

Abstract
Amyloid β protein (Aβ) has been associated with Alzheimer's disease (AD) because it is a major component of the extracellular plaque found in AD brains. Increased Aβ levels correlate with the cognitive decline observed in AD. Sporadic AD cases are thought to be chiefly associated with lack of Aβ clearance from the brain, unlike familial AD which shows increased Aβ production. Aβ aggregation leading to deposition is an essential event in AD. However, the factors involved in Aβ aggregation and accumulation in sporadic AD have not been completely characterized. This review summarizes studies that have examined the factors that affect Aβ aggregation and toxicity. By necessity these are studies that are performed with recombinant-derived or chemically synthesized Aβ. The studies therefore are not done in animals but in cell culture, which includes neuronal cells, other mammalian cells and, in some cases, non-mammalian cells that also appear susceptible to Aβ toxicity. An understanding of Aβ oligomerization may lead to better strategies to prevent AD.
Keywords: Alzheimer's disease, Abeta, oligomerization/aggregation, peptide toxicity
Introduction
Alzheimer's disease (AD) belongs to a large cohort of diseases characterized by amyloidoses. Extracellular senile plaques, the foremost pathophysiological hallmark of AD are composed of a dense core of amyloid fibrils associated with degenerating neu-rites, astrocytes and astrocytic processes [1]. Amyloid β protein (Aβ) is the main protein component of senile plaques [2]. Extracellular Aβ is generated by proteolytic processing of amyloid precursor protein (APP) by β-secretase followed by γ-secretase at the cell surface. In the AD brain, there appears to be an apparent failure in regulating the production and clearance of Aβ, leading to increased levels of Aβ and consequently aggregation and neuro-toxicity (Fig. 1). Studies using mouse models, cell culture, synthetic Aβ and biophysical methods have shown a strong correlation between increased levels of Aβ leading to acute dementia in AD [3–10]. Due to Aβ's ability to bind several different molecules and attain multiple physical states [11], our understanding of the key neurotoxic mechanism(s) causing cognitive decline in AD is incomplete. It is also increasingly believed that cognitive decline in AD is a result of multiple toxicity mechanisms of different Aβ forms.
1.
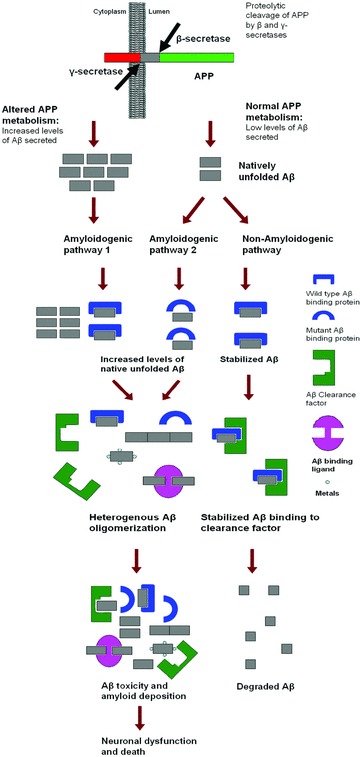
Possible events leading to Aβ accumulation and neurotoxicity.
The onset of AD supports an age-dependent dichotomous model including familial early onset cases (10% of all AD cases) and late onset cases. Increased production of Aβ is a feature of early onset AD and can be caused by mutations observed in APP and γ-secretase complex [12]. Three fully penetrant (autosomal dominant) genetic mutations (presenilin 1, presenilin 2 [components of 7-secretase complex] and APP) have been described for early onset AD. These mutations either alter APP metabolism or the nature of secreted Aβ. Thus the mechanism of amyloid plaque formation is primarily driven by increased local concentration of Aβ or due to the intrinsic aggregating property of the mutant Aβ form.
For late onset, which comprises 90% of AD cases, apolipoprotein E is the only genetic risk factor observed with moderate penetrance. However, senile extracellular amyloid plaques are observed in the majority of the late onset AD cases indicating that the mechanism of neurodegeneration could be similar. Although, late onset AD cases do not show signs of increased production of Ap, it has been suggested that reduced degradation of Aβ by neprilysin and insulin-degrading enzyme [13, 14] and reduced perivascular drainage [15] may describe the elevated levels of Aβ in the AD brain. Several molecular mechanisms for clearance of Aβ have been demonstrated, including microglial clearance [16]via the macrophage scavenger receptor [17], receptor for advanced glycation end products [18], low-density lipoprotein receptor-related protein internalization and degradation of Aβ complexes with apolipoprotein E and a2-macroglobulin [19, 20].
Amyloid structure
Amyloidoses are associated with the misfolding of a native protein into a cytotoxic form, which occurs in parallel with, or as an alternative to physiological folding. This is followed by deposition in tissues in bundles of β-sheet fibrillar protein. The fibrillar form of proteins is a structure dominated by hydrogen bonding between the amino and the carbonyl groups of the main chain, rather than by specific interactions of the side chains observed in globular proteins [21]. Amyloid proteins also exhibit the ability to form multiple conformations [22]. According to the ‘folding energy landscape theory’, protein folding follows a funnel-like pathway in which the conformational intermediates progressively merge into a final species with minimum free energy and maximum stability [23]. However, in amyloid formation, at a minimum energy similar to that of the native protein state, the polypeptide acquires an alternative and relatively stable ‘misfolded state’ which is prone to aggregation [24]. The native structures and amino acid sequences of the proteins associated with amyloid diseases have been found to vary considerably; however, amyloid fibrils isolated from different sources share a common ultrastructure [25].
The capacity to form fibrillar amyloid structures is not exclusive to a specific group of proteins but is generic to all polypeptide chains [26]. α-helical proteins forming amyloid fibrils under appropriate in vitro conditions [27] and amyloid aggregates of non-pathogenic proteins cause toxicity to neuronal cells [28] implying that amyloid formation does not exclusively depend on the intrinsic nature of the protein.
