’-GTT, TTG, CTG, GAT, CCA, CTG, GT-3’ for ryanodine receptor (RyR); forward 5’-GTT, CCA, AGA, GGC, AGC, TGA, AC-3’, and reverse 5’-GAC, GTA, GGG, TGG, GTC, TTT, GA-3’ for calsequestrin (CASQ).
Statistics
Data are expressed as mean ± standard error (SE). One-way ANOVA tests were performed for comparisons of the means among different groups. The chi-square test was used for the comparison of VF incidence among different groups. A P-value of less than 0.05 was considered statistically significant.
Results
Following an ischaemic VF inducing protocol, 80% of normoxic rats developed cardiac ischaemic VF, whereas the arrhythmia seldom (16.6%) occurred in the hearts of CIHH rats (Fig. 1). Stimulation or inhibition of TSPO with FGIN-1–27 or 49-Cl-DZP affected the fatal arrhythmia incidence in normoxic rats. However, FGIN-1–27 could not reverse CIHH-mediated antiarrhythmic effects.
Fig 1.
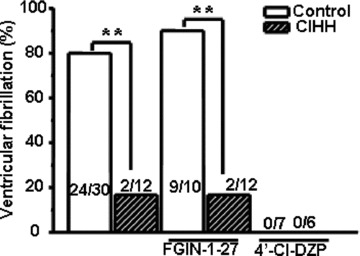
CIHH-mediated antiarrhythmic effects involve translocator protein (TSPO). TSPO stimulation promoted ischaemic VF in normoxic rats, but did not affect the antiarrhythmic effects by CIHH. CIHH: chronic intermittent hypobaric hypoxia, FGIN-1–27: N,N-dihexyl-2-(4-fluorophenyl)indole-3-acetamide; 4’-Cl-DZP: 4-chlorodiazepam, **P, 0.01.
As illustrated in Figure 2A and B, ischaemic VF was positively linked to TSPO activity alterations; specifically, ischaemic VF occurrence accompanied by stepped-up elevation of the protein activity. Of note, TSPO activity was higher in CIHH rats than in normoxic ones under basal conditions, and was preserved during ischaemia (Fig. 2B and C). Further analysis indicated that ischaemia did not modify the TSPO mRNA expression in normoxic rats. However, the level of TSPO expression was higher in CIHH rats than in normoxic ones under basal conditions, and was preserved throughout ischaemia (Fig. 2D). This evidence strongly supports the idea that CIHH blunts or resets the sensitivity of TSPO to ischaemic stress and further contributes to antiarrhythmia.
Fig 2.
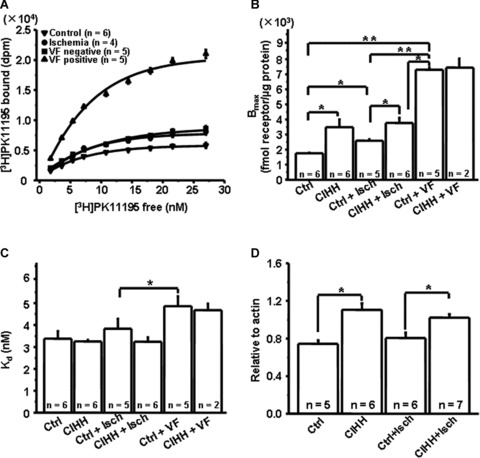
Modulations of CIHH on translocator protein (TSPO). (A) Representative binding curves for TSPO in normoxic rat hearts that underwent stress-induced ventricular fibrillation (VF) protocol. (B) Stepwise elevation of Bmax was positively linked to ischaemic VF. CIHH up-regulated and preserved TSPO throughout normal and low-flow perfusion. (CIHH: chronic intermittent hypobaric hypoxia; *P, 0.05; **P, 0.01). (C) Regulation of Kd (equilibration dissociation constant) by ischaemia and CIHH (*P, 0.05). (D) Modification of TSPO mRNA expression by ischaemia and CIHH (*P, 0.05).
Figure 3 showed that [Ca21]i was progressively elevated in normoxic cardiomyocytes during ischaemia, but no significant Ca overload was observed in CIHH cardiomyocytes. Stimulation of TSPO with FGIN-1–FGIN-27 also failed to disrupt intracellular Ca stabilization by CIHH. Inhibition of the protein with 4′-Cl-DZP abolished [Ca21]i overload in cardiomyocytes from both normoxic and CIHH rat hearts (Fig. 3B). In addition, CIHH preserved Ca kinetics including Ca transient decay rate and amplitude during myocardial ischaemia (Fig. 3C and D), which was consistent with the stabilization of TSPO activity and expression (Fig. 2).
