

Abstract
The p53 tumour suppressor protein lies at the crossroads of multiple cellular response pathways that control the fate of the cell in response to endogenous or exogenous stresses and inactivation of the p53 tumour suppressor signalling pathway is seen in most human cancers. Such aberrant p53 activity may be caused by mutations in the TP53 gene sequence producing truncated or inactive mutant proteins, or by aberrant production of other proteins that regulate p53 activity, such as gene amplification and overexpression of MDM2 or viral proteins that inhibit or degrade p53. Recent studies have also suggested that inherited genetic polymorphisms in the p53 pathway influence tumour formation, progression and/or response to therapy. In some cases, these variants are clearly associated with clinico-pathological variables or prognosis of cancer, whereas in other cases the evidence is less conclusive. Here, we review the evidence that common polymorphisms in various aspects of p53 biology have important consequences for overall tumour susceptibility, clinico-pathology and prognosis. We also suggest reasons for some of the reported discrepancies in the effects of common polymorphisms on tumourigenesis, which relate to the complexity of effects on tumour formation in combination with other oncogenic changes and other polymorphisms. It is likely that future studies of combinations of polymorphisms in the p53 pathway will be useful for predicting tumour susceptibility in the human population and may serve as predictive biomarkers of tumour response to standard therapies.
Keywords: p53, polymorphism, MDM2, R72P SNP
Introduction
TP53 is well known as the most commonly mutated gene in human cancer [1]. The gene product is a 53-kD phosphoprotein of 393 amino acids, forming five highly conserved regions and four functional domains [2]. In response to a stress signal, the p53 protein is activated by post-translational modifications. These modifications stabilize p53, intracellular levels rise and p53 is activated as a transcription factor to direct stress-specific transcriptional response programs, leading to cell cycle arrest, cell senescence or apoptosis. p53 acts as a central node in the cellular network of stress responses, with multiple divergent inputs that regulate p53 activity and multiple outputs from activated p53 [3, 4]. The mutations of TP53 gene seen in human cancers result in an inactive protein which is unable to initiate appropriate stress responses, leading to both enhanced tumouri-genic potential and impaired responses to therapeutic agents. In addition to mutations in the TP53 gene coding sequence, p53 activity may also be decreased by alterations of genes that act as regulators of p53 activity. MDM2 is one major regulator, which degrades p53 and the importance of this regulatory pathway is underscored by the observation of overexpression of MDM2 by gene amplification in a significant proportion of human cancers [5]. The ability of another member of the p53 family, p73, to interact with and partially compensate for loss of p53 activity is another emerging factor of importance for p53 function and has been suggested to be important for tumour response to some genotoxic therapies [6].
It is therefore conceivable that the existence of natural variants of p53 and other proteins that influence p53 activity could be linked with the development of tumours in the human population and could represent predictive biomarkers for preventive and early intervention strategies. A total of 38 polymorphisms in the TP53 gene are referred to in SNP500 Cancer Database [7] (http://snp500cancer.nci.nih.gov/home_1.cfm). Most of these natural variants are localized in introns, although polymorphisms also exist in exons of TP53. Some of these TP53 polymorphisms have been analysed in large populations (Fig. 1) whereas other polymorphisms have been described rarely and their significance remains to be determined. In addition, polymorphisms in genes that regulate p53 or are regulated by p53 have been described. Here, we review the evidence that polymorphisms in the p53 pathway are important determinants of cancer predisposition in the human population.
1.
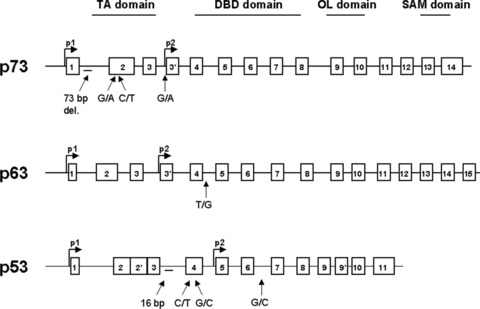
TP53, TP63 and TP73 gene structure and the positions of the most important polymorphisms.
SNPs in TP53 exons altering amino-acid sequence
The substitution of proline (codon CCG) to serine (codon TCG) at residue 47 (P47S), and the substitution of arginine (codon CGC) to proline (codon CCC) at residue 72 (R72P), both localized in exon 4, are among the most intensively studied polymorphisms found in the coding regions of TP53 which alter the amino-acid sequence. Felley-Bosco et al. first identified single nucleotide polymorphism (SNP) P47S and found that polymorphic variant S47 is very rare in African Americans and undetectable in Caucasians [8]. In vitro studies have shown that the S47 variant is not as good a substrate for the nearby phosphorylation at S46 by p38 mitogen-activated protein kinase (MAPK) and has impaired pro-apoptotic ability due to a decreased transcriptional activation of p53AIP1 and p53-upregulated modulator of apoptosis (PUMA) [9]. Although these data suggest that individuals with this polymorphism will be at higher risk for tumour development and have a relatively impaired response to therapy, there are as yet no firm data in large population studies that confirm these suggestions, partly due to the rarity of the polymorphism.
The codon R72P SNP results in a structural change of the protein [10, 11]. Further, this polymorphism in exon 4 is located in the proline-rich domain, which was shown to be important for the apoptotic function of p53 (Table 1) [12]. A significant difference in P72 allele frequency was found between a Nigerian population (African Black) and a Swedish population (Western Europe), which were 17% and 63%, respectively. In contrast, no difference was found between populations living on the same geographical latitude [13]. This variation in frequency from the equator to higher latitudes suggests a selection pressure upon these two forms of p53 protein. By comparison with chimpanzee DNA, it is evident that the P72 allele is the older allele, currently present at a higher frequency in Africa, while the R72 allele arose later in Caucasians and Asians.
1.
The roles of R72P polymorphism in cancer
| Phenomenon | p53 status | Mechanism | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| Apoptosis | Wt Wt | The R72 allele correlates with more efficient induction of apoptosis compared to the P72 variant. | [15, 20] | |||||
| Degradation | R72 allele has greater capacity to interact with MDM2. | [15] | ||||||
| R72 was found more efficiently targeted for degradation by the E6 protein of HPV16. | [22] | |||||||
| Gain of function | mut | The R72 allele is preferentially mutated and these mutants confer stronger affinity for interaction with p73 followed by its inactivation. | [16–18] | |||||
| Response to chemotherapy | Wt | R72 allele cases have higher response rates and longer survival than those with P72. | [20] | |||||
| P72 variant is more frequent and tumours are less sensitive to apoptosis-inducing treatment. | [32] | |||||||
| mut | Cancers expressing R72 mutants have lower response rates than those expressing P72 mutants. | [16] | ||||||
| R72 mutants tend to confer resistance to anticancer drugs, but it is not an universal phenomenon and depends on the mutation and drug utilization. | [128] | |||||||
| Reparation | Wt | P72 is more efficient than R72 in specifically activating several p53-dependent DNA-repair target genes. | [21] | |||||
Several lines of evidence indicate that this polymorphism can play a role in apoptosis and cancer formation in humans. This is supported by observations that iASPP, a specific cellular inhibitor of p53 that binds to this proline-rich domain of p53, interacts more strongly with the P72 variant leading to inhibition of apoptosis [14]. In contrast, Dumont et al. showed that stronger interaction of the R72 variant with the nuclear-export protein CRM1 leads to enhanced nuclear export and accumulation in mitochondria resulting in release of cytochrome c into the cytosol [15]. Additionally they suggested that the R72 form had a higher affinity for MDM2 than the P72 form resulting in enhanced ubiquitination by this ubiquitin ligase [15]. The ability of mutant p53 proteins to bind p73 and inhibit its apoptotic activity in tumours is another important factor (Table 1). Interestingly, it seems that the interaction of p53 with p73 is influenced by a common TP53 R72P polymorphism, since the binding of mutant p53 with R72 variant is stronger in comparison with the P72 variant and generates mutants with strong ‘gain of function’[16, 17]. Moreover Marin et al. reported a higher frequency of TP53 mutations on the R72 allele compared with the P72 allele in different squamous cell cancers [17]. This proposal is supported by Tada et al., who analysed mutations on the R72 allele in tumours from different tissues. Interestingly, they found a preferential selection of the R72 allele in cancers with recessive TP53 mutants (mutants that do not inactivate wild-type TP53 in a dominant negative manner). It was suggested that recessive TP53 mutants achieve a selective growth advantage by an R72-dependent inactivation of TP73, whereas the dominant negative TP53 mutants inactivate the remaining wild-type TP53 allele in an R72-independent manner [18]. Additional report indicate that ovarian cancer patients with the P72 allele and non-missense sequence variants or missense sequence variants affecting L2 or L3 of the p53 protein have significantly poorer disease-specific survival compared to those with wild-type or other sequence [19]. This is tightly linked with the fact that the two allelic forms of mutant p53 confer different cellular resistance to anticancer agents, linking that effect to the outcome of chemotherapy in carcinomas expressing mutant p53 (Table 1) [16, 17]. However, the in vitro response of cells exposed to anticancer agents is strongly influenced by this SNP in wild-type p53 as well [20]. To address the in vivo biological significance of the different molecular properties of the R72 and P72 variants, the responses of a well-defined series of patients with head and neck cancer to chemo-radiotherapy regimens were analysed. In this cohort of patients, the response rates and the survival (both overall and progression free) were significantly higher in cases retaining a wild-type p53 R72 allele [20]. The wild-type p53 P72 variant transcriptionally activates several p53-dependent target genes involved in DNA repair better than the p53 R72 form [21]. Concomitantly, cells expressing the p53 P72 form were able to repair DNA damage much more efficiently than the p53 R72-expressing cells.