Mechanism of amyloid aggregation
Fibril formation is considered to be an aggregation pathway originating from a high entropic barrier and a thermodynamically unfavourable event [29]. The aggregation of Aβ is initiated by a conformational change from random coil or α-helix into a β-strand, quite similar to prion diseases. Hydrophobic interactions are eventually maximized by β-sheet conformation [29, 30]. Aβ aggregation and fibril formation are nucleation dependent and the kinetics of fibril formation are determined by nucleation and fibril elongation rate [31]. Although, the formation of nuclei is thermo-dynamically unfavourable, the addition of monomeric molecules to the existing nuclei is favourable, and occurs by perpendicular hydrogen bonding to the axis of the amyloid nucleus [11].
A considerable number of environmental factors as well as some intrinsic properties of proteins can work in concert to cause amyloidogenesis (Fig. 1). Some of these factors, particularly those involved in AD, are listed in Table 1. These factors can influence the thermodynamic stability of the various accessible conformations of the protein potentially causing amyloidogenesis. Although, protein aggregation and amyloid formation have been thought to be cytotoxic, recent studies have identified novel biological functions for amyloidogenic protein fibrils in bacteria, fungi and even mammals [32]. Emerging evidence has indicated that rather than amyloidogenic aggregates, the oligomeric intermediates could be the toxic entities, which also have been observed in AD [33, 34]. Thus understanding the mechanism and factors causing Aβ aggregation and stability of the oligomeric intermediates have become more important.
1.
Pathogenic protein misfolding
| Causes/factors | Misfolded protein/disease | Reference |
|---|---|---|
| Nature of the protein: (ageing, Hydrophobicity) | Ageing: transthyretin protein in senile systematic amyloidoses Hydrophobicity: Aβ, Prion proteins, | [35][36][37] |
| Concentration dependence | Aβ in Alzheimer's disease (AD) | [38] |
| Mutations in amino acid sequences in the protein | Hereditary amyloidosis APP in AD | [39][4] see Table 2 |
| Mutations in associating proteins | β2-Microglobulin mutations | [40] |
| Chemical modifications of the protein | Protonation of AfS Oxidative modifications of Aβ | [2][41][42] |
| Protein folding machinery (Chaperones, heat shock proteins) | Aβ in AD Alpha synuclein in Parkinson disease | [43][44] |
| Altered proteolysis or turnover of precursor protein | Mutations in Amyloid precursor protein in AD Presenilin mutations in AD | [4,5][45,46] |
| Decreased clearance | Aβ in AD Alpha synuclein in Parkinson disease | [47][48] |
| Time of incubation | Aβ in AD | [49] |
| Temperature and ionic strength | Aβ in AD | [50] |
| Local interacting factors (other proteins, metals, osmolytes) | Aβ and Metals Osmolytes and prions ApoE and AfS | [51], [52], [53][47,54,55] |
Numerous causes/mechanism of Aβ aggregation have been observed previously. Although many studies have demonstrated the mechanism of Aβ aggregation, there are certain drawbacks in regard to the poor correlation of in vivo conditions to the controlled environment in an in vitro analysis. For example, Aβ assembly occurs in a very complex and dynamic environment; characterized by the presence of different proteins, membranes, metal ions etc., while in vitro experiments are done with extremely simplified conditions that may bias toward amyloid aggregation. Very little mechanistic/structural information is available regarding the exact conformational change and mechanism of Aβ aggregation caused by local environmental factors in AD brain.
Aβ: a natively unfolded protein?
The folding of proteins into their correct three-dimensional structure is critically important for their biological activity and normal functioning of the cell. With the human proteome reaching a size of more than 100,000 proteins, it is clearly evident that protein folding occurs in a crowded and sensitive environment highly prone to aggregation [26]. However, cellular systems have evolved to evade unfavourable protein aggregation. Negative selection has been observed to avoid alternating polar and hydrophobic residues that favour a β-sheet structure [56]. It is also suggested that residues showing increased vulnerability to aggregation are preferably located in different regions of the sequence from those that determine native protein folding, termed ‘kinetic partitioning’[57]. Apart from amino acid sequence selection, biological systems have developed molecular chaperones and degradation mechanisms to control the rate of formation of unfavourable structures [58].
Therefore it becomes obvious that apart from intrinsic properties of protein to aggregate, failure of the protein folding machinery could also play a crucial role in amyloidogenesis [43]. Mutations in Aβ sequences (Table 2) have been identified in familial forms of AD which either increase the propensity to aggregate, or decrease the stability of the native state. However, no similar genetic evidence has been identified for the more prevalent sporadic AD, although widespread neuritic amyloid plaques are observed in majority of the sporadic AD cases [1]. This indicates that progressive Aβ aggregation in sporadic AD could be associated with Aβ binding and clearance factors [47].
2.
