

Abstract
The utility of various synthetic peptides has been investigated in clinical trials of the treatment of cancers, infectious diseases and endocrine diseases. In the process of functional gene screening with in silico analysis for molecules with angiogenic properties, we generated a small peptide, angiogenic peptide (AG)-30, that possesses both antimicrobial and pro-inflammatory activities. AG-30 has an α-helix structure with a number of hydrophobic or net positively charged amino acids and a propensity to fold into amphipathic structures. Indeed, AG-30 exhibited antimicrobial activity against various bacteria, induced vascular endothelial cell growth and tube formation in a dose-dependent manner and increased neovascularization in a Matrigel plug assay. As a result, AG-30 up-regulated expression of angiogenesis-related cytokines and growth factors for up to 72 hrs in human aortic endothelial cells. To further evaluate the angiogenic effect of AG-30 in vivo, we developed a slow-release AG-30 system utilizing biodegradable gelatin microspheres. In the ischaemic mouse hind limb, slow-release AG-30 treatment results in an increase in angiogenic score, an increase in blood flow (as demonstrated by laser Doppler imaging) and an increase in capillary density (as demonstrated by immunostaining with anti-CD31 antibody). These data suggest that the novel peptide, AG-30, may have therapeutic potential for ischaemic diseases.
Keywords: angiogenesis, antimicrobial, peptide, slow release, ischaemic disease
Introduction
Advances in analytic techniques have allowed for the characterization of synthetic peptides, and the utility of various synthetic peptides has been investigated in clinical trials of the treatment of cancers, infectious diseases and endocrine diseases. Synthetic peptides have been widely used for clinical trials to treat intractable human diseases such as cancers, infectious diseases and endocrine diseases [1–3]. Unlike retrovirus vectors, synthetic peptides have little chance to integrate or recombinate to host genome, and can be analysed for purity and fidelity of sequencing using well-established analytical techniques [2].
One class of antimicrobial peptides possesses both antimicrobial and proinflammatory activities and can modulate human immune system activity [4–6]. These peptides exhibit potent killing of a broad range of micro-organisms and serve as a first-line defence system as well as interact with host repair and adaptive immune responses [7], and levels of these peptides are increased in the context of inflammation and injury [8, 9]. Major antimicrobial peptides in mammals include the family of defensins and cathelicidin peptides. Antimicrobial peptides of the cathelicidin family consist of an amphipathic a-helix with positive charged residues separate from hydrophobic residues. These structures appear to be critical for the ability of these peptides to interact with the negatively charged microbial membrane in the initiation of antimicrobial effects and may also induce growth and migration of endothelial cells to achieve angiogenesis.
Studies have demonstrated that molecules, PR-39 and LL-37, have potential for the treatment of ischaemic cardiovascular diseases [10, 11]. PR-39 is an anti-microbial peptide of the porcine cathelicidin family and induces angiogenesis by inhibiting the ubiquitin-proteasome-dependent degradation of HIF-1α[10]. The human cathelicidin family molecule, LL-37/hCAP18, is a 37-amino acid peptide beginning with two leucine residues that forms a linear amphipathic α-helix and that possesses a broad spectrum of antimicrobial activity [11]. Further, LL-37/hCAP18 acts as a chemoattractant of neutrophils, monocytes and T cells and promotes angiogenesis by binding to a G-protein-coupled receptor upstream of the PLC-γ/PKC/NFkB, ERK-1 and 2 MAPK, and the PI3K/Akt pathways [12]. Thus, some antimicrobial peptides can also stimulate angiogenesis.
We recently screened a human cDNA library in an attempt to isolate angiogenic factors and successfully identified a powerful and potent angiogenic cDNA clone (p3743) [13]. Further analysis of the angiogenic clone facilitated the identification of a core sequence of EC growth activity. Interestingly, in silico analysis showed that the peptide from the core sequence (MLSLIFLHRLKSMRKRLDRKLRLWHRKNYP) was predicted to form an a-helical structure with a high percentage hydrophobic residues, a structure that is characteristic of various antimicrobial peptides [4–6]. Of note, some antimicrobial peptides (i.e. LL37 or PR39) may possess pleiotropically hormonal properties (e.g. induction of angiogenesis) as well as antibacterial action. Thus, the goal of the present study was to evaluate the potential angiogenic effect of an antimicrobial-like peptide.
Materials and methods
Peptide synthesis and circular dichroism (CD) spectroscopy analysis
Synthetic AG-30 (NH2 -MLSLIFLHRLKSMRKRLDRKLRLWHRKNYP-COOH) and control peptide (NH2 - RSLEGTDRFPFVRLKNSRKLEFKDIKGIKR-COOH) were purchased from Peptide Institute, Inc. (Osaka, Japan). Control peptide and LL-37 were synthesized as per a previous report [12] and purchased from SIGMA Genosys (Hokkaido, Japan).
CD data were acquired with Jasco J-820 Spectropho-tometer using a 1-mm path length cuvette [14]. Spectra were collected for samples of 50 μM AG-30 and control (Ctrl) peptide in 20 mM phosphate buffer at pH 7.5 and 37°C, with and without 1-mM 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) or 1 mM 1-palmitoyl-2-oleoyl-sn-glycero-3-[phosphor-rac-(1-glycerol)] (sodium salt) (POPG) liposome. POPC and POPG were purchased from SIGMA-ALDRICH (St. Louis, MO, USA) and Avanti Polar Lipids (Alabaster, AL, USA), respectively.
Minimal inhibitory concentration (MIC) assays
MIC assays were conducted as previously described [15]. P. aeruginosa (PA) (ATCC27853), S. aureus (SA) (ATCC29213) and E. coli (EC) (ATCC25922) were grown in Mueller-Hinton broth (MHB) (Becton Dickison and Co., Sparks, MD, USA). Serial twofold dilutions of peptide were added to 1 ml of medium containing each type of bacteria (PA, SA and EC) at 1 × 105 CFU/ml. The tubes were incubated at 37°C with vigorous shaking for 16 hrs. The MIC was determined as the lowest peptide concentration that prevented visible growth of bacteria.