In other work, Storey et al. presented results indicating that p53 R72 is significantly more susceptible to degradation by the high-risk HPV (human papillomavirus) E6 protein in comparison with p53 P72 [22]. Further small-scale population analysis of women in Italy revealed that this increased susceptibility to degradation by E6 is associated with higher incidence of cervical cancer in HPV-infected women who were homozygous for the R72 variant [22]. Although there have been several reports worldwide, the question whether [23] or not [24–26] p53 R72P polymorphism represents a significant risk factor in the development of cancer associated with HPV remains uncertain.
A number of studies have attempted to determine if there is an association between codon 72 polymorphic variants of TP53 and increased risk for particular types of cancer (Table 2). These studies vary in their conclusions, as some report increased risk associated with the P72 allele for certain cancer types and others fail to show any significant associations. For example, a strong association between the R/R genotype and breast cancer was reported in Turkish patients [27]. Similarly, Langerod et al. reported association of R72 allele with TP53 somatic mutations resulting in a growth advantage of breast carcinoma cells with this allele in a Norwegian population [28], and Kalemi et al.reported the R/R genotype as a risk factor for breast cancer in a Greek population [29]. In contrast, Tommiska et al. as well as Khadang et al. did not observe any association between TP53 codon 72 variants and breast cancer risk [30, 31]. These discrepancies can be explained by suggesting a more complex role of p53 R72P polymorphism in carcinogenesis. On the one hand, owing to R72 possessing a more potent apoptotic activity in comparison to P72, individuals with this SNP will have higher cancer susceptibility compared to those carrying R72. On the other hand, in tumours with p53 mutations, mutant p53 encoded by the R72 allele may bind to and inhibit p73 pro-apoptotic activity. Therefore, in tumours with wild-type p53 the P72 variant is more frequent and tumours are less sensitive to apoptosis-inducing treatment, whereas the R72 variant exerts its effects mainly in tumours with p53 mutations. This proposal is supported by studies of head and neck tumours bearing p53 mutations, with the R72 allele conferring resistance to chemotherapy and leading to shorter time of survival [32]. Additional reasons for these discrepancies could be that the biological impact of this SNP is influenced by other important factors, such as heterogeneity of p53 mutations, tissue specificity and genetic background, that act together with other unidentified factors in the selection process. For example, several epidemiological studies have reported an association between the presence of a P/P genotype and the risk for lung adenocarcinoma development [33–35]. Interestingly, an R/R genotype confers increased risk of squamous cell carcinoma in individuals deficient in a metabolizing enzyme involved in the detoxification of tobacco carcinogens [34]. This is consistent with the preferential mutation of the p53 R72 allele found in squamous tumours [17]. On the basis of these studies, it can be argued that the presence of a P/P genotype affects overall lung cancer development because of its poorer apoptotic properties. However, the absence of detoxifying enzymes may lead to a longer exposure to active carcinogens and the selection for mutant p53 that carries the R72 polymorphism, which would more effectively block the p73 safeguard apoptotic function [36].
2.
Discussed TP53 polymorphisms in cancer epidemiology
| Polymorphism of TP53 gene | Cancer | Population / other comments | Allele or Genotype | Association with cancer risk / OR (95% CI) | Ref. | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R72P | |||||||||||||||||||
| Breast | Finnish | No | [30] | ||||||||||||||||
| Familial: 0.94 (0.80–1.09) | |||||||||||||||||||
| Unselected: 0.96 (0.82–1.12) | |||||||||||||||||||
| Iranian | No | [31] | |||||||||||||||||
| / | 1.1 (0.84–1.44) | ||||||||||||||||||
| Greek | RR | Yes | [29] | ||||||||||||||||
| / | 6.66 (2.63–16.9) (P= 0.0001) | ||||||||||||||||||
| Turkish | RR | Yes | [27] | ||||||||||||||||
| 3.05 (1.19–7.8) | |||||||||||||||||||
| (P = 0.017) | |||||||||||||||||||
| Chinese | R | Yes | [21] | ||||||||||||||||
| / | (P < 0.001) | ||||||||||||||||||
| Japanese | PP | Yes | [129] | ||||||||||||||||
| ER positive | 2.04 (P= 0.04) | ||||||||||||||||||
| Cervical | Peruvian | No | [25] | ||||||||||||||||
| Japanese | No | [26] | |||||||||||||||||
| Portuguese | / | No | [24] | ||||||||||||||||
| Indian | RR | Yes | [23] | ||||||||||||||||
| 2.59 (1.18–5.67) (P= 0.038) | |||||||||||||||||||
| SCCHN | non-Hispanic whites | No | [38] | ||||||||||||||||
| // | 1.04 (0.75–1.44) | ||||||||||||||||||
| Lung | Caucasian | RP PP | Yes | [60] | |||||||||||||||
| / | 1.34 (1.03–1.74) 1.47 (0.90–2.40) | ||||||||||||||||||
| Japanese | PP | Yes | [130] | ||||||||||||||||
| 2.2 (1.3–3.9) | |||||||||||||||||||
| (P= 0.005) | |||||||||||||||||||
| Glioma | Portuguese | No | [41] | ||||||||||||||||
| 1.07 (0.66–1.72) (P= 0.765) | |||||||||||||||||||
| American | P | Yes | [131] | ||||||||||||||||
| (P < 0.001) | |||||||||||||||||||
| PIN3 Ins 16bp | |||||||||||||||||||
| Breast | Unknown | / | No | [50] | |||||||||||||||
| Portuguese | A2A2 | Yes Familial: 4.4 (1.60–12.00) Sporadic: 3.88 (1.18–12.8) | [49] | ||||||||||||||||
| Blader | American | No | [52] | ||||||||||||||||
| Gastric | Korean | No | [51] | ||||||||||||||||
| (P= 0.1659) | |||||||||||||||||||
| Ovarian | German | A2A2 | Yes 8.64 (2.97–25.16) | [48] | |||||||||||||||
| Lung | Caucasian | A1/A2 A2/A2 | Yes 1.59 (1.17–2.15) 1.63 (0.72–3.72) | [60] | |||||||||||||||
| PIN6 G13494A | |||||||||||||||||||
| Breast | Caucasian | No | [54] | ||||||||||||||||
| Ovarian | German | Yes | [53] | ||||||||||||||||
| 1.93 (1.27–2.91) | |||||||||||||||||||
| Ovarian | Caucasian | Yes | [54] | ||||||||||||||||
| PIN6 G13964C | |||||||||||||||||||
| Breast | Australian | No | [57] | ||||||||||||||||
| Poland | No | [58] | |||||||||||||||||
| American | Yes | [56] | |||||||||||||||||
| (P= 0.0003) | |||||||||||||||||||
While correlations between cancer risk and the codon 72 polymorphism are somewhat inconsistent, there are much firmer associations with cancer progression, age of onset, overall survival, etc. For example, survival analysis of 888 unselected breast cancer patients revealed that patients with P72 homozygous genotype had significantly poorer survival than patients with other genotypes (P= 0.003). This effect on survival was independent of p53 expression in the tumours and multivariate analysis showed that P72 homozygous genotype was overall an independent prognostic factor (risk ratio of death, 2.1; 95% CI, 1.4–3.3; P= 0.001) [30].
Interestingly, a recent study analysed 93 patients diagnosed with Hereditary Nonpolyposis Colorectal Cancer; these individuals had germline mutations in the mismatch repair genes MLH1 or MSH2. Individuals with at least one copy of the P72 allele had a median age of onset for disease 13 years earlier than individuals who were homozygous for the R72 allele [37]. Analyses by other groups have revealed an 11 year earlier age of onset in individuals with at least one P72 allele for oral cancer, and a 6-year earlier age of onset for squamous carcinoma of the head and neck [38]. These trends may be explained by the increased apoptotic potential of the R72 variant, as well as the findings that individuals with the R/R genotype have higher response rates and better survival after receiving chemo- and radiation therapy for advanced head and neck cancer [20] and for cancers of the breast and lung [30, 39, 40]. These data are in contrast with recently published results of Lima-Ramos et al., who determined the impact of R72P SNP on patient survival and therapeutic response in 171 glioma patients in Portugal. They did not observe an association of TP53 R72P polymorphism with glioma risk and overall patient survival. However a significant association was observed in glioma patients harbouring the P72 allele and their response to adjuvant therapy [41].
Polymorphism in TP53 introns
There is growing evidence that mechanisms of gene regulation, including mutations in enhancers, splice donor and acceptor sites of introns, and promoter elements, could play an important role in the regulation of gene expression, including p53 [42, 43]. In particular, mutations in intron sequences may initiate aberrant pre-messenger RNA splicing, producing mRNA that may be translated into a defective protein. Furthermore, introns are often involved in regulation of gene expression and DNA-protein interactions and mutations in intron sequences may affect these functions. Therefore the identification of mutations in non-coding sequences that result in aberrant gene expression may provide useful tools for the detection of patients with risk of the development of cancer.
The IARC TP53 Mutation Database lists 29 common polymorphisms in the non-coding region of TP53, of which a 16bp duplication in intron 3 localized at nucleotide 11951 (+16) [44, 45], G to A transversion in intron 6 at nucleotide 13494 (G13494A) often reported as MspI[46] or BstNI/NciI polymorphisms [47] and a G to C transversion in intron 6 at nucleotide 13964 (G13964C) [44, 45] have been suggested to impact on the function or level of expression of p53 to date. Studies of the association between these intronic polymorphisms and cancer predisposition have revealed inconsistent results. For example, although Runnebaum et al. noted an eightfold elevated risk for ovarian cancer in patients with the intron 3 variant [48] and Costa et al. suggest this polymorphism as a risk modifier in sporadic and familial breast cancer [49], other studies found no such association [50–52].