Mutations affecting Aβ aggregation
| Mutations | Aggregation/toxicity |
|---|---|
| Flemish (A21G)[59] | Short 2 h incubation shows elevated apoptosis, slower aggregation than wild-type [60] |
| Arctic (E22G)[61] | Increased rate of fibril formation [62] |
| Dutch (E22Q)[63] | Causes higher amount of apoptosis at physiological concentrations with 24 h incubation, faster aggregation than wild-type, increased rate of fibril formation [60, 62] |
| Italian (E22K)[64] | Aggregates rapidly [65] |
| Iowa (D23N)[66] | Aggregates rapidly [65] |
| Pyroglutamate-modified AβN3(pE)[67] | AβN3(pE)-40/42 peptides shows resistance to degradation by cultured astrocytes [68] |
Proteins are required to possess fast folding kinetics and high stability to minimize the risk of protein aggregation in the cell [69]. But certain proteins may require higher-order structural disorders mediated by binding to ligands to fulfil their function [70, 71]. Such proteins have greater structural plasticity to favour ligand binding and could be classified under the emerging class of ‘natively unfolded proteins’[72]. The presence of natively unfolded proteins in the cell is believed to provide a simple solution to having large intermolecular interfaces for diverse ligand binding and regulation, and smaller protein, genome and cell sizes [73]. There is an increasing belief that amyloidogenic proteins could be natively unfolded proteins [74]. Aβ is known to bind a large array of extracellular and cell-associated ligands (Table 3, Fig. 1). However, Aβ interaction with different molecules has not been clearly understood in relevance to its biological function or pathology. Although not experimentally proved, Aβ shows characteristics of a ‘natively unfolded protein’. Under normal conditions, Ap could be bound to a ligand essential for a normal function and occurrence of Aβ aggregation in sporadic AD may be a result of absence or structural disorder of its ligand. Thus it is suggested that the clinical manifestations observed in AD could be either the toxic property of the pre-fibrillar intermediate or loss of function of native Aβ.
3.
Aβ interacting molecules
| Cofactor | Reference |
|---|---|
| Metals: | |
| Copper | [2] |
| Zinc | [51] |
| Aluminium | [75] |
| Iron | [76] |
| CSF and plasma proteins: | |
| Albumin | [77] |
| Lipoprotein: ApoE | [55] |
| Insulin | [78] |
| Serum amyloid P | [79] |
| Other plasma proteins: IgG, IgA, IgM β1-Antitrypsin, Transferrin, β2-Macroglobulin, β1-Antichymotrypsin Antithrombin III, Transthyretin and Fibrinogen | [80] |
| Cell Surface Receptors: | |
| Transforming growth factor β receptor | [81] |
| Insulin receptor | [82] |
| NMDA receptor | [83] |
| p75 neurotrophin receptor | [84] |
| Receptor for advanced glycation End products (RAGE) | [85] |
| Formyl peptide receptor-like 1 | [86] |
| Amyloid precursor protein | [87] |
| Scavenger receptors SR-A, SR-BI | [17] |
| α7nicotinic acetylcholine receptor (α7nAChR) | [88] |
| CD47, CD36, β6p1-integrin | [89] |
| Serpin-enzyme complex receptor (SEC-R) | [90] |
| Integrin β1 | [91] |
| HSP | [92] |
| Intracellular proteins: | |
| AfS binding alcohol dehydrogenase | [93] |
| Chaperone proteins | [94] |
| 20S proteasome | [95] |
| Extracellular matrix proteins: | |
| Heparin sulphate | [96] |
| Agrin | [97] |
| Laminin, | [98] |
| Collagen-like Alzheimer amyloid plaque component CLAC. | [99] |
| Others: | |
| Membrane lipids | [100] |
| Chondroitin sulphate-derived Monosaccharides and Disaccharides | [101] |
| Cholesterol | [102] |
Ambiguities in synthetic Aβ studies
The dynamic nature of Aβ in physiological conditions has been a major concern in determining the mechanism of Aβ toxicity. Since aggregating Aβ is neurotoxic, cell loss and Aβ deposition are considered to correlate with the severity of disease symptoms. However, several studies predict that events preceding neuronal cell death may provide a better explanation to the progressive decline in cognition in AD [103–105]. There are robust correlations between the levels of soluble Aβ and the extent of synaptic loss and severity of cognitive impairment [33, 34]. In these studies, the term soluble Aβ describes all isoforms of Aβ that remain in the supernatant following high speed centrifugation (>100,000 × g) of tissue extracts. Although these studies have not identified a specific assembly form(s) of soluble Aβ, it is clearly implied that non-fibrillar assemblies are the main cause of synaptic dysfunction leading to cognitive decline in AD. Identifying a particular Aβ species as the main cause of synaptic loss has posed serious difficulties owing to the heterogenic nature of Aβ. Monomeric Aβ has the ability to associate into higher-ordered aggregates depended on several interdependent factors. Thus, it is difficult to unequivocally attribute toxicity to a discrete species. Consequently, much confusion has resulted regarding the variable behaviour of different peptide stocks [106, 107]. Table 4 refers to the literature describing a range of Aβ isoforms. Not all descriptions are unique, but it is clear that many isoforms have been observed, ranging from monomers to oligomers of various sizes to protofibrils, fibrils and aggregates.
4.
Different isoforms of Aβ
| Aβ isoform |
|---|
| Soluble dimers [108], tetramers and oligomers [2] |
| Non-amyloidogenic amorphous aggregates [109] |
| Amyloidogenic fibrils [109] |
| Fibrillar aggregates [50] |
| Amyloid proto-fibrils [31] |
| Amyloid derived diffusible ligands [110] |
| Soluble non-fibrillar [111] |
| Hexamer, nonamer, dodecamer, Aβ*56 (56-kD soluble Aβ assembly) [112] |
Apart from the propensity of Aβ42 to generate multiple conformations, it possesses oxidative [113], hydrolytic [114], and surfactant properties [115]. It is also clear that different Aβ assemblies can possess distinct toxicity mechanisms [116] in different cell lines, even in yeast [111].
Formation of amyloid plaques
Aβ deposits in the brain are usually referred to as senile plaques. Aβ plaques can also be observed in cognitively normal individuals [117–119]. The major variation between Aβ deposits in normal individuals and those found in AD patients is their distribution [118]. In addition to Aβ, other proteins accumulate within senile plaques, including apolipoprotein E (apoE), α2-macroglobulin, interleukins, components of the complement system, α2-macroglobulin receptor, low-density lipoprotein receptor-related protein, collagenous Alzheimer amyloid component [120–124] and also dystrophic neu-rites, reactive astrocytes, and microglial cells [121, 125].