Cell cultures
HAECs (human aortic endothelial cells) and HASMCs (human aortic smooth muscle cells) (passage 3) were purchased from Clonetics Corp. (Palo Alto, CA, USA) and were maintained in endothelial basal medium (EBM-2 medium) supplemented with 5% fetal bovine serum (FBS) and endothelial growth supplement, as described previously [16] or smooth muscle medium supplemented with 5% FBS and smooth muscle growth supplement.
Cell viability and migration assay
HAECs and HASMCs (103 cells/well) were seeded on 96-well collagen I-coated plates the day before transfection. Cell viability of HAECs and HASMCs were measured using the MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] Assay. On the first, second and fourth day (fifth day for HASMCs) after transfection, 10 μl of CellTiter 96 One Solution Reagent (Promega, Madison, WI, USA) was added to each well, and absorbance at 490 nm was measured.
HAEC chemokinetic migration was assayed using a modified Boyden chamber, as previously described [17]. 106 cells/ml of HAEC suspended in 50-μl EBM2 medium containing either AG-30, LL-37 or control peptide (10 μg/ml) were added to the upper chamber. After 24-hr incubation, the membrane was removed. The cells on the lower side of the membrane were stained with Diff-Quick (Sysmex, Hyogo, Japan). The number of cells was counted in eight randomly chosen fields under ×100 magnification.
Chemotactic migration of HAEC in response to AG-30 was also assessed using a modified Boyden chamber as previously described [18]. In brief, AG-30 was added in different concentrations (0.1, 1.0 and 10 μg/ml) in the lower chambers, and HAEC (106 cells/ml in 50 μl) suspended in EBM2 medium (1% BSA and no growth factor added) were added to the upper chambers. After 4-hr incubation, the membrane was removed and the migrated cells were counted as described above.
Tube formation assay
HAEC tube formation assay was conducted in triplicate in a 24-well plate using an Angiogenesis Kit (Kurabo, Osaka, Japan), as per the manufacturer's instructions. Human endothelial and fibroblast cells in the kit were cultured in Optimized Medium supplemented with 1% FBS, followed by daily treatment with AG-30 peptide (0.1, 1, 10 μg/ml), LL-37 peptide (1 and 10 μg/ml) or control peptide (1, 10 μg/ml). Seven days later, cells were stained with anti-human CD31 monoclonal antibody. Stained cells were photographed, and tubule-like structures in the images were analysed by an Angiogenesis Image Analyzer (Kurabo, Osaka, Japan).
In vivo Matrigel plug assay
Two different types of Matrigel plug assays were performed as previously described [17]. First, growth factor-depleted Matrigel (0.5 ml, BD Biosciences, Franklin Lakes, NJ, USA) was mixed with 40 U/ml of heparin (Aventis Pharma, Tokyo, Japan) and either AG-30 peptide (10 μg/ml), control peptide (10 μg/ml) or no peptide. The mixture was then injected subcutaneously into C57BL/6 male mice obtained from Oriental Bio Science Co., Ltd. (Kyoto Japan). After 7 days, the mice were humanely killed, and the plugs were recovered and fixed in methanol. For immunostaining, sections were incubated with monoclonal anti-CD31 (PECAM-1) antibody (1:100 dilution, BD Pharmigen, San Diego, CA, USA) and anti-α-smooth muscle actin antibody (1:400 dilution, SIGMA, Saint Louis, MO, USA) overnight at 4°C, and then incubated in Alexa Flour 488 and 546 secondary antibodies (1:500, Molecular Probes, Eugene, OR, USA) for 2 hrs before photography. Next, the directed in vitro angiogenesis assay (DIVAA) (Trevigen, Inc., Gaithersburg, MD, USA) was performed essentially as per the manufacture's instructions. In brief, sterilized DIVAA angioreactors were filled with 18 μl of Basement Membrane Extract (BME) with AG-30 peptide (10 μg/ml) or control peptide (10 μg/ml). The angioreactors were implanted subcutaneously into the dorsal area of C57BL/6 male mice (6 weeks of age) and maintained for 15 days. All experimental protocols were approved by the Osaka University Graduate School of Medicine Standing Committee on Animals.
Microarray analysis
HAEC RNA samples that had been stimulated with or without AG-30 peptide (10 μg/ml) after 24 and 72 hrs were extracted, and the quality of these samples was examined using the Agilient 2100 Bioanalyzer (Agilient Technologies, Palo Alto, CA, USA). RNA amplification and labelling was performed following the LRIFLA protocol (Agilent Low RNA Input Fluorescent Linear Amplification Kit Protocol, version 2.0. Agilient Technologies, Palo Alto, CA, USA). AG-30 stimulated and non-stimulated cDNA samples were labelled by Cyanine 3- and 5-labelled CTP (cystidine 5-triphosphate) respectively. Hybridization was performed according to instructions provided by the Agilent oligonucleotide microarray hybridization user manual (Agilient Technologies, Palo Alto, CA, USA). Finally, 0.75 μg of labelled cDNA was applied to a Whole Human Genome Oligonucleotide Microarray and then hybridized. The arrays were scanned by the Agilent dual-laser DNA microarray scanner and analysed by Feature Extraction software.
Real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
After treatment with AG-30 peptide (10 μg/ml), HAEC RNA was extracted at 72-hr time points using the RNeasy Mini kit (QIAGEN, Valencia, CA, USA). Complementary DNA was synthesized using the Thermo Script RT-PCR System (Invitrogen, Carlsbad, CA, USA). Relative gene copy numbers of interleukin-8 (IL-8: Hs00174103), angiopoietin-2 (Ang-2: Hs00169867) Jagged 1 (Jag-1:Hs00164982), insulin-like growth factor-1 (IGF-1: Hs00153126), vascular endothelial growth factor (VEGF: Hs00173626) and GAPDH (Hs99999905) were quantified by real-time qRT-PCR using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA). The absolute number of gene copies was normalized using GAPDH and standardized by a sample standard curve. Results are expressed as fold-increase relative to the non-stimulants for copy numbers of each mRNA.