Clinical study of 310 German Caucasian ovarian cancer patients and 364 healthy controls for association of intron 6 G13494A polymorphism showed that the individuals who carried the rare MspI allele have an overall 1.93-fold increased risk (95% CI 1.27–2.91) of ovarian cancer [53]. Similarly Mavridou et al. described a statistically significant difference in the distribution of the G13494A polymorphism in intron 6 between healthy Caucasian control subjects and patients with ovarian cancer, but found no difference between control subjects and patients with breast cancer [54]. In contrast, in a small set of samples, Peller et al. reported a statistically significant association between the G13494A polymorphism and the risk of breast and colon cancers [55].
In case of G13964C polymorphism, Lehman et al. detected the rare C allele in 3 of 42 American hereditary breast cancer patients, compared to 0 of 171 control sporadic breast cancer patients [56]. Moreover, they demonstrated prolonged in vitro survival in response to cisplatin treatment in lymphoblastoid cell lines derived from the heterozygous GC patients and showed decreased chemotherapy-induced apoptosis [56]. Immunohistochemical analysis of breast tumours from these patients revealed high levels of mutant p53 protein, suggesting a functional mutation in the TP53 gene. However, contradictory data were reported by Marsh et al., who found the G and C variants at the same frequency in Australian hereditary breast (ovarian) cancer syndrome patients (3/71) and in healthy control individuals (5/143) [57]. Similarly, there was no evidence of the association of the 13964C variant with a high risk of cancer development in a series of Polish cancer families previously screened for germline mutations in p53 [58].
Linkage between the polymorphisms
Haplotype is a set of single nucleotide polymorphisms on a single chromatid that are statistically associated. Knowledge of the accumulated number of variants on the same chromosome may provide additional information about changes in p53 function because there could be additive or multiplicative changes in function. Interestingly, several studies have found that these polymorphisms in exon 4, introns 3 and 6 are in strong linkage disequilibrium. In vitro experiments revealed that cell lines with at least one variant allele at all three polymorphic sites had a statistically significantly reduced DNA repair capacity in comparison with cell lines with all wild-type alleles. It remains to be more precisely determined whether the effect of the combined polymorphisms occurs also in vivo, however, reduced DNA repair capacity and apoptotic responses have been linked to an increased risk of lung cancer [59, 60]. Further it was also determined that the p53 hap-lotypes were associated with risk for lung [60–62], colorectal [63], breast [33, 64] and other cancers [65, 66].
Generally these data highlight the value of examining multiple polymorphisms in TP53 gene, which could be critical for risk of developing tumours, risk of progression, and prognostic effects. For example, occurrence of the multivariant alleles from TP53 exon 4 and intron 3 results in a significantly poorer prognosis in non-small cell lung cancer [67], and both TP53 polymorphisms P72 and 16bp insertion are associated with higher incidence of lymph node metastases [49]. Further studies are needed to determine the mechanism by which exon 4 and introns 3 and 6 variants modify cancer risk.
Silent mutations and SNPs in TP53
Silent mutations are a potentially interesting group of TP53 gene polymorphisms. These SNPs change one codon for another synonymous one, and for this reason do not affect the structure of the gene product. The predominant view is that these synonymous mutations are effectively neutral and consequently they play no role in carcinogenesis. Nevertheless it has been proposed that silent mutations are not necessarily neutral [68, 69]. Silent mutations may affect the function of the gene in many different ways. The most classical view is related to the different usage of synonymous codons resulting in different translational rate [70, 71]. Another possible explanation is that alterations in mRNA splicing are an important cause of genetic diseases, and in particular they are relatively common in cancer [72]. Point mutations may either destroy or create donor or acceptor splicing sites or alter mRNA folding which can influence splicing, processing or translational control and regulation. Splicing mutations that fall in splicing junctions (donor or acceptor sites) are relatively rare in TP53, accounting for less than 1% of the database [73]. In some cases the mutations altering the splicing site are silent, such as the case of the silent mutation at codon 125 of TP53 (ACG→ACA), which has been experimentally demonstrated to generate aberrant transcripts [74]. Exonic splicing enhancers are the second type of splicing signals whose alteration is more likely to be associated with genetic disorders. Splicing enhancers are degenerate motifs located inside exons that are recognized by splicing factors that are critical for exonic recognition. Nucleotide mutations inside these motifs may disrupt splicing by directly inactivating the ability of the spliceosome machinery to recognize them, leading to exon skipping. It has been estimated that up to 50% of point mutations responsible for genetic diseases cause aberrant splicing [75–77]. It was shown that silent mutations, especially those mutations that were always found as singlet and never as a doublet or as a multiplet, tend to be preferentially located inside candidate exonic splicing enhancers motifs [68]. Moreover in TP53, all exonic CpG sequences are methylated. This epigenetic modification is correlated with frequent G:C→A:T transitions [78]. Interestingly, it was found that silent mutations are non-random, mostly involving G:C→A:T transitions (62%) as well [79]. It is also the case of the polymorphism in codon 36 (CCG→CCA) in TP53 gene described by Felix et al.[80]. This polymorphism has been described in several publications; however, no association with a specific phenotype has been described so far.
Polymorphisms in other members of the p53 family
The discovery of the p53-related proteins p63 and p73, which are able to regulate the cell cycle and apoptosis after DNA damage, leads to evaluation of the status of a network that contains p53, p73 and p63 in predicting prognosis and response to chemotherapy.
p63 is actually the eldest evolutionary conserved member of the p53 family phylogenetically [81]. The TP63 gene is commonly overexpressed in certain human cancers, but mutations are very rare. Inherited mutations cause various developmental syndromes of the cranio-facial and skeletal abnormalities [82], but polymorphisms that associate with cancer incidence have not been described. In tumours, mutant p53 can enhance p63 survival functions and inhibit p63 apoptotic functions in a mutant-specific manner, although the influence of the TP53R72P variant in this process has not been tested (unlike the effects on TP73 noted below).
The human TP73 gene is composed of 15 exons and its expression is complicated by the presence of at least seven alternatively spliced C-terminal isoforms and of at least four alternatively spliced N-terminal isoforms [83]. Extensive molecular analyses of p73 in various tumours suggested that mutations in the TP73 gene are rare; however, loss of heterozygosity at the TP73 locus has been reported at various frequencies in different tumours. Furthermore the expression of the TP73 gene is commonly altered in human malignancies, implying that this gene may have important roles in tumourigenesis [84, 85].
TP73 gene can be transcribed from two distinct promoters, driving the expression of p53-like proteins containing the transac-tivation domain (TAp73), and proteins lacking TA, called ATAp73. The TAp73 isoforms are able to bind specifically to DNA through p53 response elements (RE) and activate transcription of target genes. Like p53, such activation can induce cell cycle arrest or apoptosis [83]. ANp73 acts as a potent transdominant inhibitor of TAp63, TAp73 and wild-type p53 [86]. Thus, theoretically, p73 expression can be either pro- or anti-oncogenic, depending on whether anti- or pro-apoptotic (ATAp73 or TAp73, respectively) isoforms are expressed. In keeping with the theory, there is evidence for oncogenic or tumour suppressor functions of TP73 in different human tumours [87].
It is unknown whether the alteration of p73 expression has any genetic basis such as sequence variations or polymorphisms. At least 20 single-nucleotide polymorphisms have been identified in TP73 (some in exons and others in introns), but none cause an amino acid change. Four polymorphisms have been suggested to alter TP73 gene expression: a 73 bp deletion in intron 1 and three single nucleotide polymorphisms: two in exon 2 and one in promoter 3 (Fig. 1). Whereas the promoter 3 variant did not have a significant biological effect, there are several studies indicating that the two SNPs localized at position 4 (G to A) and 14 (C to T) in the 5’-untranslated region of exon 2 of TP73 gene may be associated with an increased risk of certain types of cancer. These two polymorphisms are in complete linkage disequilibrium with one another and form a polymorphism referred to as the AT and GC alleles. This polymorphism lies upstream of the initiating codon AUG of exon 2, a region which may theoretically form a stemloop structure that could potentially affect gene expression through alteration of the efficiency of translation initiation [88]. To test the hypothesis that the TP73 G4C14 to A4T14 polymorphism is associated with risk of cancers, several hospital based case-control studies have been done (Table 3). These studies provide evidence that TP73AT variant is significantly associated with an increased risk of squamous cell carcinoma of the head and neck (SCCHN), endometrial cancer and lung cancer [89–91]. On the other hand, some studies suggest that the AT variants possess a protective role against cancer development [92, 93] and a study of 526 breast cancer patients showed an association of AT variant with long-term survival [94]. Although inconsistent, these results raise the possibility that analysis of TP73 polymorphism may provide useful prognostic information for cancer patients. Nevertheless, additional independent studies are needed to confirm these findings.
3.