Apart from extracellular accumulation, Aβ is also known to form insoluble pools intracellularly (reviewed in [126]). Recent studies have confirmed the build up of intracellular Aβ in neuronal cells as an early event in AD pathogenesis [127, 128] which precedes formation of amyloid plaques in the brain [129]. Intracellular Aβ has been postulated to originate from the result of intracellularly localized APP proteolysis [130–132] or by receptor associated uptake of extracellularly secreted Aβ[133–137]. As the majority of Aβ is secreted, it is suggested that APP proteolysis predominantly occurs at the cell membrane or cleaved Aβ is rapidly secreted [126]. Previous studies also have shown that intra-neuronal Aβ is observed only in transgenic models based on APP overexpression [138, 139] and not in wild-type. Thus it seems like that the origin of intracellular Aβ is largely by uptake from extracellular media. Intracellular Aβ is also implicated in synaptic dysfunction and associated cognitive decline [140]. However, the nature of intracellular Aβ assembly, mechanism of action and its relevance to AD pathology needs to be addressed.
Role of Aβ in AD pathogenesis
Memory impairment including the loss of the ability to form and retain new episodic memories is the hallmark of early stages of AD. Cognitive impairment is often attributed to synaptic dysfunction and neuronal cell loss particularly in the cells interconnecting the hippocampal formation with the associating structures crucial for memory [141, 142]. Depleted neurotransmitters [142], 25 to 35% decrease in the synapses [143, 144] and quantitative correlations of postmortem cytopathology with cognitive deficits indicate that synaptic loss is more robustly correlated than the numbers of plaques or tangles, or extent of cortical gliosis [145].
Lesne et al.[112] study implicated a unique and novel Aβ iso-form (Ap*56: 56-kD soluble Aβ assembly) as the key neurotoxic Aβ isoform responsible for cognitive decline in APP overexpress-ing Tg2576 mice, based on its abundance, stability and occurrence during memory decline. However, a more recent study [108] has identified Aβ dimers from the soluble extract of AD cerebral cortex tissues. They also specifically attribute Aβ dimers to the loss of long-term potentiation, enhanced long-term depression, reduced dendritic spine density in normal rodent hippocampus and memory disruption of a learned behaviour in normal rats. Whether it is Aβ dimer or Aβ*56, the different toxic Aβ species identified might reflect differences in the toxicity assays and Aβ detection methods used. Even though the increase in soluble Aβ levels and aggregation in the brain has been consistently observed as the main indicator of cognitive decline, the localization of the Aβ accumulation in the brain has not been specified.
A recent study has elaborated the extraction of Aβ from different anatomical compartments (extracellular soluble, intracellular soluble, membrane associated and extracellular insoluble) [146]. It identified membrane-associated and intracellular Aβ in the temporal neocortex of AD patients to be more closely related to AD symptoms than other measured Aβ species. This study has addressed the very important aspect of Aβ localization and accumulation in AD pathogenesis.
It therefore becomes essential to identify and systematically categorize the factors responsible for Aβ's native form and pathological aggregation based on its relevance to physiological role or synaptic failure in AD.
Conclusion
Aβ plays the central role in neurodegeneration in AD. It is widely accepted that Aβ has a wide array of biological activities and affinities, which have not been definitively mapped to its native or pathological role in the brain. Although, there has been much progress in understanding the role of Aβ in AD, there are several important questions still unanswered including the physiological nature and function of APP/Aβ in the normal ageing brain, how Aβ contributes to the vascular defects observed in AD, what are the genetic risk factors associated with late onset AD, what causes vulnerability to Aβ toxicity, is AD caused by loss of native Aβ function or by its pathogenic form and are AD symptoms caused by the cumulative effect of different toxic Aβ forms or exclusive to a particular form. With Aβ emerging as one of the primary targets for immunotherapy and target based drug design for AD, it becomes essential to gain further insight into Aβ caused cognitive decline in AD.
References
- 1.Wisniewski HM, Terry RD. Reexamination of the pathogenesis of the senile plaque. Prog Neuropathol. 1973;11:1–26. [Google Scholar]
- 2.Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neuro-fibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–63. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH. The precursor of Alzheimer's disease amyloid β protein resembles a cell surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 4.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, Van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–6. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 5.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A. Amyloid [J protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–2. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 6.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL. Amyloid (J-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–5. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 7.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid β-peptide. Trends Mol Med. 2001;7:548–54. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 8.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the (β-amyloid precursor protein intracellular domain in vivo. PNAS. 2003;100:4162–7. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–82. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 10.Shen CL, Murphy RM. Solvent effects on self-assembly of β-amyloid peptide. Biophys J. 1995;69:640–51. doi: 10.1016/S0006-3495(95)79940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasegawa K, Yamaguchi I, Omata S, Gejyo F, Naiki H. Interaction Between Aβ(1–42) and Aβ(1–40) in Alzheimer's [J-amyloid fibril formation in vitro. Biochemistry. 1999;38:15514–21. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]