Preparation of gelatin microspheres incorporating AG-30
The carboxyl groups of gelatin with an isoelectric point of 5.0 (MW 100,000) were prepared from pig skin by an acid process (Nitta gelatin Inc., Osaka, Japan) and were chemically converted by introducing amino groups for cationization of gelatin as previously described [19]. To impregnate AG-30 into gelatin microspheres, 20 μl of PBS containing 100 or 500 μg of AG-30 or control peptide was placed onto 2 mg of freeze-dried gelatin microspheres, followed by incubation for 24 hrs at 4°C. Before injection into the ischaemic hindlimb, 80 μl of PBS was added to this solution.
Evaluation of in vivo degradation of gelatin microspheres
Gelatin microspheres were radioiodinated using [125I] Bolton-Hunter reagent [20]. The 125I-labelled gelatin microspheres (2 mg/200 1 PBS/mouse) were subcutaneously injected into the back of 6- to 8-week-old C57BL/6 mice (Japan SLC, Inc., Hamamatsu, Japan). At 3, 7, 14 and 21 days after injection, the mouse skin and muscle containing the gelatin microspheres were excised in order to measure their radioactivity on a gamma counter. The radioactivity ratio of the sample to the gelatin microspheres injected initially was measured to express the percentage of remaining radioactivity in the gelatin microspheres. AG-30 was also radioiodinated and gelatin microspheres incorporating 100 μg of 125I-labelled AG-30 were prepared similarly and subcutaneously injected to mice at the injection volume of 200 μl. The percentage of remaining radioactivity in AG-30 was similarly calculated. All animal experiments were performed in accordance with the Institutional Guidelines of Kyoto University on Animal Experimentation.
Mouse hind limb ischaemic model and evaluation of angiogenesis
Wild-type C57Bl/6J mice (8-week old, male) were anaesthetized with keta-mine chloride (80 mg/kg) and xyladine sulfate (8 mg/kg) subcutaneously and unilateral hind limb ischaemia was induced as described previously [21]. The solution containing AG-30 or control peptide was carefully injected into the mouse ischaemic limb with a 26-gauge needle. Three separate injections of cells (intramuscularly into the ischaemic limb, near both the proximal and distal arterial stumps) were performed. All experimental protocols were approved by the Medicine Standing Committee on Animals of the Osaka University Graduate School.
Blood flow was assessed by laser Doppler imaging (LDI: Moor Instruments, Devon, United Kingdom) before and on post-treatment days 7, 14, 21 and 28 as described previously [21]. Mice were assigned to one of four groups: injection of AG-30 (500 μg) with gelatin, AG-30 (100 μg) with gelatin, control peptide (500 μg) with gelatin and only AG-30 (500 μg). Capillary density within the ischaemic thigh adductor skeletal muscles was analysed to obtain specific evidence of vascularity at the level of the microcirculation. After fixation in cold acetone (−20°C for 15 min.) capillary endothelial cells (EC) were identified by immunohistochemical staining with anti-mouse PECAM mAb (Pharmingen, San Diego, CA, USA).
Statistical analysis
All values are expressed as mean ± S.E.M. Analysis of variance with subsequent Fisher's PLSD test was employed to determine the significance of differences in multiple comparisons.
Results
Novel angiogenic peptide, AG-30
Review of the protein databases (Blast in the NCBI database) failed to identify any known proteins with homology to this antimicrobial-like peptide, which we subsequently designated as AG (angiogenic peptide)-30. Structural analysis by the AGADIR program suggested that AG-30 forms an a-helix structure (predicted value: 21.17%) and an amphipathic structure in which most positively charged amino acids are localized to one side of the molecule with most hydrophobic amino acids localized to the other side of the molecule (see hydrophobic cluster analysis [HCA] plot [22] in Fig. 1A). To follow and give a better impression of the environment of each amino acid widely separated by the unfolding of the cylinder, hydrophobic residues were encircled, demonstrating sets of adjacent hydrophobic residues in hydrophobic clusters (Fig. 1B). Of note, there were very few proline or glycine residues that would otherwise interrupt the α-helical structure. The structure of this peptide is reminiscent of that of some antimicrobial peptides that directly interact with bacterial membranes.
1.
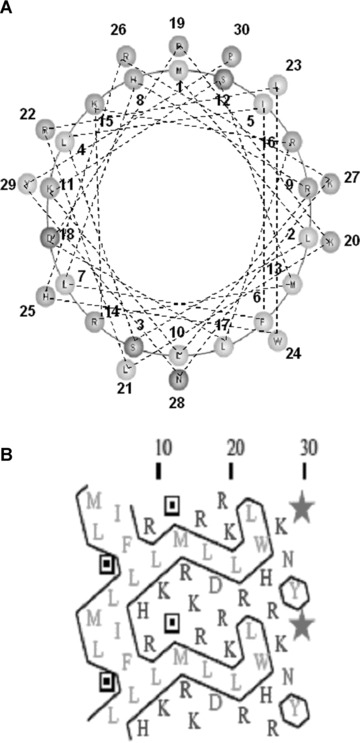
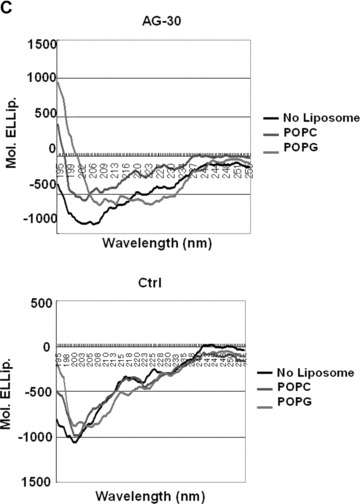
Conformation analysis of AG-30. (A) Schematic wheel plot of amino acid distribution of AG-30 in alpha-helical structure (http://www.nmr.cabm.rutgers.edu/bioinformatics/Proteomic_tools/Helical_wheel/). The hydrophobic amino acids are represented in gold, positive charged amino acids in green, negative charged amino acids in red-purple, proline in red and the other amino acids in blue-purple. (B) Protein sequence analysis on an alpha-helical 2D pattern using an HCA plot (http://bioserv.rpbs.jussieu.fr/RPBS/cgi-bin/Ressource.cgi?chznlg=fr&chzn_rsrc=HCA). The hydrophobic residues are represented in yellow-green and encircled, and different symbols used for P (*), and S (???). The positively charged amino acids are represented in purple, and the negatively charged amino acid (D) or other amino acids (N) are represented in red. (C) Secondary structure analysis of AG-30 and control peptide (Ctrl) by circular dichroism spectroscopy. CD spectra of AG-30 (50 μmol/L) and control peptide (50 μmol/L) without liposome (black line), with POPC (blue line) and with POPG (red line). Molar ellipticities, which indicates dimensions degrees decilitres mol–1 decimeter−1, were plotted against wavelength from 195 to 255 nm.