Discussed TP73 polymorphisms in cancer epidemiology
| Polymorphism | Comments | Ref. | ||
|---|---|---|---|---|
| G4C14 to A4T14 | ||||
| Prognosis | 708 SCCHN patients and 1229 cancer-free controls. Genotypes (GC/AT + AT/AT) were associated with significantly increased risk for SCCHN (OR = 1.33, 95% CI = 1.10–1.60). | [89] | ||
| 1054 lung cancer patients and 1139 cancer-free controls. Genotypes (GC/AT + AT/AT) were associated with significantly increased risk for lung cancer (OR = 1.32, 95% CI = 1.10–1.59). | [90] | |||
| 114 endometrial cancer patients and 442 controls. Association between the TP73 AA genotype and an increased risk of endometrial cancer (OR = 2.82, 95% CI = 1.36–5.82). | [91] | |||
| Protective role of AT variant | 84 oesophageal cancers (25 squamous and 59 adenocarcinoma) and 152 cancer-free controls. | [92] | ||
| AT/AT homozygotes were less prevalent in the cancer population (1.2%) compared to controls (9.9%) (P < 0.02), OR = 0.11 (95% CI = 0.02–0.6). | ||||
| 425 lung cancer patients and 588 cancer-free controls. AT haplotypes were less common in the cancers (P= 0.0018). Compared to the TP73 GC/GC homozygotes, both the AT/AT variant homozygotes and GC/AT heterozygotes were associated with significantly decreased risk (OR = 0.45, 95% CI = 0.26–0.80 and OR = 0.70, 95% CI = 0.53–0.92, respectively). | [93] | |||
| Association with cancer risk | Study of 526 breast cancer patients with a median follow-up of 7.3 years.GC/GC genotype was associated with worse clinical outcome (P= 0.02).GC/GC genotype remained an independent indicator of poor prognosis (disease-free survival: HR = 1.82, P= 0.003 and overall survival: HR = 1.99, P= 0.004) in multivariate analysis. | [94] | ||
| Intron 1 del 73 bp | ||||
| Tumour and normal tissue from 81 colorectal cancer patients. 73 bp deletion was found in at least one allele in 40.7% of the patients, its presence was associated with advanced stages (P= 0.03), vascular invasion (P= 0.02), and lymph node metastases (P= 0.04). Statistical association was found between the presence of the deletion in hemi- or homozygosis and low levels of the TAp73 suppressor isoforms. | [96] | |||
| 45 colorectal and 43 breast cancer patients and 34 healthy controls. Allele with Intron 1 del 73 bp was significantly associated with increased risk of tumour (P= 0.045). | [95] | |||
A novel regulatory region of p73 expression has been identified in a 1 kb stretch of intron 1 upstream of the initiating ATG codon in exon 2. This region contains six sites for binding of the tran-scriptional repressor ZEB1 (δEF1), whose inhibition leads to increased expression of p73. Interestingly the 73 bp deletion in intron 1 is localized just between −489 and −417 of this negatively regulated fragment and was observed to be statistically more frequent in individuals with a diagnosis of breast or colorectal carcinomas compared to healthy controls [95]. An expression-based analysis of primary tumour material suggests that the 73 bp deletion in the first intron of the TP73 gene and different expression levels of ZEB1 and p300 may act in concert to affect the ratio of TAp73/ATAp73 forms, favouring p73 oncogenic variants (Fig. 2A). In addition, up-regulation of p73 oncogenic isoforms predicts a poor prognosis based on its relationship with advanced tumour stage [87, 96]. However, it is still unclear whether the presence of the deletion increases tumour susceptibility, or whether the effects are seen only on prognosis and further large-scale studies are required. In addition, the influence of this polymorphism on p53 responses are unclear, but might for example be expected to diminish even further the relatively decreased pro-apoptotic ability of the P72 allele of TP53, due to an increase in ΔNp73/p73 ratios, enhancing tumour susceptibility. As noted above, in tumours with mutant p53, an opposite effect on prognosis may be seen. To date no studies have investigated these possibilities.
2.
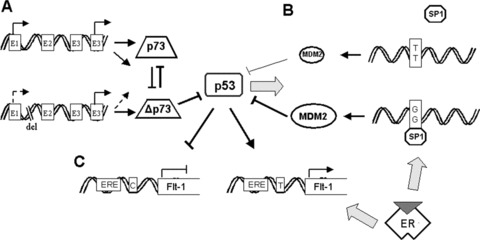
Polymorphisms in p53 pathways. (A) Role of p73 polymorphism. The presence of 73 bp deletion in the first intron of the TP73 gene is associated with lower levels of p73 but do not affect expression of Δp73 resulting in increased ratio of TAp73/ATAp73 forms, favouring p73 oncogenic variants. (B) Role of MDM2 SNP309. Single nucleotide polymorphism 309 in a region of the MDM2 promoter increases the affinity of the transcriptional activator Sp1, which along with active oestrogen receptors (ER) and genotoxic stress, was found to attenuate the p53 pathways resulting in the acceleration of tumour formation. (C) Role of RE polymorphisms. C to T SNP at the Flt-1 promoter creates a p53 binding site resulting in p53-dependent up-regulation of Flt-1 transcription in human cells. Active oestrogen receptor signalling considerably enhances this effect.
Polymorphisms in p53 target genes
There is clear evidence that increases in the levels of the MDM2 protein attenuate p53 function, leading to cancer formation [97]. A SNP at nucleotide 309 which lies in intron 1 and is within the MDM2 promoter was found to be associated with the attenuation of the p53 pathway and with the acceleration of tumour formation in humans [98]. The SNP changes a T to a G (the G allele is designated SNP309, and occurs at high frequency) creating a higher affinity Sp1 binding site, which is suggested to increase MDM2 mRNA levels (Fig. 2B). In most publications, the association of MDM2 SNP309 with cancer was connected with the Li-Fraumeni syndrome or with p53 mutational status [99–101]. Studies associating SNP309 with sporadic cancer risk have provided conflicting results: either no increased risk of breast and ovarian cancer was observed [102, 103] or accelerated tumour and hormone-dependent manner formation was seen in a gender-specific manner [104, 105]. In our study, we could find no association with SNP309 and cancer risk in a series of 158 breast, 17 endometrium, 13 cervix and 45 ovarian cancers. Moreover, we could not find an association of SNP309 with increased MDM2 mRNA, in contrast to previous findings in a small panel of cell lines that indicated an average eightfold higher level of MDM2 mRNA in GG genotype cells [106], nor could we find an increase in MDM2 protein levels in primary tumour material [107]. These data are similar to those seen in mantle cell lymphomas [108]. Thus, neither primary human lymphomas nor the common epithelial malignancies that we studied show a significant role for SNP309 in regulating MDM2 protein levels. We also showed that p53 protein levels were unaffected by SNP309. In summary, the results show no association between SNP309 with breast and other cancer risks, and the presence of G alleles does not increase MDM2 or decrease p53 protein levels in primary human tumours. Thus, the role of MDM2 SNP309 outside of Li-Fraumeni patients is unclear.
A major downstream component of the p53 tumour suppressor pathway is p21 af1, which inhibits progression of the cell cycle from G1 to S phase by inhibiting cyclin-dependent kinases [109]. Mutations in p21 a are extremely rare; however, several polymorphisms have been described. The polymorphism at codon 31 results in a non-conservative amino-acid change from serine to arginine (S31R). Interestingly, this SNP appears to occur more frequently in cancer patients manifesting wild-type p53 [110]. The frequency of the R allele varies dramatically between major ethnic groups and functional analyses revealed that codon 31 polymorphism does not affect the structure or function of the protein [111]. More recently, Su et al. reported that the codon 31 polymorphism alters mRNA expression of p21 Waf1[112]. A number of studies have been performed to examine the involvement of the p21 Waf1 codon 31 polymorphism on cancer susceptibility. Nevertheless, conclusions are inconsistent, as approximately half of these studies support the idea that this polymorphism correlates with cancer risk while others refute this claim [113]. Several studies also examined whether the codon 31 polymorphism may influence cancer prognosis and progression. Studies on a Taiwanese population demonstrated that individuals carrying the S/S genotype had a shorter post-operative survival compared to those with the S/R or R/R genotype [114]. On the other hand, this polymorphism was not found to affect survival in a population of Caucasian Australian breast cancer patients [115].
The S31R polymorphism is also linked to a polymorphism in the 3’UTR of the p21 Waf1 gene. Functionally, the 3’UTR polymorphism may also influence p21 Wa mRNA stability and the codon 31 polymorphism combined with the polymorphism in the 3’UTR may contribute jointly to the development of squamous cell carcinoma of the head and neck, prostate adenocarcinoma and breast tumours [110, 116].
Considering the fact that p53 binds and transcriptionally regulates several hundreds of genes, it could be expected that many of these genes harbour genetic polymorphisms influencing their function. To labour all of them is outside the remit of this review, therefore only several other examples are mentioned: (i) PIG3 pentanucleotide microsatellite repeat, which leads to increased p53 inducibility [117]; (ii) 5’ untranslated region of the BAX pro-moter G(-248)A SNP was reported to be associated with both reduced expression of BAX and altered susceptibility to chronic lymphocytic leukaemia [118, 119]; (iii) BCL2SNP C(-838)A in the promoter region is responsible for decrease prostate cancer risk [120]; (iv) several SNPs were also reported in FAS gene to be associated with increased risk of breast, cervical, melanoma, lung and other cancers [121–123].
Polymorphisms in p53 response elements
The main functions of p53 as a tumour suppressor lie in the ability to transcriptionally regulate genes through binding to the p53 response element. A computational search of SNPs in putative p53 REs in promoter regions identified hundreds of SNPs that alter the sequence of p53 REs [124]. Functional analysis of a small subset of these, including genes with known roles in tumourigen-esis showed that the variant SNPs render the REs less able to mediate p53-induced transcription. Thus, there are numerous genes that are involved in mediating p53 pathway responses that are functionally polymorphic for p53 activation [125]. For example, a C-to-T polymorphism in the VEGF receptor/Flt-1 promoter creates a p53 RE in the less common Flt-1T variant (6% of the population) and only this variant induces expression after p53 activation (Fig. 2C) [126]. Further studies have also demonstrated the critical role for oestrogens identifying the second regulatory sequence within the Flt-1 promoter harbouring a partial response element for oestrogen receptors upstream of the p53 binding site [127]. Their finding suggested that this polymorphism could influence tumour formation/progression in a hormone-sensitive manner. There are no studies to date that have investigated the epidemiology of these SNPs in human cancers, but it seems likely that their influence on the ability to respond to wild-type p53 will affect cancer risk and response to treatment [125].