- 12.Jonghe CD, Esselens C, Singh SK, Craessaerts K, Serneels S, Checler F, Annaert W, Van Broeckhoven C, Strooper BD. Pathogenic APP mutations near the -secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Human Mol Genet. 2001;10:1665–71. doi: 10.1093/hmg/10.16.1665. [DOI] [PubMed] [Google Scholar]
- 13.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 14.Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-(J peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA. 2003;100:6221–6. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid (3 accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol. 1998;153:725–33. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular β amyloid in rat brains injected with Alzheimer β-amyloid. Am J Pathol. 1992;140:1389–99. [PMC free article] [PubMed] [Google Scholar]
- 17.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid β-protein via a scavenger receptor. Neuron. 1996;17:553–65. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 18.Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte W, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM. Amyloid-β peptide-receptor for advanced glycation end product interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflamma-tory pathway in Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:5296–301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan J, Galindo MF, Miller RJ, Reardon CA, Getz GS, LaDu MJ. Isoform-specific effect of apolipoprotein E on cell survival and β-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. J Neurosci. 1998;18:195–204. doi: 10.1523/JNEUROSCI.18-01-00195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urmoneit B, Prikulis I, Wihl G. Cerebrovascular smooth muscle cells internalize Alzheimer amyloid β protein via a lipoprotein pathway: implications for cerebral amyloid angiopathy. Lab Invest. 1997;77:157–66. [PubMed] [Google Scholar]
- 21.Dobson CM. Getting out of shape. Nature. 2002;418:729–30. doi: 10.1038/418729a. [DOI] [PubMed] [Google Scholar]
- 22.Perutz MF. Amyloid fibrils: mutations make enzyme polymerize. Nature. 1997;385:773–5. doi: 10.1038/385773a0. [DOI] [PubMed] [Google Scholar]
- 23.Dobson CM, Karplus M. The fundamentals of protein folding: bringing together theory and experiment. Curr Opin Struct Biol. 1999;9:92–101. doi: 10.1016/s0959-440x(99)80012-8. [DOI] [PubMed] [Google Scholar]
- 24.Schultz CP. Illuminating folding intermediates. Nat Struct Biol. 2000;7:7–10. doi: 10.1038/71197. [DOI] [PubMed] [Google Scholar]
- 25.Sunde M, Blake C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv Protein Chem. 1997;50:123–59. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- 26.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–32. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 27.Pertinhez TA, Bouchard M, Tomlinson EJ, Wain R, Ferguson SJ, Dobson CM, Smith LJ. Amyloid fibril formation by a helical cytochrome. FEBS Lett. 2001;495:184–6. doi: 10.1016/s0014-5793(01)02384-5. [DOI] [PubMed] [Google Scholar]
- 28.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–11. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 29.Lansbury JrPT. A reductionist view of Alzheimer's disease. Acc Chem Res. 1996;29:317–21. [Google Scholar]
- 30.Tycko R. Insights into the amyloid folding problem from solid state NMR. Biochemistry. 2003;42:3151–9. doi: 10.1021/bi027378p. [DOI] [PubMed] [Google Scholar]
- 31.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-pro-tein fibrillogenesis. Detection of a protofib-rillar intermediate. J BiolChem. 1997;272:22364–72. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 32.Kelly JW, Balch WE. Amyloid as a natural product. J Cell Biol. 2003;161:461–2. doi: 10.1083/jcb.200304074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid (J peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush Al, Masters CL. Soluble pool of A(J amyloid as a determinant of severity of neu-rodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 35.Saraiva MJ. Transthyretin amyloidosis: a tale of weak interactions. FEBS Lett. 2001;498:201–3. doi: 10.1016/s0014-5793(01)02480-2. [DOI] [PubMed] [Google Scholar]
- 36.Kim W, Hecht MH. Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer's A(J42 peptide. Proc Natl Acad Sci USA. 2006;103:15824–9. doi: 10.1073/pnas.0605629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zbilut JP, Webber CL, Colosimo A, Giuliani A. The role of hydrophobicity patterns in prion folding as revealed by recurrence quantification analysis of primary structure. Protein Eng. 2000;13:99–104. doi: 10.1093/protein/13.2.99. [DOI] [PubMed] [Google Scholar]
- 38.Verdone G, Corazza A, Viglino P. The solution structure of human (J2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2002;11:487–99. doi: 10.1110/ps.29002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buxbaum JN, Tagoe CE. The genetics of the amyloidoses. Annu Rev Med. 2000;51:543–69. doi: 10.1146/annurev.med.51.1.543. [DOI] [PubMed] [Google Scholar]
- 40.Chiba T, Hagihara Y, Higurashi T, Hasegawa K, Naiki H, Goto Y. Amyloid fibril formation in the context of full-length protein. J Biol Chem. 2003;278:47016–24. doi: 10.1074/jbc.M304473200. [DOI] [PubMed] [Google Scholar]
- 41.Dyrks T, Dyrks E, Hartmann T, Masters CL, Beyreuther K. Amyloidogenicity of A4 and (JA4-bearing APP fragments by metal catalysed oxidation. J Biol Chem. 1992;267:18210–7. [PubMed] [Google Scholar]
- 42.Palmblad M, Westlind-Danielsson A, Bergquist J. Oxidation of methionine 35 attenuates formation of amyloid β-peptide 1–40 oligomers. J Biol Chem. 2002;277:19506–10. doi: 10.1074/jbc.M112218200. [DOI] [PubMed] [Google Scholar]
- 43.Yoo BC, Kim SH, Cairns N, Fountoulakis M, Lubec G. Deranged expression of molecular chaperones in brains of patients with Alzheimer's disease. Biochem Biophys Res Commun. 2001;280:249–58. doi: 10.1006/bbrc.2000.4109. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang W, Moore DJ, Dawson TM, Dawson VL, Chiosis G, Cookson MR, Cai H. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci. 2008;28:3384–91. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.ADCG. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11:219–22. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- 46.Cruts M, Van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, Hofman A, Van Broeckhoven C. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of prese-nile Alzheimer disease. Hum Mol Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- 47.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. PNAS. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24:1888–96. doi: 10.1523/JNEUROSCI.3809-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabel C. Assembly and aggregation properties of synthetic Alzheimer's a4/β amyloid peptide analogs. J Biol Chem. 1992;267:546–54. [PubMed] [Google Scholar]