Antimicrobial effect of AG-30
To examine whether AG-30 may form a helix structure, CD analysis was performed. AG-30 and the control peptide were subjected to structural analysis in phosphate buffer (PB), and no helical structure was observed for either AG-30 or the control peptide. Importantly, after the addition of POPC liposomes (mimicking a zwitterionic eucaryotic membrane) or POPG liposomes (mimicking a bacterial membrane) in PBS, AG-30 showed a helical propensity in membrane mimetic solvent, especially in POPG. Upon combining AG-30 and liposomes, the negative molar ellip-ticy, suggestive of a coiled shape, changed to a recognizable positive value, indicative of a helical structure in the region of 185–195 nm (Fig. 1C). The region from 195 to 225 nm also retained a characteristic shape for a coil without liposomes and a helical structure in POPG.
AG-30 showed antimicrobial activity against P. aeruginosa (MIC: 5 μg/ml), E. coli (MIC: 40 μg/ml), and S. aureus (MIC: 20 μg/ml) (Table 1) through a lytic’ action by attaching to cationic peptides. Addition of exogenous CaCl2 or MgCl2 (3 mmol/L or 6 mmol/L) completely attenuated the antimicrobial effect of AG-30 against P. aeruginosa (data not shown). These results suggest that AG-30 transforms its structure to α-helix when it encounters bacterial membrane, thereby acting as antimicrobial peptide by disrupting the bacterial membrane.
1.
Minimal inhibitory concentrations (MIC*: μg/ml) of AG-30 and LL-37
| AG30 | LL37 | Control | |
|---|---|---|---|
| E. coli (ATCC 25922) | 40.0 | 5.00 | >80 |
| P. aeruginosa (ATCC 27853) | 5.00 | 2.50 | >10 |
| S. aureus (ATCC 29213) | 20.0 | >80 | >80 |
MIC was defined as the lowest concentration of peptide that inhibited the bacterial visible growth after incubation for 16 hrs at 37°C with vigorous shaking.
Angiogenic property of AG-30
Some antibacterial peptides also possess angiogenic properties. In a human endothelial cell viability assay, AG-30 increased MTS activity in a dose-dependent (from 0.1 to 1.0 μg/ml) manner, and was more potent than LL-37 (Fig. 2A). Treatment with AG-30 also increased vascular smooth muscle cell viability in a dose-dependent manner (from 0.1 to 1.0 μg/ ml), as assessed by MTS assay (Fig. 2B). Human cell lines, such as HEK293, SAS (tongue cancer), HuH7 (hepatoma) and HeLa (cervical cancer) did not show significant response in viability in response to AG-30 stimulation (data not shown). In addition, chemokinetic cell migration significantly increased in response to AG-30 (which was more potent than LL-37) but not in response to control peptide (Fig. 2C). We have also addressed the chamotactic action of AG-30 by the addition of AG-30 in the lower chambers and HAEC in the upper chambers. As a result, the treatment of AG-30 with HAEC showed not only chemokinetic migration but also chemotactic action (Fig. 2D), although the chemokinetic action of AG-30 was stronger than chemotactic action. Further, tube formation was greater in response to AG-30 treatment when compared with LL-37 or control peptide treatment (Fig. 2E). These data demonstrate that AG-30 was superior to LL-37 in terms of inducing endothelial cell growth, migration and tube formation.
2.
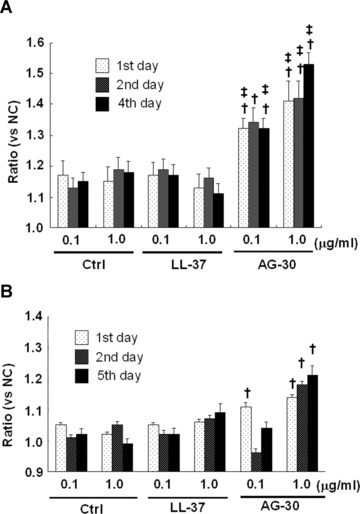
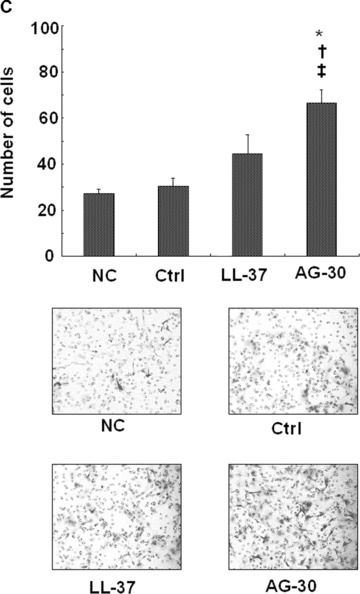
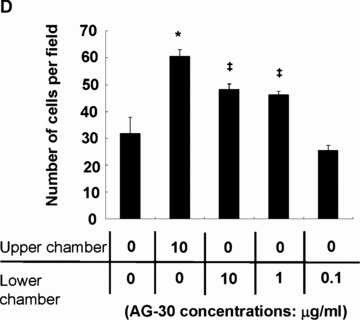
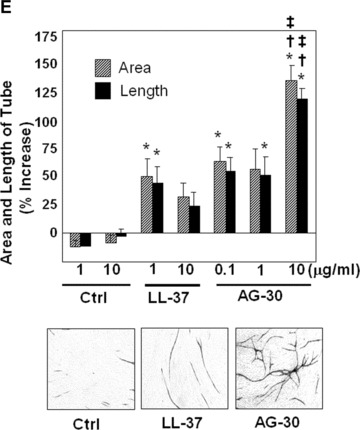
Effect of AG-30 peptide on human aortic endothelial cells (HAEC) and human aortic smooth muscle cells (HASMC). (A) MTS assay with HAEC on days 1, 2 and 4. (B) MTS assay with HASMC on days 1, 3, and 5. (C) Chemokinetic migration assay with HAEC. Lower panel shows representative pictures of each group. (D) Chemotactic migration assay by the addition of AG-30 in the lower chambers (0.1, 1.0 and 10 μg/ml). *P < 0.01 versus NC, ‡P < 0.05 versus NC. n= 8 per group. (E) Tube formation, quantified as area and length. Lower panel shows representative pictures of each group. ‘Ctrl’ indicates treatment with control peptide. ‘AG-30’ indicates treatment with AG-30 peptide (10 μg/ml). ‘LL-37’ indicates treatment with LL-37 peptide (10 μg/ml). Results are expressed as fold-increase relative to the effect of NC (no treatment) for MTS assay and percentage increase to the effect of NC for migration and tube formation, respectively. *P < 0.05 versus NC, †P < 0.05 versus Ctrl, ‡P < 0.05 versus LL-37. n= 8 per group.