Concluding remarks
One of the goals of translational oncology is to identify the molecular markers predictive of treatment outcome. Such predictive biomarkers would be of particular value for treatment regimens, such as combined modality therapy, which are associated with severe morbidity and substantial expense, yet produce complete response in only a subset of patients. As such, it is important to find the molecular genetic determinants of treatment outcome to facilitate identification of patients most likely to benefit from such treatments.
p53, a prominent tumour suppressor protein, is a master regulator that targets over a hundred genes for transcriptional up-regulation or repression through sequence-specific interactions with DNA response elements. The precise mechanism by which p53 is inactivated during tumourigenesis may influence its tumour suppressor activity in different ways or to different extents and influence the resulting tumour phenotype. Functionally important polymorphisms in the TP53 gene exist, as well as the SNPs in p53 response elements of many target genes. The discovery of these individual differences has implications for variation in human responses to environmental stresses, risk of disease, and responsiveness to drug therapies. The data we present in this review suggest that analysis of polymorphism in p53 is one potentially useful marker. Confirmation of the usefulness of analysis of this SNP as a biomarker of treatment outcome should prompt serious consideration of its use in the routine work-up of patients prior to treatment decisions being made. However, as a consequence of the complexity of the p53 pathway, there is a need to perform large-scale studies and use multivariate analyses to identify independent SNPs that are of most value. It is to be hoped that such investigations, together with similar studies of other known genetic variations or relevance to cancer, will inform clinical practice to help in the prevention/early detection of cancer and allow better informed decisions for individualized treatment options.
Acknowledgments
We would like to thank Eva Michalova for manuscript preparation. This study was financial supported by grants MSMT LC06035 and MZ0MOU2005.
References
- 1.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–6. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 2.Harris CC, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med. 1993;329:1318–27. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Jin S, Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114:4139–40. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- 5.Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39:1476–82. doi: 10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 2005;96:729–37. doi: 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Packer BR, Yeager M, Burdett L, Welch R, Beerman M, Qi L, Sicotte H, Staats B, Acharya M, Crenshaw A, Eckert A, Puri V, Gerhard DS, Chanock SJ. SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res. 2006;34:D617–21. doi: 10.1093/nar/gkj151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Felley-Bosco E, Weston A, Cawley HM, Bennett WP, Harris CC. Functional studies of a germ-line polymorphism at codon 47 within the p53 gene. Am J Hum Genet. 1993;53:752–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Dumont P, Della Pietra A, Shetler C, Murphy ME. The codon 47 polymorphism in p53 is functionally significant. J Biol Chem. 2005;280:24245–51. doi: 10.1074/jbc.M414637200. [DOI] [PubMed] [Google Scholar]
- 10.Harris N, Brill E, Shohat O, Prokocimer M, Wolf D, Arai N, Rotter V. Molecular basis for heterogeneity of the human p53 protein. Mol Cell Biol. 1986;6:4650–6. doi: 10.1128/mcb.6.12.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matlashewski GJ, Tuck S, Pim D, Lamb P, Schneider J, Crawford LV. Primary structure polymorphism at amino acid residue 72 of human p53. Mol Cell Biol. 1987;7:961–3. doi: 10.1128/mcb.7.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baptiste N, Friedlander P, Chen X, Prives C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene. 2002;21:9–21. doi: 10.1038/sj.onc.1205015. [DOI] [PubMed] [Google Scholar]
- 13.Beckman G, Birgander R, Sjalander A, Saha N, Holmberg PA, Kivela A, Beckman L. Is p53 polymorphism maintained by natural selection? Hum Hered. 1994;44:266–70. doi: 10.1159/000154228. [DOI] [PubMed] [Google Scholar]
- 14.Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, Smith P, Gasco M, Crook T, Lu X. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet. 2006;38:1133–41. doi: 10.1038/ng1879. [DOI] [PubMed] [Google Scholar]
- 15.Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–65. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 16.Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, Yulug I, Merlano M, Numico G, Comino A, Attard M, Reelfs O, Gusterson B, Bell AK, Heath V, Tavassoli M, Farrell PJ, Smith P, Lu X, Crook T. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell. 2003;3:387–402. doi: 10.1016/s1535-6108(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 17.Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, James N, McGregor JM, Harwood CA, Yulug IG, Vousden KH, Allday MJ, Gusterson B, Ikawa S, Hinds PW, Crook T, Kaelln WG., Jr A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet. 2000;25:47–54. doi: 10.1038/75586. [DOI] [PubMed] [Google Scholar]
- 18.Tada M, Furuuchl K, Kaneda M, Matsumoto J, Takahashl M, Hlral A, Mltsumoto Y, Iggo RD, Moriuchi T. Inactivate the remaining p53 allele or the alternate p73? Preferential selection of the Arg72 polymorphism in cancers with recessive p53 mutants but not transdomi-nant mutants. Carcinogenesis. 2001;22:515–7. doi: 10.1093/carcin/22.3.515. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Krlngen P, Krlstensen GB, Holm R, Baekelandt MM, Olivier M, Skomedal H, Halnaut P, Trope CG, Abeler VM, Nesland JM, Borresen-Dale AL, Helland A. Effect of the codon 72 polymorphism (c.215G>C, p.Arg72Pro) in combination with somatic sequence variants in the TP53 gene on survival in patients with advanced ovarian carcinoma. Hum Mutat. 2004;24:21–34. doi: 10.1002/humu.20055. [DOI] [PubMed] [Google Scholar]
- 20.Sullivan A, Syed N, Gasco M, Bergamaschl , Trigiante G, Attard M, Hlller L, Farrell PJ, Smith P, Lu X, Crook T. Polymorphism in wild-type p53 modulates response to chemotherapy in vitro and in vivo. Oncogene. 2004;23:3328–37. doi: 10.1038/sj.onc.1207428. [DOI] [PubMed] [Google Scholar]
- 21.Slddlque M, Sabapathy K. Trp53-dependent DNA-repair is affected by the codon 72 polymorphism. Oncogene. 2006;25:3489–500. doi: 10.1038/sj.onc.1209405. [DOI] [PubMed] [Google Scholar]
- 22.Storey A, Thomas M, Kallta A, Harwood C, Gardlol D, Mantovanl F, Breuer J, Leigh IM, Matlashewskl G, Banks L. Role of a p53 polymorphism in the development of human papillomavirus-associated cancer. Nature. 1998;393:229–34. doi: 10.1038/30400. [DOI] [PubMed] [Google Scholar]
- 23.Mltra S, Mlsra C, Singh RK, Panda CK, Roychoudhury S. Association of specific genotype and haplotype of p53 gene with cervical cancer in India. J Clin Pathol. 2005;58:26–31. doi: 10.1136/jcp.2004.019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santos AM, Sousa H, Catarlno R, Plnto D, Perelra D, Vasconcelos A, Matos A, Lopes C, Medeiros R. TP53 codon 72 polymorphism and risk for cervical cancer in Portugal. Cancer Genet Cytogenet. 2005;159:143–7. doi: 10.1016/j.cancergencyto.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Klug SJ, Wilmotte R, Santos C, Almonte M, Herrero R, Guerrero I, Caceres E, Pelxoto-Gulmaraes D, Lenolr G, Halnaut P, Walboomers JM, Munoz N. TP53 polymorphism, HPV infection, and risk of cervical cancer. Cancer Epidemiol Biomarkers Prev. 2001;10:1009–12. [PubMed] [Google Scholar]
- 26.Ueda M, Hung YC, Teral Y, Kanda K, Takehara M, Yamashlta H, Yamaguchl H, Akise D, Yasuda M, Nishiyama K, Ueki M. Glutathione S-transferase GSTM1, GSTT1 and p53 codon 72 polymorphisms in human tumor cells. Hum Cell. 2003;16:241–51. doi: 10.1111/j.1749-0774.2003.tb00158.x. [DOI] [PubMed] [Google Scholar]
- 27.Buyru N, Tigli H, Dalay N. P53 codon 72 polymorphism in breast cancer. Oncol Rep. 2003;10:711–4. [PubMed] [Google Scholar]
- 28.Langerod A, Bukholm IR, Bregard A, Lonning PE, Andersen TI, Rognum TO, Meling GI, Lothe RA, Borresen-Dale AL. The TP53 codon 72 polymorphism may affect the function of TP53 mutations in breast carcinomas but not in colorectal carcinomas. Cancer Epidemiol Biomarkers Prev. 2002;11:1684–8. [PubMed] [Google Scholar]