- 50.Stine WB, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-(J peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–22. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 51.Bush Al, Multhaup G, Moir RD, Williamson TG, Small DH, Rumble B. A novel zinc (II) binding site modulates the function of the βA4 amyloid protein precursor of Alzheimer's disease. J Biol Chem. 1993;268:16109–12. [PubMed] [Google Scholar]
- 52.Nandi PK, Bera A, Sizaret PY. Osmolyte trimethylamine N-oxide converts recombinant α-helical prion protein to its soluble [J-structured form at high temperature. J Mol Biol. 2006;362:810–20. doi: 10.1016/j.jmb.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 53.Yang D, Yip CM, Huang THJ, Chakrabartty V, Fraser PE. Manipulating the amyloid-(J aggregation pathway with chemical chaperones. J Biol Chem. 1999;274:32970–4. doi: 10.1074/jbc.274.46.32970. [DOI] [PubMed] [Google Scholar]
- 54.Dolev I, Michaelson DM. A nontransgenic mouse model shows inducible amyloid-β (Aβ peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. PNAS. 2004;101:13909–14. doi: 10.1073/pnas.0404458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holtzman DM. Role of apoE/Aβ interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci. 2001;17:147–55. doi: 10.1385/JMN:17:2:147. [DOI] [PubMed] [Google Scholar]
- 56.Broome BM, Hecht MH. Nature disfavours sequences of alternating polar and non-polar amino acids: implications for amyloidogenesis. J Mol Biol. 2000;296:961–8. doi: 10.1006/jmbi.2000.3514. [DOI] [PubMed] [Google Scholar]
- 57.Chiti F. Kinetic partitioning of protein folding and aggregation. Nature Struct Biol. 2002;9:137–43. doi: 10.1038/nsb752. [DOI] [PubMed] [Google Scholar]
- 58.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 59.Hendriks L, Van Duijn CM, Cras P, Cruts M, Van Hul W, Van Harskamp F, Warren A, Mclnnis MG, Antonarakis SE, Martin JJ. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β-amyloid precursor protein gene. Nat Genet. 1992;1:218–21. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 60.Singh SK, Julliamsa A, Nuydensk R, Ceuterick C, Labeur C, Serneels S, Vennekens K, Van Osta P, Geerts H, De Strooper B, Van Broeckhoven C. In vitro studies of Flemish, Dutch, and wild-type [J-amyloid provide evidence for two-staged neurotoxicity. Neurobiol Dis. 2002;11:330–40. doi: 10.1006/nbdi.2002.0529. [DOI] [PubMed] [Google Scholar]
- 61.Nilsberth C, Westlincl-Danielsson A, Eckman C, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced A(3 protofibril formation. Nat Neurosci. 2001;4:887–93. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 62.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-(J peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–53. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 63.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, Van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–6. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 64.Tagliavini F, Rossi G, Padovani A, Magoni M, Andora G, Sgarzi M, Bizzi A, Savoiardo M, Carella F, Morbin M, Giaccone G, Bugiani O. A new BPP mutation related to hereditary cerebral hemorrhage. Alzheimer's Rep. 1999:S28. [Google Scholar]
- 65.Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, Shirasawa T. Synthesis, aggregation, neurotoxicity, and secondary structure of various Aβ 1–42 mutants of familial Alzheimer's disease at positions 21–23. Biochem Biophys Res Commun. 2002;294:5–10. doi: 10.1016/S0006-291X(02)00430-8. [DOI] [PubMed] [Google Scholar]
- 66.Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 67.Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid β protein in Alzheimer's disease. J Biol Chem. 1992;267:17082–6. [PubMed] [Google Scholar]
- 68.Russo C, Violani E, Salis S, Venezia V, Dolcini V, Damonte G, Benatti U, D’Arrigo C, Patrone E, Carlo P, Schettini G. Pyroglutamate-modified amyloid β-peptides-AβN3(pE)-strongly affect cultured neuron and astrocyte survival. J Neurochem. 2002;82:1480–9. doi: 10.1046/j.1471-4159.2002.01107.x. [DOI] [PubMed] [Google Scholar]
- 69.Carrell RW, Lomas DA. Conformational disease. Lancet. 1997;350:134–8. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 70.Love JJ, Li X, Chung J, Dyson HJ, Wright PE. The LEF-1 highmobility group domain undergoes a disorder-to-order transition upon formation of a complex with cognate DNA. Biochemistry. 2004;43:8725–34. doi: 10.1021/bi049591m. [DOI] [PubMed] [Google Scholar]
- 71.Sunde M, McGrath KC, Young L, Matthews JM, Chua EL, Mackay JP, Death AK. TC-1 is a novel tumorigenic and natively disordered protein associated with thyroid cancer. Cancer Res. 2004;64:2766–73. doi: 10.1158/0008-5472.can-03-2093. [DOI] [PubMed] [Google Scholar]
- 72.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 73.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem Sci. 2003;28:81–5. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 74.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 75.Fasman GD, Perczel A, Moore CD. Solubilization of β-amyloid-(1–42)-peptide: reversing the β-sheet conformation induced by aluminum with silicates. PNAS. 1995;92:369–71. doi: 10.1073/pnas.92.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ. Deposition of iron and β-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem. 2006;99:438–49. doi: 10.1111/j.1471-4159.2006.04079.x. [DOI] [PubMed] [Google Scholar]