Fig 3.
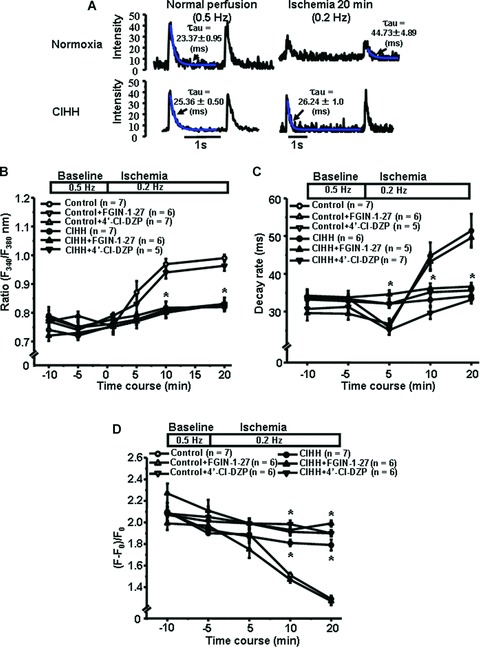
Translocator protein (TSPO) contributes to [Ca2+]i maintenance during ischaemia in CIHH rats. (A) Typical calcium transients both in normoxic and CIHH cardiomyocytes during normal perfusion and simulated ischaemia. Blue lines indicate the decay constants of calcium transients. (B) Quantitative analysis of relative fluorescence ratio with Ca indicator Fura-2 AM in cardiomyocytes. (C) Decay rate of Ca transients. (D) Amplitude of Ca transients. Upper panel of (C), (D) and (E) indicate the protocol for simulated ischaemia; CIHH, chronic intermittent hypobaric hypoxia; F, maximal fluorescence intensity at a transient peak; F0, minimal fluorescence intensity during cardiomyocyte diastole; *P, 0.05, compared to Control.
As indicated in Figure 4A and B, SERCA2a and RyR mRNA expression in normoxic rat hearts were up-regulated during ischaemia; however, they were preserved at a higher level in CIHH rat hearts throughout normal perfusion and ischaemia. CASQ, another important SR Ca-buffering protein, regulates SR Ca release by forming a functional complex with RyR and other SR proteins [15]. Our study demonstrated that the metabolic compromise did not modify the expression of CASQ mRNA (Fig. 4C). We also found that TSPO activation did not further modify the expression of RyR, SERCA2a and CASQ during ischaemia; its inhibition preserved their expressions in normoxic rat hearts. Otherwise, CIHH up-regulated and stabilized their expression throughout normal perfusion and ischaemia, which matched with the changes of TSPO activity and expression.
Fig 4.
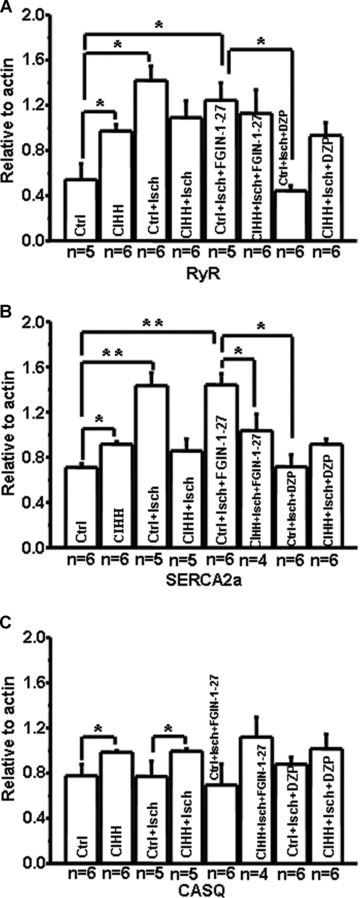
Chronic intermittent hypobaric hypoxia (CIHH) preserved Ca-handling protein expression. (A, B) Myocardial ischaemia up-regulated RyR and SERCA2a mRNA expression in normoxic rats; CIHH up-regulated RyR and SERCA2a mRNA expression under normal conditions, but maintained it at the similar level during ischaemia. Stimulation or inhibition of TSPO did not promote the ischaemic modification of RyR and SERCA2a both in normoxic and CIHH rats. (C) Myocardial ischaemia did not change CASQ mRNA expression in normoxic rat. CIHH up-regulated CASQ mRNA expression under basal conditions and maintained it at a similar level during myocardial ischaemia. RyR: ryanodine receptor; SERCA2a: Ca-ATPase; CASQ: calsequestrin; Ctrl: control; Isch: ischaemia; DZP: 4′-Cl-DZP; *P < 0.05, **P < 0.01).