- 29.Kalemi TG, Lambropoulos AF, Gueorguiev M, Chrisafi S, Papazisis KT, Kotsis A. The association of p53 mutations and p53 codon 72, Her 2 codon 655 and MTHFR C677T polymorphisms with breast cancer in Northern Greece. Cancer Lett. 2005;222:57–65. doi: 10.1016/j.canlet.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 30.Tommiska J, Eerola H, Heinonen M, Salonen L, Kaare M, Tallila J, Ristimaki A, Von Smitten K, Aittomaki K, Heikkila P, Blomqvist C, Nevanlinna H. Breast cancer patients with p53 Pro72 homozygous genotype have a poorer survival. Clin Cancer Res. 2005;11:5098–103. doi: 10.1158/1078-0432.CCR-05-0173. [DOI] [PubMed] [Google Scholar]
- 31.Khadang B, Fattahi MJ, Talei A, Dehaghani AS, Ghaderi A. Polymorphism of TP53 codon 72 showed no association with breast cancer in Iranian women. Cancer Genet Cytogenet. 2007;173:38–42. doi: 10.1016/j.cancergencyto.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–12. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Weston A, Pan CF, Ksieski HB, Wallenstein S, Berkowitz GS, Tartter PI, Bleiweiss IJ, Brower ST, Senie RT, Wolff MS. p53 haplotype determination in breast cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:105–12. [PubMed] [Google Scholar]
- 34.Liu G, Miller DP, Zhou W, Thurston SW, Fan R, Xu LL, Lynch TJ, Wain JC, Su L, Christiani DC. Differential association of the codon 72 p53 and GSTM1 polymorphisms on histological subtype of non-small cell lung carcinoma. Cancer Res. 2001;61:8718–22. [PubMed] [Google Scholar]
- 35.Fan R, Wu MT, Miller D, Wain JC, Kelsey KT, Wiencke JK, Christiani DC. The p53 codon 72 polymorphism and lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:1037–42. [PubMed] [Google Scholar]
- 36.Robles AI, Linke SP, Harris CC. The p53 network in lung carcinogenesis. Oncogene. 2002;21:6898–907. doi: 10.1038/sj.onc.1205563. [DOI] [PubMed] [Google Scholar]
- 37.Jones JS, Chi X, Gu X, Lynch PM, Amos CI, Frazier ML. p53 polymorphism and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Clin Cancer Res. 2004;10:5845–9. doi: 10.1158/1078-0432.CCR-03-0590. [DOI] [PubMed] [Google Scholar]
- 38.Shen H, Zheng Y, Sturgis EM, Spitz MR, Wei Q. P53 codon 72 polymorphism and risk of squamous cell carcinoma of the head and neck: a case-control study. Cancer Lett. 2002;183:123–30. doi: 10.1016/s0304-3835(02)00117-9. [DOI] [PubMed] [Google Scholar]
- 39.Nelson HH, Wilkojmen M, Marsit CJ, Kelsey KT. TP53 mutation, allelism and survival in non-small cell lung cancer. Carcinogenesis. 2005;26:1770–3. doi: 10.1093/carcin/bgi125. [DOI] [PubMed] [Google Scholar]
- 40.Xu Y, Yao L, Ouyang T, Li J, Wang T, Fan Z, Lin B, Lu Y, Xie Y. p53 Codon 72 polymorphism predicts the pathologic response to neoadjuvant chemotherapy in patients with breast cancer. Clin Cancer Res. 2005;11:7328–33. doi: 10.1158/1078-0432.CCR-05-0507. [DOI] [PubMed] [Google Scholar]
- 41.Lima-Ramos V, Pacheco-Figueiredo L, Costa S, Pardal F, Silva A, Amorim J, Lopes JM, Reis RM. TP53 codon 72 polymorphism in susceptibility, overall survival, and adjuvant therapy response of gliomas. Cancer Genet Cytogenet. 2008;180:14–9. doi: 10.1016/j.cancergencyto.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Lozano G, Levine AJ. Tissue-specific expression of p53 in transgenic mice is regulated by intron sequences. Mol Carcinog. 1991;4:3–9. doi: 10.1002/mc.2940040103. [DOI] [PubMed] [Google Scholar]
- 43.Varley JM, McGown G, Thorncroft M, White GR, Tricker KJ, Kelsey AM, Birch JM, Evans DG. A novel TP53 splicing mutation in a Li-Fraumeni syndrome family: a patient with Wilms’ tumour is not a mutation carrier. Br J Cancer. 1998;78:1081–3. doi: 10.1038/bjc.1998.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lazar V, Hazard F, Bertin F, Janin N, Bellet D, Bressac B. Simple sequence repeat polymorphism within the p53 gene. Oncogene. 1993;8:1703–5. [PubMed] [Google Scholar]
- 45.Hillebrandt S, Streffer C, Demidchik EP, Biko J, Reiners C. Polymorphisms in the p53 gene in thyroid tumours and blood samples of children from areas in Belarus. Mutat Res. 1997;381:201–7. doi: 10.1016/s0027-5107(97)00169-3. [DOI] [PubMed] [Google Scholar]
- 46.McDaniel T, Carbone D, Takahashi T, Chumakov P, Chang EH, Pirollo KF, Yin J, Huang Y, Meltzer SJ. The MspI polymorphism in intron 6 of p53 (TP53) detected by digestion of PCR products. Nucleic Acids Res. 1991;19:4796. doi: 10.1093/nar/19.17.4796-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chumakov PM, Jenkins JR. BstNI/NciI polymorphism of the human p53 gene (TP53. Nucleic Acids Res. 1991:19. doi: 10.1093/nar/19.24.6969-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Runnebaum IB, Tong XW, Konig R, Zhao H, Korner K, Atkinson EN, Kreienberg R, Kieback DG, Hong Z. p53-based blood test for p53PIN3 and risk for sporadic ovarian cancer. Lancet. 1995;345:994. doi: 10.1016/s0140-6736(95)90745-9. [DOI] [PubMed] [Google Scholar]
- 49.Costa S, Pinto D, Pereira D, Rodrigues H, Cameselle-Teijeiro J, Medeiros R, Schmitt F. Importance of TP53 codon 72 and intron 3 duplication 16bp polymorphisms in prediction of susceptibility on breast cancer. BMC Cancer. 2008;8:32. doi: 10.1186/1471-2407-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campbell IG, Eccles DM, Dunn B, Davis M, Leake V. p53 polymorphism in ovarian and breast cancer. Lancet. 1996;347:393–4. doi: 10.1016/s0140-6736(96)90569-3. [DOI] [PubMed] [Google Scholar]
- 51.Kim JM, Lee OY, Lee CG, Kwon SJ, Kim KS, Moon W, Koh DH, Lee HL, Yoon BC, Choi HS, Hahm JS, Lee MH, Lee DH. [p53 Codon 72 and 16-bp duplication polymorphisms of gastric cancer in Koreans] Korean J Gastroenterol. 2007;50:292–8. [PubMed] [Google Scholar]
- 52.Lancaster JM, Brownlee HA, Wiseman RW, Taylor J. p53 polymorphism in ovarian and bladder cancer. Lancet. 1995;346:182. doi: 10.1016/s0140-6736(95)91239-8. [DOI] [PubMed] [Google Scholar]
- 53.Wang-Gohrke S, Weikel W, Risch H, Vesprini D, Abrahamson J, Lerman C, Godwin A, Moslehi R, Olipade O, Brunet JS, Stickeler E, Kieback DG, Kreienberg R, Weber B, Narod SA, Runnebaum IB. Intron variants of the p53 gene are associated with increased risk for ovarian cancer but not in carriers of BRCA1 or BRCA2 germline mutations. Br J Cancer. 1999;81:179–83. doi: 10.1038/sj.bjc.6690669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mavridou D, Gornall R, Campbell IG, Eccles DM. TP53 intron 6 polymorphism and the risk of ovarian and breast cancer. Br J Cancer. 1998;77:676–7. doi: 10.1038/bjc.1998.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peller S, Kopilova Y, Slutzki S, Halevy A, Kvitko K, Rotter V. A novel polymorphism in intron 6 of the human p53 gene: a possible association with cancer predisposition and susceptibility. DNA Cell Biol. 1995;14:983–90. doi: 10.1089/dna.1995.14.983. [DOI] [PubMed] [Google Scholar]
- 56.Lehman TA, Haffty BG, Carbone CJ, Bishop LR, Gumbs AA, Krishnan S, Shields PG, Modali R, Turner BC. Elevated frequency and functional activity of a specific germ-line p53 intron mutation in familial breast cancer. Cancer Res. 2000;60:1062–9. [PubMed] [Google Scholar]
- 57.Marsh A, Spurdle AB, Turner BC, Fereday S, Thorne H, Pupo GM, Mann GJ, Hopper JL, Sambrook JF, Chenevix-Trench G. The intronic G13964C variant in p53 is not a high-risk mutation in familial breast cancer in Australia. Breast Cancer Res. 2001;3:346–9. doi: 10.1186/bcr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fiszer-Maliszewska L, Kazanowska B, Kusnierczyk P, Manczak M, Niepieklo W, Pochron-Zeman B, Nowakowska B. Is p53 intronic variant G13964C associated with predisposition to cancer? J Appl Genet. 2003;44:547–52. [PubMed] [Google Scholar]
- 59.Wei Q, Cheng L, Amos CI, Wang LE, Guo Z, Hong WK, Spitz MR. Repair of tobacco carcinogen-induced DNA adducts and lung cancer risk: a molecular epidemiologic study. J Natl Cancer Inst. 2000;92:1764–72. doi: 10.1093/jnci/92.21.1764. [DOI] [PubMed] [Google Scholar]
- 60.Wu X, Zhao H, Amos CI, Shete S, Makan N, Hong WK, Kadlubar FF, Spitz MR. p53 Genotypes and Haplotypes Associated With Lung Cancer Susceptibility and Ethnicity. J Natl Cancer Inst. 2002;94:681–90. doi: 10.1093/jnci/94.9.681. [DOI] [PubMed] [Google Scholar]
- 61.Birgander R, Sjalander A, Rannug A, Alexandrie AK, Sundberg MI, Seidegard J, Tornling G, Beckman G, Beckman L. P53 polymorphisms and haplotypes in lung cancer. Carcinogenesis. 1995;16:2233–6. doi: 10.1093/carcin/16.9.2233. [DOI] [PubMed] [Google Scholar]