The in vivo angiogenic activity of AG-30 has been evaluated by an established mouse Matrigel plug assay [23]. Matrigel, containing either control peptide or AG-30, was subcutaneously injected into male C57BL/6 mice. Seven days later, neovessels containing intact red blood cells were more numerous in mice that received Matrigel plug containing AG-30 than in mice receiving Matrigel plug containing control peptide. The increased presence of neovessels in the AG-30-treated group relative to the other groups was confirmed by immunostaining with anti-CD31 and anti-a-smooth muscle actin antibody (Fig. 3A). Further, these results were reproduced using another type of matrigel assay (DIVAA), and quantification by FITC-lectin staining showed that treatment with AG-30 significantly increased neovessels with a potency that was superior to that of LL-37 (Fig. 3B). In combination with the results demonstrated above, these data suggest that AG-30 induces angiogenesis via increased endothelial cell and smooth muscle cell migration and invasion.
3.
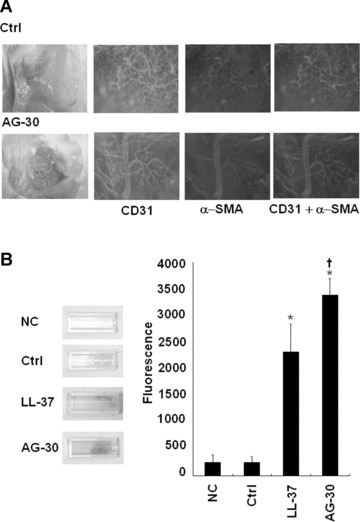
Effect of AG-30 peptide on in vivo angiogenesis evaluated by Matrigel plug assay and directed in vivo angiogenesis assay (DIVAA). (A) Representative pictures of the Matrigel with AG-30 peptide or control peptide (10 μg/ml). Matrigel plugs were stained with anti-CD31 antibody (green) or anti-α-smooth muscle antibody (red), and capillary like structures were photographed ×100 magnification) under a fluorescent microscope. (B) Left panel shows that representative pictures of angioreactors implanted in mice, and the right panel shows the quantification of FITC-lectin positive cells that were treated with control peptide (10 μg/ml), LL-37 (10 μg/ml), AG-30 (10 μg/ml) or no treatment (NC). *P < 0.05 versus NC, †P < 0.05 versus Ctrl, ‡P < 0.05 versus LL-37. n= 3–4 per group.
Microarray and real-time PCR Analysis in AG-30-treated endothelial cells
To confirm the mechanism of AG-30-induced angiogenesis, differences in gene expression were assessed by microarray analysis (total 43,941 genes). At 6, 24 and 72 hrs after the start of treatment, 47 genes (0.001%), 61 genes (0.13%) and 287 genes (0.65%) were up-regulated and 2 genes (0.00005%), 297 genes (0.67%) and 288 genes (0.66%), were down-regulated, respectively. Subsequent cluster analysis with GeneSpring software revealed at least a twofold increase in angiopoietin 2 (Ang-2), IL8, JAG1, IGF-1, ETS variant gene 7, and chemokine ligand 2 (CCL2) and chemokine receptor 4 (CXCR4) (data not shown). Further, up-regulation of these angiogenesis-related genes occurred in a time-dependent manner for most genes and was maintained for at least 72 hrs (Table 2). We further carried out quantitative RT-PCR of these angiogenesis-related molecules in triplicates to confirm if they would be up-regulated or not. Indeed, we confirmed the increased expression of Ang-2 by 6.24-fold increases in average, IL-8 by 8.45-fold, Jag-1 by 2.82-fold, VEGF by 3.28-fold and IGF-1 by 2.82-fold, respectively, at 72 hrs after AG-30 stimulation (Fig. 4). These results suggest that these angiogenesis-related genes are actually up-regulated and may synergistically induce angiogenesis after the treatment with AG-30.
2.
Cluster analysis using GeneSpring software to detect a twofold increased or decreased in gene expression at 72 hrs after AG-30 treatment
| 6 hrs | 24 hrs | 72 hrs | ||
|---|---|---|---|---|
| Angiopoietin 2 | 1.03 | 2.18 | 8.52 | NM_001147 |
| Angiopoietin-like 4, transcript variant | 0.80 | 1.64 | 5.46 | NM_139314 |
| Interleukin 8 | 1.00 | 1.45 | 3.92 | NM_000584 |
| Jagged 1 | 1.73 | 1.31 | 3.90 | NM_000214 |
| Epiregulin | 1.27 | 1.72 | 2.45 | NM_001432 |
| Vascular endothelial growth factor | 1.36 | 0.87 | 2.07 | NM_003376 |
| Insulin-like growth factor | 0.85 | 1.17 | 2.07 | NM_00061 |
| Neuropilin-1 soluble isoform 11 | 0.15 | 0.21 | 0.20 | AF280547 |
4.