- 77.Biere AL, Dagger BO, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. Amyloid β-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271:32916–22. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 78.Larson JL, Miranker AD. The mechanism of insulin action on islet amyloid polypep-tide fiber formation. J Mol Biol. 2004;335:221–31. doi: 10.1016/j.jmb.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 79.Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. PNAS. 1995;92:4299–303. doi: 10.1073/pnas.92.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bohrmann B, Tjernberg L. Endogenous proteins controlling amyloid β-peptide polymerization. J Biol Chem. 1999;274:15990–5. doi: 10.1074/jbc.274.23.15990. [DOI] [PubMed] [Google Scholar]
- 81.Mousseau DD, Chapelsky S. A direct interaction between transforming growth factor (TGF)-(is and amyloid-(J protein affects fibrillogenesis in a TGF-β receptor-independent manner. J Biol Chem. 2003;278:38715–22. doi: 10.1074/jbc.M304080200. [DOI] [PubMed] [Google Scholar]
- 82.Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins RN. Alzheimer's β-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:1–5. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-fl protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of β-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest. 1997;100:2333–40. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yan SD. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 86.Le Y, Gong W, Tiffany HL, Tumanov A, Nedospasov S, Shen W, Dunlop NM, Gao JL, Murphy PM, Oppenheim JJ, Wang JM. Amyloid B42 activates a Gprotein-coupled chemoattractant receptor, FPR-like-1. J Neurosci. 2001;21:1–5. doi: 10.1523/JNEUROSCI.21-02-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Panegyres PK. The functions of the amyloid precursor protein gene. Rev Neurosci. 2001;12:1–39. doi: 10.1515/revneuro.2001.12.1.1. [DOI] [PubMed] [Google Scholar]
- 88.Wang HY, Li W, Benedetti NJ, Lee DH. α7 nicotinic acetylcholine receptors mediate β-amyloid peptide-induced tau protein phosphorylation. J Biol Chem. 2003;278:31547–53. doi: 10.1074/jbc.M212532200. [DOI] [PubMed] [Google Scholar]
- 89.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar [J-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boland K, Behrens M, Choi D, Manias K, Perlmutter DH. The serpin-enzyme complex receptor recognizes soluble, nontoxic amyloid-(J peptide but not aggregated, cytotoxic amyloid-(J peptide. J Biol Chem. 1996;271:18032–44. doi: 10.1074/jbc.271.30.18032. [DOI] [PubMed] [Google Scholar]
- 91.Sabo S, Lambert MP, Kessey K, Wade W, Krafft G, Klein WL. Interaction of (J-amyloid peptides with integrins in a human nerve cell line. Neurosci Lett. 1995;184:25–8. doi: 10.1016/0304-3940(94)11159-g. [DOI] [PubMed] [Google Scholar]
- 92.Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of [J-amyloid provides a structural basis for the immunopathology of Alzheimer's disease. J Biol Chem. 1998;273:29719–26. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 93.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 94.Fonte V, Kapulkin V, Taft A, Fluet A, Friedman D, Christopher D. Interaction of intracellular amyloid peptide with chaper-one proteins. PNAS. 2002;99:9439–44. doi: 10.1073/pnas.152313999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gregori L, Hainfeld JF, Simon MN, Goldgaber D. Binding of amyloid β protein to the 20 S proteasome. J Biol Chem. 1997;272:58–62. doi: 10.1074/jbc.272.1.58. [DOI] [PubMed] [Google Scholar]
- 96.Ancsin JB, Kisilevsky R. The heparin/heparan sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J Biol Chem. 1999;274:7172–81. doi: 10.1074/jbc.274.11.7172. [DOI] [PubMed] [Google Scholar]
- 97.Cotman SL, Halfter W, Cole GJ. Agrin binds to β-amyloid (Aβ, accelerates Aβ fibril formation, and is localized to A(3 deposits in Alzheimer's disease brain. Mol Cell Neurosci. 2000;15:183–98. doi: 10.1006/mcne.1999.0816. [DOI] [PubMed] [Google Scholar]
- 98.Castillo GM, Lukito W, Peskind E, Raskind M, Kirschner DA, Yee AG, Snow AD. Laminin inhibition of β-amyloid protein (Aβ) fibrillogenesis and identification of an Aβ binding site localized to the globular domain repeats on the laminin a chain. J Neurosci Res. 2000;62:451–62. doi: 10.1002/1097-4547(20001101)62:3<451::AID-JNR15>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 99.Soderberg L, Kakuyama H. Characterization of the Alzheimer's disease-associated CLAC protein and identification of an amyloid β-peptide-binding site. J Biol Chem. 2005;280:1007–15. doi: 10.1074/jbc.M403628200. [DOI] [PubMed] [Google Scholar]
- 100.Ciccotosto GD, Tew D, Curtain CC, Smith D, Carrington D, Masters CL, Bush AL, Cherny RA, Cappai R, Barnham KJ. Enhanced toxicity and cellular binding of a modified amyloid peptide with a methionine to valine substitution. J Biol Chem. 2004;279:42528–34. doi: 10.1074/jbc.M406465200. [DOI] [PubMed] [Google Scholar]
- 101.Fraser PE, Darabie AA, McLaurin J. Amyloid-β interactions with chondroitin sulfate-derived monosaccharides and dis-accharides. J Biol Chem. 2001;276:6412–9. doi: 10.1074/jbc.M008128200. [DOI] [PubMed] [Google Scholar]
- 102.Chochina SV, Avdulov NA, Igbavboa U, Cleary JP, O’Hare EO, Wood WG. Amyloid |i-peptide1–40 increases neuronal membrane fluidity: role of cholesterol and brain region. J Lipid Res. 2001;42:1292–7. [PubMed] [Google Scholar]
- 103.Cummings BJ, Pike CJ, Shankle R, Cotman CW. β-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging. 1996;17:921–33. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 104.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–98. doi: 10.1016/0197-4580(95)00013-5. [DOI] [PubMed] [Google Scholar]
- 105.Katzman R. Medical progress: Alzheimer's disease. New England J Med. 1986;314:964–73. doi: 10.1056/NEJM198604103141506. [DOI] [PubMed] [Google Scholar]
- 106.Brining SK. Predicting the in vitro toxicity of synthetic β-amyloid (1–40) Neurobiol Aging. 1997;18:581–9. doi: 10.1016/s0197-4580(97)00153-x. [DOI] [PubMed] [Google Scholar]
- 107.May PC, Gitter BD, Waters DC, Simmons LK, Becker GW, Small JS, Robison PM. β-Amyloid peptide in vitro toxicity: lot-to-lot variability. Neurobiol Aging. 1992;13:605–12. doi: 10.1016/0197-4580(92)90064-5. [DOI] [PubMed] [Google Scholar]
- 108.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lorenzo A, Yankner BA. (J-Amyloid neuro-toxicity requires fibril formation and is inhibited by Congo red. PNAS. 1994;91:12243–7. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bharadwaj P, Waddington L, Varghese J, Macreadie IG. A new method to measure cellular toxicity of non-fibrillar and fibrillar Alzheimer's Aβ using yeast. J Alzheimer's Dis. 2008;13:1–4. doi: 10.3233/jad-2008-13204. [DOI] [PubMed] [Google Scholar]