- 62.Khaliq S, Hameed A, Khaliq T, Ayub Q, Qamar R, Mohyuddin A, Mazhar K, Qasim-Mehdi S. P53 mutations, polymorphisms, and haplotypes in Pakistani ethnic groups and breast cancer patients. Genet Test. 2000;4:23–9. doi: 10.1089/109065700316435. [DOI] [PubMed] [Google Scholar]
- 63.Sjalander A, Birgander R, Athlin L, Stenling R, Rutegard J, Beckman L, Beckman G. P53 germ line haplotypes associated with increased risk for colorectal cancer. Carcinogenesis. 1995;16:1461–4. doi: 10.1093/carcin/16.7.1461. [DOI] [PubMed] [Google Scholar]
- 64.Sjalander A, Birgander R, Hallmans G, Cajander S, Lenner P, Athlin L, Beckman G, Beckman L. p53 polymorphisms and haplotypes in breast cancer. Carcinogenesis. 1996;17:1313–6. doi: 10.1093/carcin/17.6.1313. [DOI] [PubMed] [Google Scholar]
- 65.Biros E, Kalina I, Kohut A, Bogyiova E, Salagovic J, Sulla I. Allelic and haplotype frequencies of the p53 polymorphisms in brain tumor patients. Physiol Res. 2002;51:59–64. [PubMed] [Google Scholar]
- 66.Birgander R, Sjalander A, Zhou Z, Fan C, Beckman L, Beckman G. p53 polymorphisms and haplotypes in nasopharyngeal cancer. Hum Hered. 1996;46:49–54. doi: 10.1159/000154325. [DOI] [PubMed] [Google Scholar]
- 67.Boldrini L, Gisfredi S, Ursino S, Lucchi M, Greco G, Mussi A, Donati V, Fontanini G. Effect of the p53 codon 72 and intron 3 polymorphisms on non-small cell lung cancer (NSCLC) prognosis. Cancer Invest. 2008;26:168–72. doi: 10.1080/07357900701788023. [DOI] [PubMed] [Google Scholar]
- 68.Lamolle G, Marin M, Alvarez-Valin F. Silent mutations in the gene encoding the p53 protein are preferentially located in conserved amino acid positions and splicing enhancers. Mutat Res. 2006;600:102–12. doi: 10.1016/j.mrfmmm.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 69.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–8. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 70.Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T. Codon usage and tRNA genes in eukaryotes: correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis. J Mol Evol. 2001;53:290–8. doi: 10.1007/s002390010219. [DOI] [PubMed] [Google Scholar]
- 71.Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986;14:7737–49. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64:7647–54. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 73.Holmila R, Fouquet C, Cadranel J, Zalcman G, Soussi T. Splice mutations in the p53 gene: case report and review of the literature. Hum Mutat. 2003;21:101–2. doi: 10.1002/humu.9104. [DOI] [PubMed] [Google Scholar]
- 74.Warneford SG, Witton LJ, Townsend ML, Rowe PB, Reddel RR, Dalla-Pozza L, Symonds G. Germ-line splicing mutation of the p53 gene in a cancer-prone family. Cell Growth Differ. 1992;3:839–46. [PubMed] [Google Scholar]
- 75.Gorlov IP, Gorlova OY, Frazier ML, Amos CI. Missense mutations in cancer suppressor gene TP53 are colocalized with exonic splicing enhancers (ESEs) Mutat Res. 2004;554:175–83. doi: 10.1016/j.mrfmmm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 76.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–98. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 77.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–71. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pfeifer GP. p53 mutational spectra and the role of methylated CpG sequences. Mutat Res. 2000;450:155–66. doi: 10.1016/s0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- 79.Kouidou S, Malousi A, Maglaveras N. Methylation and repeats in silent and nonsense mutations of p53. Mutat Res. 2006;599:167–77. doi: 10.1016/j.mrfmmm.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 80.Felix CA, Brown DL, Mitsudomi T, Ikagaki N, Wong A, Wasserman R, Womer RB, Biegel JA. Polymorphism at codon 36 of the p53 gene. Oncogene. 1994;9:327–8. [PubMed] [Google Scholar]
- 81.Finlan LE, Hupp TR. p63: the phantom of the tumor suppressor. Cell Cycle. 2007;6:1062–71. doi: 10.4161/cc.6.9.4162. [DOI] [PubMed] [Google Scholar]
- 82.Barrow LL, Van Bokhoven H, Daack-Hirsch S, Andersen T, Van Beersum SE, Gorlin R, Murray JC. Analysis of the p63 gene in classical EEC syndrome, related syndromes, and non-syndromic orofacial clefts. J Med Genet. 2002;39:559–66. doi: 10.1136/jmg.39.8.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muller M, Schleithoff ES, Stremmel W, Melino G, Krammer PH, Schilling T. One, two, three–p53, p63, p73 and chemosensitivity. Drug Resist Updat. 2006;9:288–306. doi: 10.1016/j.drup.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 84.Casciano I, Mazzocco K, Boni L, Pagnan G, Banelli B, Allemanni G, Ponzoni M, Tonini GP, Romani M. Expression of DeltaNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death Differ. 2002;9:246–51. doi: 10.1038/sj.cdd.4400993. [DOI] [PubMed] [Google Scholar]
- 85.Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, Chalas E, Moll UM. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J Exp Med. 2002;196:765–80. doi: 10.1084/jem.20020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–86. [PubMed] [Google Scholar]
- 87.Coates PJ. Regulating p73 isoforms in human tumours. J Pathol. 2006;210:385–9. doi: 10.1002/path.2080. [DOI] [PubMed] [Google Scholar]
- 88.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 89.Li G, Sturgis EM, Wang LE, Chamberlain RM, Amos CI, Spitz MR, El-Naggar AK, Hong WK, Wei Q. Association of a p73 exon 2 G4C14-to-A4T14 polymorphism with risk of squamous cell carcinoma of the head and neck. Carcinogenesis. 2004;25:1911–6. doi: 10.1093/carcin/bgh197. [DOI] [PubMed] [Google Scholar]
- 90.Li G, Wang LE, Chamberlain RM, Amos CI, Spitz MR, Wei Q. p73 G4C14-to-A4T14 polymorphism and risk of lung cancer. Cancer Res. 2004;64:6863–6. doi: 10.1158/0008-5472.CAN-04-1804. [DOI] [PubMed] [Google Scholar]
- 91.Niwa Y, Hirose K, Matsuo K, Tajima K, Ikoma Y, Nakanishi T, Nawa A, Kuzuya K, Tamakoshi A, Hamajima N. Association of p73 G4C14-to-A4T14 polymorphism at exon 2 and p53 Arg72Pro polymorphism with the risk of endometrial cancer in Japanese subjects. Cancer Lett. 2005;219:183–90. doi: 10.1016/j.canlet.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 92.Ryan BM, McManus R, Daly JS, Carton E, Keeling PW, Reynolds JV, Kelleher D. A common p73 polymorphism is associated with a reduced incidence of oesophageal carcinoma. Br J Cancer. 2001;85:1499–503. doi: 10.1054/bjoc.2001.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu Z, Miao X, Ma H, Tan W, Wang X, Lu D, Wei Q, Lin D, Shen H. Dinucleotide polymorphism of p73 gene is associated with a reduced risk of lung cancer in a Chinese population. Int J Cancer. 2005;114:455–60. doi: 10.1002/ijc.20746. [DOI] [PubMed] [Google Scholar]
- 94.Li H, Yao L, Ouyang T, Li J, Wang T, Fan Z, Fan T, Dong B, Lin B, Xie Y. Association of p73 G4C14-to-A4T14 (GC/AT) polymorphism with breast cancer survival. Carcinogenesis. 2007;28:372–7. doi: 10.1093/carcin/bgl153. [DOI] [PubMed] [Google Scholar]
- 95.Pena C, Garcia JM, Dominguez G, Silva J, Garcia V, Carcereny E, Vargas J, Provencio M, Espana P, Bonilla F. Intronic deletion affecting a negative regulatory region of TP73 is related to breast and colorectal carcinomas. Genes Chromosomes Cancer. 2004;39:257–62. doi: 10.1002/gcc.10322. [DOI] [PubMed] [Google Scholar]
- 96.Dominguez G, Pena C, Silva J, Garcia JM, Garcia V, Rodriguez R, Cantos B, Citores MJ, Espana P, Bonilla F. The presence of an intronic deletion in p73 and high levels of ZEB1 alter the TAp73/DeltaTAp73 ratio in colorectal carcinomas. J Pathol. 2006;210:390–7. doi: 10.1002/path.2066. [DOI] [PubMed] [Google Scholar]
- 97.Toledo F, Wahl GM. Regulating the p53 pathway: in vitrohypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 98.Bond GL, Hu W, Levine A. A single nucleotide polymorphism in the MDM2 gene: from a molecular and cellular explanation to clinical effect. Cancer Res. 2005;65:5481–4. doi: 10.1158/0008-5472.CAN-05-0825. [DOI] [PubMed] [Google Scholar]
- 99.Bougeard G, Baert-Desurmont S, Tournier I, Vasseur S, Martin C, Brugieres L, Chompret A, Bressac-de Paillerets B, Stoppa-Lyonnet D, Bonaiti-Pellie C, Frebourg T. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet. 2006;43:531–3. doi: 10.1136/jmg.2005.037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Menin C, Scaini MC, De Salvo GL, Biscuola M, Quaggio M, Esposito G, Belluco C, Montagna M, Agata S, D’Andrea E, Nitti D, Amadori A, Bertorelle R. Association between MDM2-SNP309 and age at colorectal cancer diagnosis according to p53 mutation status. J Natl Cancer Inst. 2006;98:285–8. doi: 10.1093/jnci/djj054. [DOI] [PubMed] [Google Scholar]