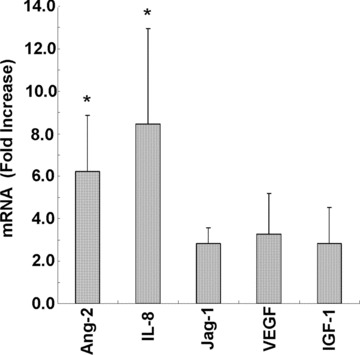
Real-time quantitative reverse transcriptase polymerase chain reaction of angiopoietin-2 (Ang-2), Interleukin-8 (IL-8), Jagged 1 (Jag-1), vascular endothelial growth factor (VEGF) and insulin-like growth factor-1 (IGF-1) transcripts in HAEC at 72 hrs after treatment with AG-30. Results are expressed as fold-increase relative to the non-stimulants for copy numbers of each mRNA. *P < 0.05 versus Ctrl. n= 3 per group.
Effect of slow-release AG-30 in a mouse ischaemic hind limb model
Finally, we evaluated the angiogenic effect of AG-30 in a mouse ischaemic hind limb model. To achieve sustained administration of AG-30 over a local area, a slow-release system was developed using cationic microspheres. As AG-30 includes a number of cationic amino acids, an anionic gelatin might be suitable for slow release of AG-30. However, a part of the gelatin might bind to AG-30 and interfere with its angiogenic effect. Thus, we used PI (iso-electric point) = 5 gelatin, which has been used for the delivery of recombinant FGF-2 in clinical trials of therapeutic angiogenesis. Characterization of the slow release profile of AG-30 (Fig. 5A) revealed that the PI = 5 gelatin slowly dissolved and disappeared in mice within approximately 14 days and that AG-30 was rapidly released within the first 3 days.
5.
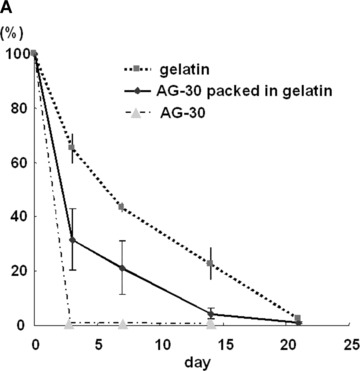
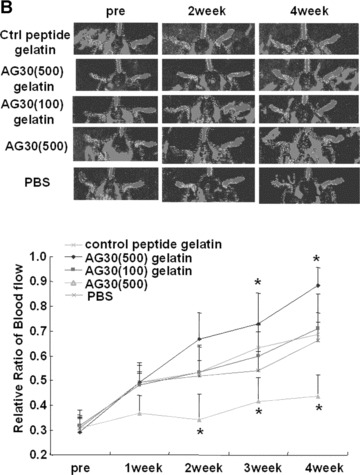
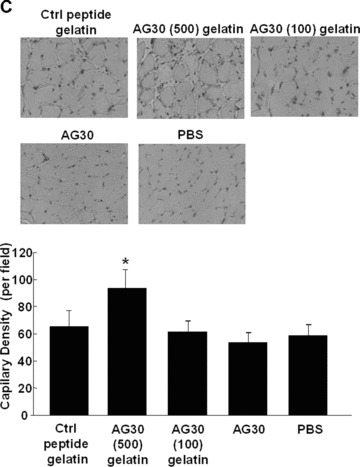
Angiogenic effect of AG-30 in mouse ischaemic hind limb model. (A) Slow release profile of 125I-labelled AG-30 with/without gelatin microspheres or 125I-labelled gelatin at 3, 7, 14 and 21 day. (B) Evaluation of angiogenic effect by measuring mouse hind limb blood flow at pre-treatment (pre), 1, 2, 3 and 4 week. Ischaemic hind limb mouse were treated with gelatin/AG-30 (500 μg or 100 μg), gelatin/control peptide (500 μg), only AG-30 (500 μg) or PBS. Upper panel shows representative pictures of LDI images of hind limb blood flow at pre-, 2 and 4 weeks after treatment. *P < 0.05 versus control peptide gelatin. (C) Evaluation of capillary density of the AG-30-treated ischaemic hind limb. Upper panel shows representative photomicrographs (×400) of tissue immunostained with anti-CD31 antibody. Lower panel shows number of capillary per field. *P < 0.05 versus control peptide gelatin. n= 5–8 per group.
At 10 days after unilateral ligation of the femoral artery of the mouse hind limb, blood flow was measured by LDI. Figure 5B shows representative LDI images of hind limb blood flow at 0, 14 and 28 days after injection. Although serial LDI examination revealed natural recovery of hind limb blood flow in the control peptide group, injection of AG-30 (500 μg) with gelatin resulted in more rapid recovery of the ratio of ischaemic(normal blood flow and a significantly sustained increase, but injection of AG-30 (100 μg) with gelatin or only AG-30 did not.
Figure 5C shows representative photomicrographs of tissue immunostained with anti-CD31 antibody. Quantitative analysis revealed that the capillary density was significantly increased in the AG-30 (500 μg) with gelatin group when compared with other groups.
Discussion
The present study demonstrated that a novel small peptide, AG-30, induced angiogenesis by promoting the expression of diverse angiogenic molecules. AG-30 was predicted to belong to a family of antimicrobial peptides that function to protect the host organisms against a large variety of invading pathogens [4–6]. These peptides are usually highly amphipathic molecules with hydrophobic and hydrophilic moieties segregating into distinct domains on the molecule surface.