Discussion
There are two major findings in this study. First, CIHH blunted or reset the sensitivity of TSPO to ischaemic stress, and further prevented ischaemic VF. Second, preservation of TSPO by CIHH contributed to the maintenance of [Ca2+]i and Ca-handling proteins such as RyR and SERCA2a during ischaemia.
Substantial evidence demonstrates that the TSPO is immediately up-regulated by acute stress, and conversely, kept normal during chronic stress, suggesting that the abrupt alteration of TSPO activity may contribute to the induction of the acute stress response [6]. Recently, the relation between abrupt stimulation of TSPO and ischaemia/reperfusion (I/R) ventricular arrhythmias has also been confirmed [9, 10]. In this study, although the TSPO activity was up-regulated by CIHH under basal conditions, it was kept at a similar level during myocardial ischaemia, with the result that VF seldom occurred in CIHH rats. Blunt sensitivity of cardiac TSPO to ischaemic stress by CIHH may confer cardioprotection. These findings further enhanced the point that abrupt and excessive elevation of the TSPO activity contributed to ischaemic VF initiation, although, conversely, preservation of its activity protected the heart from the arrhythmias.
Intermittent hypoxia or CIHH, a model of adaptation to mild hypoxia, has been perceived to enhance the power of stress-limiting systems, mitochondrial oxygen utilization and cardiac tolerance to severe hypoxia, as well as further conferred cardioprotection [16–18]. As reported here, CIHH conferred resistance to ischaemic VF, which may involve the blunted response of myocardial TSPO to transient pacing episodes in the setting of ischaemia, in that TSPO agonists could not reverse CIHH-mediated antiarrhythmic effects (Fig. 1). It is known that TSPO is an important sensor to stresses, and that it regulates mitochondrial oxygen utilization [4]. Thus, this finding also supports the notion that TSPO may be an important stress-limiting system in the heart.
Intracellular [Ca2+]i overload is recognized as the key event in triggering VF and preventing its reversal [1, 19, 20]. In addition, dysfunction of the sarcoplasmic reticulum (SR) calcium release and uptake increases ventricular automaticity and arrhythmia initiation [1, 19]. Previous studies have demonstrated that adaptation to hypoxia protected cardiomyocytes from ischaemia reperfusion [Ca2+]i overload [16–18]. Here, analysis of [Ca2+]i and Ca kinetics indicated that CIHH also preserved them during ischaemia, and that TSPO stimulation could not reverse such effects, which may partially account for CIHH’s antiarrhythmic effects.
Consistent with previous reports [21], our data indicated that cardiac metabolic insult up-regulated the expression of RyR and SERCA2a. CIHH up-regulated their expression under basal conditions, but stabilized them during myocardial ischaemia. TSPO stimulation with FGIN-1–FGIN-27 did not change CIHH-mediated stabilization of RyR and SERCA2a expression. It is difficult for us to determine the cause-effect of changes in SR gene expression and [Ca2+]i during ischaemia. However, modulations on these SR Ca-handling proteins by CIHH might contribute to a better heart performance during ischaemia and to arrhythmias prevention, which is consistent with previous reports that stabilization of SERCA2a restores contractile function and reduces ventricular arrhythmias during I/R [22]. Although alterations of these SR proteins transcript levels cannot completely reflect their functional status, it seems reasonable that the stabilized SR proteins during cardiac ischaemia in CIHH hearts may contribute to the preservation of [Ca2+]i and may be partially responsible for the antiarrhythmic effects. Here, we cannot exclude the possibility that CIHH has effects on molecules other than SR proteins.
In summary, preservation of TSPO may mediate antiarrhythmic effects by CIHH, which partially involves the preservation of [Ca2+]i and the stabilization of RyR and SERCA2a expression. This finding provides insight into the merit of CIHH regarding its utility in patients with coronary artery diseases.
Acknowledgments
This work was supported by the National Science Fund for Distinguished Young Scholars (30425016), the ‘973’ Programme Fund of China (2007CB512100), the ‘863’ Programme Fund of China (2007AA02Z438), the Programme Fund for Outstanding Medical Academic Leader of Shanghai, China, the Yangze Scholars Programme Fund by the Ministry of Education, China, the Programme Fund for Innovative Research Team by the Ministry of Education, China (all to Yi-Han Chen), and the ‘973’ Programme Fund of China (2006CB504100, to Zhao-Nian Zhou).