- 112.Lesne S, Koh TM, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 113.Pogocki D, Schneich C. Redox properties of Met(35) in neurotoxic β-amyloid peptide. A molecular modeling study. Chem Res Toxicol. 2002;15:408–18. doi: 10.1021/tx0101550. [DOI] [PubMed] [Google Scholar]
- 114.Brzyska M, Bacia A, Elbaum D. Oxidative and hydrolytic properties of β-amyloid. Eur J Biochem. 2001;268:3443–54. doi: 10.1046/j.1432-1327.2001.02248.x. [DOI] [PubMed] [Google Scholar]
- 115.Soreghan B, Kosmoski J, Glabe C. Surfactant properties of Alzheimer's Aβ peptides and the mechanism of amyloid aggregation. J Biol Chem. 1994;269:28551–4. [PubMed] [Google Scholar]
- 116.Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–8. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42:1681–8. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 118.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 119.Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging. 1991;12:295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 120.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, III, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7611–5. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McGeer PL, Walker DG, Akiyama H, Yasuhara O, McGeer EG. Involvement of microglia in Alzheimer's disease. Neuropathol Appl Neurobiol. 1994;20:191–2. [PubMed] [Google Scholar]
- 122.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–6. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 123.Strauss S, Bauer J, Ganter U, Jonas U, Berger M, Volk B. Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer's disease patients. Lab Invest. 1992;66:223–30. [PubMed] [Google Scholar]
- 124.Thai DR, Schober R, Birkenmeier G. The subunits of alpha2-macroglobulin receptor/low density lipoprotein receptor-related protein, native and transformed alpha2-macroglobulin and interleukin 6 in Alzheimer's disease. Brain Res. 1997;777:223–7. doi: 10.1016/s0006-8993(97)01021-4. [DOI] [PubMed] [Google Scholar]
- 125.Yasuhara O, Kawamata T, Aimi Y, McGeer EG, McGeer PL. Two types of dystrophic neurites in senile plaques of Alzheimer disease and elderly non-demented cases. Neurosci Lett. 1994;171:73–6. doi: 10.1016/0304-3940(94)90608-4. [DOI] [PubMed] [Google Scholar]
- 126.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosc. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 127.Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal Aβ-amyloid plaques in down syndrome. Arch Pathol Lab Med. 2001:489–92. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- 128.Mori C. Intraneuronal Aβ42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- 129.Wirths O. Intraneuronal Aβ42 accumulation precedes plaque formation in β-amy-loid precursor protein and presenilin-1 double transgenic mice. Neurosci Lett. 2001;306:116–20. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- 130.Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci. 2003;116:3339–46. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- 131.Mizuguchi M, Ikeda K, Kim SU. Differential distribution of cellular forms of β-amyloid precursor protein in murine glial cell cultures. Brain Res. 1992;584:219–25. doi: 10.1016/0006-8993(92)90898-j. [DOI] [PubMed] [Google Scholar]
- 132.Xu H, Greengard P, Gandy S. Regulated formation of Golgi secretory vesicles containing Alzheimer β-amyloid precursor protein. J Biol Chem. 1995;270:23243–5. doi: 10.1074/jbc.270.40.23243. [DOI] [PubMed] [Google Scholar]
- 133.Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G. The low density lipoprotein receptor related protein 1B retains β amyloid precursor protein at the cell surface and reduces amyloid-β peptide production. J Biol Chem. 2004;279:29639–46. doi: 10.1074/jbc.M313893200. [DOI] [PubMed] [Google Scholar]
- 134.Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in the brain. Nature Med. 2003;9:907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 135.Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of β-amyloid1–42 in neurons is facilitated by the β7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 136.Yazawa H. β Amyloid peptide (Aβ42) is internalized via the G-protein coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. FASEB J. 2001;15:2454–62. doi: 10.1096/fj.01-0251com. [DOI] [PubMed] [Google Scholar]
- 137.Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Aβ42 accumulation in Amyloid model mice. J Biol Chem. 2006;281:36180–6. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]
- 138.Cabrejo L, Guyant-Marèchal L, Laquerriëre A, Vercelletto M, De La, Fourniëre F, Thomas-Antèrion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D. Phenotype associated with APP duplication in five families. Brain. 2006;129:2966–76. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- 139.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriëre A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature Genet. 2006;38:24–6. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 140.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–88. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 141.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–64. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 142.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–70. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 143.Small DH, Mok SS, Bornstein JC. Alzheimer's disease and Aβ toxicity: from top to bottom. Nature Rev Neurosci. 2001;2:595–8. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- 144.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR, Whitehouse PJ. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–42. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 145.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–64. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 146.Steinerman JR, Irizarry M, Scarmeas N, Raju S, Brandt J, Albert M, Blacker D, Hyman B, Stern Y. Distinct pools of β-amyloid in Alzheimer disease-affected brain: a clinicopathologic study. Arch Neurol. 2008;65:906–12. doi: 10.1001/archneur.65.7.906. [DOI] [PMC free article] [PubMed] [Google Scholar]