In response to a bacterial membrane, AG-30 transforms to an α-helix structure and disrupts the membrane via a lytic effect. By contrast, the eucaryotic membrane does not induce a change in AG-30 structure change into an α-helix. Cholesterol, which is an essential component in the eucaryotic cell membrane, prevents self-damage of endogenous antimicrobial peptides [24]. This differential phenomenon underlies the ability of AG-30 to kill bacteria while simultaneously promoting endothelial cell growth. Defensins and cathelicidin families are well known as antimicrobial peptides, which are related to angiogenesis. Defensin contains 6–8 cysteine residues that form characteristic disulfide bridge, and shows β-sheet structure [25], which may be different from that of AG-30. In antimicrobial properties, both peptides can cover the similar broad spectrum, however AG-30, but not defensin, can directly induce angiogenesis. Moreover, defensin may induce apoptosis of human umbilical vein endothelial cell (HUVEC) and inhibits new capillary formation [26]. Human cathelicidin LL-37, on the other hand, is a peptide which has both antimicrobial and angiogenic effects [25]. In the present study, AG-30-stimulated EC growth, cell migration, tube formation and in vivo arteriogenesis was more potent than LL-37. LL-37-mediated angiogenesis is dependent on stimulation of a G-protein-coupled receptor that is upstream of the PLC-γ/PKC/NFkB, ERK-1 and 2 MAPK, and the PI3K/Akt pathways. Another porcine cathelicidin peptide, PR-39, appears to affect angiogenesis by inhibiting the ubiquitin-proteasome-dependent degradation of HIF-1α[10]. In our study, microarray analysis demonstrated that AG-30 induced increases in IGF-1, IL-8, JAG1, CCL2, CXCR2 or Ang-2 expression, all of which may independently induce angiogenesis [27–29]. For example, IL-8 is a potent chemotactic cytokine that induces migration during angiogenesis [30] and its expression is regulated by NF-κB. Jagged-1 is a ligand of Notch signalling that regulates embryonic patterning and binary cell fate decisions and plays a critical role in mammalian embryogenesis and vascular development [31]. Although the exact mechanism is unclear, AG-30 could potentially act on several key molecules to regulate gene expression via autocrine or paracrine effects, thereby resulting in angiogenesis or vasculogenesis.
The effect of AG-30 on up-regulation of angiogenic genes appeared to be much slower than that of typical cell surface receptor-mediated processes. In fact, the AG-30-induced up-regulation of angiogenic genes was time-dependent and was maintained for at least 72 hrs. Of importance, we found that AG-30 was present in the nucleus at 24 or 48 hrs after treatment, which is a unique phenomenon when compared with other microbial peptides. Thus, AG-30 may enter the cytoplasm and nucleus in a sequential fashion to ultimately interact directly with transcription factors to regulate gene expression. We speculate that the ability of AG-30 to enter intracellular compartments may be cell type-dependent.
A slow-release system was used to stabilizing AG-30, as naked AG-30 peptide is very unstable and is easily degraded by proteases in vivo. As shown in Figure 4A, gelatin microspheres enabled the slow release of AG-30 in muscle over a period of 2 weeks in response to a single injection. One unique advantage of gelatin is its variable electrical nature (produced by altering the processing method of collagen) [32] that can be used to change its degradation rate or to modulate the physicochemical interactions between the drug and gelatin molecules [33]. The present study developed a suitable slow release system for AG-30 that resulted in therapeutic angiogenesis in a mouse ischaemic hind limb model. The release of AG-30 is driven by enzymatic degradation of carrier gelatin microspheres. Previous studies have reported the absence of inflammation, macrophage accumulation and granuloma formation around the injected site of cationized gelatin microspheres and that the degree of inflammatory reaction was dependent on the type and size of the particle [34]. Moreover, gelatin itself has been used for medical applications, and its biosafety has been proved through its extensive clinical use in surgical biomaterials. Thus, the use of this system may allow the application of AG-30 in various clinical conditions.
In summary, the present study identified a unique angiogenic and antimicrobial peptide, AG-30, with potential clinical applications. However, since small peptides are typically not stable in vivo, development of a drug delivery system to establish slow release of this peptide would be required for clinical utility.
Acknowledgments
This study was supported by a grant from the National Institute of Biomedical Innovation. We would like to thank all members of the Gene Therapy Science laboratory for critical discussion during the course of this work.
References
- 1.Lalich M, McNeel DG, Wilding G, Liu G. Endothelin receptor antagonists in cancer therapy. Cancer Invest. 2007;25:785–94. doi: 10.1080/07357900701522588. [DOI] [PubMed] [Google Scholar]
- 2.Jackson DC, Purcell AW, Fitzmaurice CJ, Zeng W, Hart DN. The central role played by peptides in the immune response and the design of peptide-based vaccines against infectious diseases and cancer. Curr Drug Targets. 2002;3:175–96. doi: 10.2174/1389450024605436. [DOI] [PubMed] [Google Scholar]
- 3.Groneberg DA, Rabe KF, Fischer A. Novel concepts of neuropeptide-based drug therapy: vasoactive intestinal polypeptide and its receptors. Eur J Pharmacol. 2006;533:182–94. doi: 10.1016/j.ejphar.2005.12.055. [DOI] [PubMed] [Google Scholar]
- 4.Ganz T. Defensins and host defense. Science. 1999;286:420–1. doi: 10.1126/science.286.5439.420. [DOI] [PubMed] [Google Scholar]
- 5.Steiner H, Hultmark D, Engstrom A, Bennich H, Boman HG. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature. 1981;292:246–8. doi: 10.1038/292246a0. [DOI] [PubMed] [Google Scholar]