- 101.Ruijs MW, Schmidt MK, Nevanlinna H, Tommiska J, Aittomaki K, Pruntel R, Verhoef S, Van't Veer LJ. The single-nucleotide polymorphism 309 in the MDM2 gene contributes to the Li-Fraumeni syndrome and related phenotypes. Eur J Hum Genet. 2007;15:110–4. doi: 10.1038/sj.ejhg.5201715. [DOI] [PubMed] [Google Scholar]
- 102.Boersma BJ, Howe TM, Goodman JE, Yfantis HG, Lee DH, Chanock SJ, Ambs S. Association of breast cancer outcome with status of p53 and MDM2 SNP309. J Natl Cancer Inst. 2006;98:911–9. doi: 10.1093/jnci/djj245. [DOI] [PubMed] [Google Scholar]
- 103.Schmidt MK, Reincke S, Broeks A, Braaf LM, Hogervorst FB, Tollenaar RA, Johnson N, Fletcher O, Peto J, Tommiska J, Blomqvist C, Nevanlinna HA, Healey CS, Dunning AM, Pharoah PD, Easton DF, Dork T, Van't Veer LJ. Do MDM2 SNP309 and TP53 R72P interact in breast cancer susceptibility? A large pooled series from the breast cancer association consortium. Cancer Res. 2007;67:9584–90. doi: 10.1158/0008-5472.CAN-07-0738. [DOI] [PubMed] [Google Scholar]
- 104.Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, Bartel F, Taubert H, Wuerl P, Hait W, Toppmeyer D, Offit K, Levine AJ. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res. 2006;66:5104–10. doi: 10.1158/0008-5472.CAN-06-0180. [DOI] [PubMed] [Google Scholar]
- 105.Bond GL, Levine AJ. A single nucleotide polymorphism in the p53 pathway interacts with gender, environmental stresses and tumor genetics to influence cancer in humans. Oncogene. 2007;26:1317–23. doi: 10.1038/sj.onc.1210199. [DOI] [PubMed] [Google Scholar]
- 106.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, Strong LC, Lozano G, Levine AJ. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 107.Krekac D, Brozkova K, Knoflickova D, Hrstka R, Muller P, Nenutil R, Vojtesek B. MDM2SNP309 does not associate with elevated MDM2 protein expression or breast cancer risk. Oncology. 2008;74:84–7. doi: 10.1159/000139135. [DOI] [PubMed] [Google Scholar]
- 108.Hartmann E, Fernandez V, Stoecklein H, Hernandez L, Campo E, Rosenwald A. Increased MDM2 expression is associated with inferior survival in mantle-cell lymphoma, but not related to the MDM2 SNP309. Haematologica. 2007;92:574–5. doi: 10.3324/haematol.10891. [DOI] [PubMed] [Google Scholar]
- 109.O’Connor PM. Mammalian G1 and G2 phase checkpoints. Cancer Sur v. 1997;29:151–82. [PubMed] [Google Scholar]
- 110.Mousses S, Ozcelik H, Lee PD, Malkin D, Bull SB, Andrulis IL. Two variants of the CIP1/WAF1 gene occur together and are associated with human cancer. Hum Mol Genet. 1995;4:1089–92. doi: 10.1093/hmg/4.6.1089. [DOI] [PubMed] [Google Scholar]
- 111.Birgander R, Sjalander A, Saha N, Spitsyn V, Beckman L, Beckman G. The codon 31 polymorphism of the p53-inducible gene p21 shows distinct differences between major ethnic groups. Hum Hered. 1996;46:148–54. doi: 10.1159/000154344. [DOI] [PubMed] [Google Scholar]
- 112.Su L, Sai Y, Fan R, Thurston SW, Miller DP, Zhou W, Wain JC, Lynch TJ, Liu G, Christiani DC. P53 (codon 72) and P21 (codon 31) polymorphisms alter in vivo mRNA expression of p21. Lung Cancer. 2003;40:259–66. doi: 10.1016/s0169-5002(03)00081-3. [DOI] [PubMed] [Google Scholar]
- 113.Pietsch EC, Humbey O, Murphy ME. Polymorphisms in the p53 pathway. Oncogene. 2006;25:1602–11. doi: 10.1038/sj.onc.1209367. [DOI] [PubMed] [Google Scholar]
- 114.Shih CM, Lin PT, Wang HC, Huang WC, Wang YC. Lack of evidence of association of p21WAF1/CIP1 polymorphism with lung cancer susceptibility and prognosis in Taiwan. Jpn J Cancer Res. 2000;91:9–15. doi: 10.1111/j.1349-7006.2000.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Powell BL, Van Staveren IL, Roosken P, Grieu F, Berns EM, Iacopetta B. Associations between common polymorphisms in TP53 and p21WAF1/Cip1 and phenotypic features of breast cancer. Carcinogenesis. 2002;23:311–5. doi: 10.1093/carcin/23.2.311. [DOI] [PubMed] [Google Scholar]
- 116.Facher EA, Becich MJ, Deka A, Law JC. Association between human cancer and two polymorphisms occurring together in the p21Waf1/Cip1 cyclin-dependent kinase inhibitor gene. Cancer. 1997;79:2424–9. doi: 10.1002/(sici)1097-0142(19970615)79:12<2424::aid-cncr19>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 117.Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30:315–20. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- 118.Fegan C, Starczynski J, Pratt G, Pepper C. The role of the bax gene polymorphism G(-248)A in chronic lymphocytic leukemia. Leukemia. 2006;20:1460–1. doi: 10.1038/sj.leu.2404280. [DOI] [PubMed] [Google Scholar]
- 119.Saxena A, Moshynska O, Sankaran K, Viswanathan S, Sheridan DP. Association of a novel single nucleotide polymorphism, G(-248)A, in the 5’-UTR of BAX gene in chronic lymphocytic leukemia with disease progression and treatment resistance. Cancer Lett. 2002;187:199–205. doi: 10.1016/s0304-3835(02)00378-6. [DOI] [PubMed] [Google Scholar]
- 120.Kidd LR, Coulibaly A, Templeton TM, Chen W, Long LO, Mason T, Bonilla C, Akereyeni F, Freeman V, Isaacs W, Ahaghotu C, Kittles RA. Germline BCL-2 sequence variants and inherited predisposition to prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:284–92. doi: 10.1038/sj.pcan.4500884. [DOI] [PubMed] [Google Scholar]
- 121.Crew KD, Gammon MD, Terry MB, Zhang FF, Agrawal M, Eng SM, Sagiv SK, Teitelbaum SL, Neugut AI, Santella RM. Genetic polymorphisms in the apoptosis-associated genes FAS and FASL and breast cancer risk. Carcinogenesis. 2007;28:2548–51. doi: 10.1093/carcin/bgm211. [DOI] [PubMed] [Google Scholar]
- 122.Ueda M, Hung YC, Terai Y, Yamaguchi H, Saito J, Nunobiki O, Noda S, Ueki M. Fas gene promoter -670 polymorphism (A/G) is associated with cervical carcinogenesis. Gynecol Oncol. 2005;98:129–33. doi: 10.1016/j.ygyno.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 123.Zhang X, Miao X, Sun T, Tan W, Qu S, Xiong P, Zhou Y, Lin D. Functional polymorphisms in cell death pathway genes FAS and FASL contribute to risk of lung cancer. J Med Genet. 2005;42:479–84. doi: 10.1136/jmg.2004.030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tomso DJ, Inga A, Menendez D, Pittman GS, Campbell MR, Storici F, Bell DA, Resnick MA. Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proc Natl Acad Sci USA. 2005;102:6431–6. doi: 10.1073/pnas.0501721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Resnick MA, Tomso D, Inga A, Menendez D, Bell D. Functional diversity in the gene network controlled by the master regulator p53 in humans. Cell Cycle. 2005;4:1026–9. doi: 10.4161/cc.4.8.1904. [DOI] [PubMed] [Google Scholar]
- 126.Menendez D, Krysiak O, Inga A, Krysiak B, Resnick MA, Schonfelder G. A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc Natl Acad Sci USA. 2006;103:1406–11. doi: 10.1073/pnas.0508103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Menendez D, Inga A, Snipe J, Krysiak O, Schonfelder G, Resnick MA. A single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol Cell Biol. 2007;27:2590–600. doi: 10.1128/MCB.01742-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vikhanskaya F, Siddique MM, Kei Lee M, Broggini M, Sabapathy K. Evaluation of the combined effect of p53 codon 72 polymorphism and hotspot mutations in response to anticancer drugs. Clin Cancer Res. 2005;11:4348–56. doi: 10.1158/1078-0432.CCR-04-1547. [DOI] [PubMed] [Google Scholar]
- 129.Noma C, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S. Association of p53 genetic polymorphism (Arg72Pro) with estrogen receptor positive breast cancer risk in Japanese women. Cancer Lett. 2004;210:197–203. doi: 10.1016/j.canlet.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 130.Sakiyama T, Kohno T, Mimaki S, Ohta T, Yanagitani N, Sobue T, Kunitoh H, Saito R, Shimizu K, Hirama C, Kimura J, Maeno G, Hirose H, Eguchi T, Saito D, Ohki M, Yokota J. Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer. 2005;114:730–7. doi: 10.1002/ijc.20790. [DOI] [PubMed] [Google Scholar]
- 131.Parhar P, Ezer R, Shao Y, Allen JC, Miller DC, Newcomb EW. Possible association of p53 codon 72 polymorphism with susceptibility to adult and pediatric high-grade astrocytomas. Brain Res Mol Brain Res. 2005;137:98–103. doi: 10.1016/j.molbrainres.2005.02.016. [DOI] [PubMed] [Google Scholar]