References
- 1.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–15. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maloney JP, Broeckel U. Epidemiology, risk factors, and genetics of high-altitude-related pulmonary disease. Clin Chest Med. 2005;26:395–404. doi: 10.1016/j.ccm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Asemu G, Papousek F, Ostádal B, et al. Adaptation to high altitude hypoxia protects the rat heart against ischemia-induced arrhythmias. Involvement of mitochondrial K(ATP) channel. J Mol Cell Cardiol. 1999;31:1821–31. doi: 10.1006/jmcc.1999.1013. [DOI] [PubMed] [Google Scholar]
- 4.Meerson FZ, Ustinova EE, Manukhina EB. Prevention of cardiac arrhythmias by adaptation to hypoxia: regulatory mechanisms and cardiotropic effect. Biomed Biochim Acta. 1989;48:S83–8. [PubMed] [Google Scholar]
- 5.O’Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiology. 2005;20:303–15. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gavish M, Bachman I, Shoukrun R, et al. Enigma of the peripheral benzodiazepine receptor. Pharmacol Rev. 1999;51:629–50. [PubMed] [Google Scholar]
- 7.Mukhin AG, Papadopoulos V, Costa E, et al. Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc Natl Acad Sci USA. 1989;86:9813–6. doi: 10.1073/pnas.86.24.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leducq N, Bono F, Sulpice T, et al. Role of peripheral benzodiazepine receptors in mitochondrial, cellular, and cardiac damage induced by oxidative stress and ischemia-reperfusion. J Pharmacol Exp Ther. 2003;306:828–37. doi: 10.1124/jpet.103.052068. [DOI] [PubMed] [Google Scholar]
- 9.Akar FG, Aon MA, Tomaselli GF, et al. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–35. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Xiao J, Liu Y, et al. Mitochondrial benzodiazepine receptors mediate cardioprotection of estrogen against ischemic ventricular fibrillation. Pharmacol Res. 2009;60:61–7. doi: 10.1016/j.phrs.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Curtis MJ. Characterisation, utilisation and clinical relevance of isolated perfused heart models of ischaemia-induced ventricular fibrillation. Cardiovasc Res. 1998;39:194–215. doi: 10.1016/s0008-6363(98)00083-2. [DOI] [PubMed] [Google Scholar]
- 12.McEnery MW, Snowman AM, Trifiletti RR, et al. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA. 1992;89:3170–4. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garber PM, Mezl VA. Cardiac mRNA levels during the development and growth of the rat. Basic Res Cardiol. 1988;83:384–91. doi: 10.1007/BF02005824. [DOI] [PubMed] [Google Scholar]
- 14.Parola AL, Stump DG, Pepperl DJ, et al. Cloning and expression of a pharmacologically unique bovine peripheral-type benzodiazepine receptor isoquinoline binding protein. J Biol Chem. 1991;266:14082–7. [PubMed] [Google Scholar]
- 15.Inesi G, De Meis L. Regulation of steady state filling in sarcoplasmic reticulum. Roles of back-inhibition, leakage, and slippage of the calcium pump. J Biol Chem. 1989;264:5929–36. [PubMed] [Google Scholar]
- 16.Xie Y, Zhu WZ, Zhu Y, et al. Intermittent high altitude hypoxia protects the heart against lethal Ca21 overload injury. Life Sci. 2004;76:559–72. doi: 10.1016/j.lfs.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Xie Y, Zhu Y, Zhu WZ, et al. Role of dual-site phospholamban phosphorylation in intermittent hypoxia-induced cardioprotection against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2005;288:H2594–602. doi: 10.1152/ajpheart.00926.2004. [DOI] [PubMed] [Google Scholar]
- 18.Zhu WZ, Xie Y, Chen L, et al. Intermittent high altitude hypoxia inhibits opening of mitochondrial permeability transition pores against reperfusion injury. J Mol Cell Cardiol. 2006;40:96–106. doi: 10.1016/j.yjmcc.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 19.Del Monte F, Lebeche D, Guerrero JL, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci USA. 2004;101:5622–7. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ter Keurs HE, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Temsah RM, Kawabata K, Chapman D, et al. Modulation of cardiac sarcoplasmic reticulum gene expression by lack of oxygen and glucose. FASEB J. 2001;15:2515–7. doi: 10.1096/fj.00-0870fje. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Dorado D, Piper HM, Eisner DA. Sarcoplasmic reticulum and mitochondria in cardiac pathophysiology. Cardiovasc Res. 2008;77:231–3. doi: 10.1093/cvr/cvm070. [DOI] [PubMed] [Google Scholar]