- 6.Izadpanah A, Gallo RL. Antimicrobial peptides. J Am Acad Dermatol. 2005;52:381–90. doi: 10.1016/j.jaad.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 7.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–95. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 8.Zasloff M. Antimicrobial peptides in health and disease. N Engl J Med. 2002;347:1199–200. doi: 10.1056/NEJMe020106. [DOI] [PubMed] [Google Scholar]
- 9.Zasloff M. Innate immunity, antimicrobial peptides, and protection of the oral cavity. Lancet. 2002;360:1116–7. doi: 10.1016/S0140-6736(02)11239-6. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Post M, Volk R, Gao Y, Li M, Metais C, Sato K, Tsai J, Aird W, Rosenberg RD, Hampton TG, Sellke F, Carmeliet P, Simons M. PR39, a peptide regulator of angiogenesis. Nat Med. 2000;6:49–55. doi: 10.1038/71527. [DOI] [PubMed] [Google Scholar]
- 11.Larrick JW, Hirata M, Zhong J, Wright SC. Anti-microbial activity of human CAP18 peptides. Immunotechnology. 1995;1:65–72. doi: 10.1016/1380-2933(95)00006-2. [DOI] [PubMed] [Google Scholar]
- 12.Koczulla R, Von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, Pfosser A, Boekstegers P, Welsch U, Hiemstra PS, Vogelmeier C, Gallo RL, Clauss M, Bals R. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest. 2003;111:1665–72. doi: 10.1172/JCI17545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishikawa T, Nakagami H, Matsuki A, Maeda A, Yo CY, Harada T, Morishita R, Tamai K, Kaneda Y. Development of high-throughput functional screening of therapeutic genes, using a hemagglutinating virus of Japan envelope vector. Hum Gene Ther. 2006;17:470–5. doi: 10.1089/hum.2006.17.470. [DOI] [PubMed] [Google Scholar]
- 14.Hunter HN, Jing W, Schibli DJ, Trinh T, Park IY, Kim SC, Vogel HJ. The interactions of antimicrobial peptides derived from lysozyme with model membrane systems. Biochim Biophys Acta. 2005;1668:175–89. doi: 10.1016/j.bbamem.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Pearson RD, Steigbigel RT, Davis HT, Chapman SW. Method of reliable determination of minimal lethal antibiotic concentrations. Antimicrob Agents Chemother. 1980;18:699–708. doi: 10.1128/aac.18.5.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagami H, Morishita R, Yamamoto K, Taniyama Y, Aoki M, Matsumoto K, Nakamura T, Kaneda Y, Horiuchi M, Ogihara T. Mitogenic and antiapoptotic actions of hepatocyte growth factor through ERK, STAT3, and AKT in endothelial cells. Hypertension. 2001;37:581–6. doi: 10.1161/01.hyp.37.2.581. [DOI] [PubMed] [Google Scholar]
- 17.Rikitake Y, Hirata K, Kawashima S, Ozaki M, Takahashi T, Ogawa W, Inoue N, Yokoyama M. Involvement of endothelial nitric oxide in sphingosine-1-phosphate-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22:108–14. doi: 10.1161/hq0102.101843. [DOI] [PubMed] [Google Scholar]
- 18.De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule-and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–74. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukunaka Y, Iwanaga K, Morimoto K, Kakemi M, Tabata Y. Controlled release of plasmid DNA from cationized gelatin hydrogels based on hydrogel degradation. J Control Release. 2002;80:333–43. doi: 10.1016/s0168-3659(02)00026-3. [DOI] [PubMed] [Google Scholar]
- 20.Bolton AE, Hunter WM. The labelling of proteins to high specific radioactivities by conjugation to a 125I-containing acy-lating agent. Biochem J. 1973;133:529–39. doi: 10.1042/bj1330529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagami H, Maeda K, Morishita R, Iguchi S, Nishikawa T, Takami Y, Kikuchi Y, Saito Y, Tamai K, Ogihara T, Kaneda Y. Novel autologous cell therapy in ischemic limb disease through growth factor secretion by cultured adipose tissue-derived stromal cells. Arterioscler Thromb Vasc Biol. 2005;25:2542–7. doi: 10.1161/01.ATV.0000190701.92007.6d. [DOI] [PubMed] [Google Scholar]
- 22.Gaboriaud C, Bissery V, Benchetrit T, Mornon JP. Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett. 1987;224:149–55. doi: 10.1016/0014-5793(87)80439-8. [DOI] [PubMed] [Google Scholar]
- 23.Rikitake Y, Kawashima S, Yamashita T, Ueyama T, Ishido S, Hotta H, Hirata K, Yokoyama M. Lysophosphatidylcholine inhibits endothelial cell migration and proliferation via inhibition of the extracellular signal-regulated kinase pathway. Arterioscler Thromb Vasc Biol. 2000;20:1006–12. doi: 10.1161/01.atv.20.4.1006. [DOI] [PubMed] [Google Scholar]
- 24.Steiner H, Andreu D, Merrifield RB. Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim Biophys Acta. 1988;939:260–6. doi: 10.1016/0005-2736(88)90069-7. [DOI] [PubMed] [Google Scholar]
- 25.Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Defensins and cathelicidins: neutrophil peptides with roles in inflammation, hyperlipidemia and atherosclerosis. J Cell Mol Med. 2005;9:3–10. doi: 10.1111/j.1582-4934.2005.tb00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chavakis T, Cines DB, Rhee JS, Liang OD, Schubert U, Hammes HP, Higazi AA, Nawroth PP, Preissner KT, Bdeir K. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18:1306–8. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- 27.Smith LE, Shen W, Perruzzi C, Soker S, Kinose F, Xu X, Robinson G, Driver S, Bischoff J, Zhang B, Schaeffer JM, Senger DR. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat Med. 1999;5:1390–5. doi: 10.1038/70963. [DOI] [PubMed] [Google Scholar]
- 28.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–80. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 29.Rabinovsky ED, Draghia-Akli R. Insulinlike growth factor I plasmid therapy promotes in vivo angiogenesis. Mol Ther. 2004;9:46–55. doi: 10.1016/j.ymthe.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 31.Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–32. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veis A. The physical chemistry of gelatin. Int Rev Connect Tissue Res. 1965;3:113–200. doi: 10.1016/b978-1-4831-6753-4.50010-7. [DOI] [PubMed] [Google Scholar]
- 33.Ikada Y, Tabata Y. Protein release from gelatin matrices. Adv Drug Deliv Rev. 1998;31:287–301. doi: 10.1016/s0169-409x(97)00125-7. [DOI] [PubMed] [Google Scholar]
- 34.Kushibiki T, Matsumoto K, Nakamura T, Tabata Y. Suppression of tumor metastasis by NK4 plasmid DNA released from cationized gelatin. Gene Ther. 2004;11:1205–14. doi: 10.1038/sj.gt.3302285. [DOI] [PubMed] [Google Scholar]