

Abstract
Myocardial necrosis triggers inflammatory changes and a complex cytokine cascade that are only incompletely understood. The chemokine receptor CCR1 mediates inflammatory recruitment in response to several ligands released by activated platelets and up-regulated after myocardial infarction (MI). Here, we assess the effect of CCR1 on remodelling after MI using Ccr1-deficient (Ccr1−/−) mice. MI was induced in Ccr1−/− or wild-type mice by proximal ligation of the left anterior descending (LAD). Mice were sacrificed and analysed at day 1, 4, 7, 14 and 21 after MI. While initial infarct areas and areas at risk did not differ between groups, infarct size increased to 20.6±8.4% of the left ventricle (LV) in wild-type mice by day 21 but remained at 11.2±1.2% of LV (P<0.05) in Ccr1−/− mice. This attenuation in infarct expansion was associated with preserved LV function, as analysed by isolated heart studies according to Langendorff. Left ventricular developed pressure was 84.5±19.8 mmHg in Ccr1−/− mice compared to 49.0±19.7 mmHg in wild-type mice (P<0.01) and coronary flow reserve was improved in Ccr1−/− mice. An altered post-infarct inflammatory pattern was observed in Ccr1−/− mice characterized by diminished neutrophil infiltration, accelerated monocyte/lymphocyte infiltration, decreased apoptosis, increased cell proliferation and earlier myofibroblast population in the infarcted tissue. In conclusion, functional impairment and structural remodelling after MI is reduced in the genetic absence of Ccr1 due to an abrogated early inflammatory recruitment of neutrophils and improved tissue healing, thus revealing a potential therapeutic target.
Keywords: myocardial infarction, myocardial remodelling, inflammation, chemokine receptor, leukocyte recruitment
Introduction
As a major cause of death in western countries, myocardial infarction (MI) due to the atherothrombotic occlusion of coronary arteries represents an irreversible necrosis of heart muscle caused by a prolonged imbalance between the oxygen supply and its metabolic demand. Myocardial necrosis is followed by an inflammatory response with complement activation and free radical generation, triggering a complex cytokine cascade and an up-regulation of chemokines [1–6]. Beyond their crucial role in coordinating leukocyte recruitment during inflammation and vascular disease [7, 8], chemokines appear to modulate the inflammatory response and to influence the myocardial healing and scar formation after MI [4, 9, 10].
A study using gene-deficient mice and bone marrow chimeras [11] revealed that Cxcr2, a receptor for murine CXCL8 orthologs, exerts opposing effects on myocardial viability during ischaemia-reperfusion with recruitment of damaging inflammatory cells prevailing over tissue-protective effects mediated in resident myocardial cells. Besides recent findings indicating that CCL2 and its receptor Ccr2 contribute to leukocyte infiltration and left ventricular remodelling after MI [12–18], little is known about the involvement of other chemokines in the progression of tissue damage after MI, although an improved understanding may reveal promising therapeutic targets.
CCR1 is a receptor for CCL3 and CCL5, chemokines released by activated platelets and presented on the endothelium to mediate inflammatory recruitment [7, 19]. The CCR1 ligands CCL3/MIP-1α[14, 17] and CCL5/RANTES [14, 17] are potent mononuclear cell chemoattractants and markedly up-regulated after MI. Moreover, myeloid progenitor inhibitory factor-1/CCL23 induces endothelial cell migration and promotes angiogenesis via CCR1 [20, 21], while the CCR1 ligand CCL7 appears to be a potent myocardial mesenchymal stem cell homing factor [22]. The purpose of this study was to test the hypothesis that Ccr1 plays an important role in myocardial remodelling and inflammatory cell recruitment after MI using Ccr1 knockout mice.
Methods
Animals and murine model of myocardial infarction
C57/B6 mice and Ccr1−/−[23] were intubated under general anaesthesia (100 mg/kg ketamine, 10 mg/kg xylacine, intraperitoneal) and positive pressure ventilation was maintained with oxygen and isofluran 0.2%, using a rodent respirator. Hearts were exposed by left thoracotomy and MI was produced by suture occlusion of the left anterior descending artery (LAD). The muscle layer and skin incision were closed with a silk suture. Animal experiments were approved by local authorities and complied with German animal protection law.
Langendorff perfusion
At the indicated time points, the mice were anaesthetized and the heart function was analysed using a Langendorff apparatus (Hugo Sachs Elektronik-Harvard Apparatus) and dedicated software (EMKA Technologies, Paris, France) under constant perfusion pressure (100 mmHg) and electrical stimulation to assure a constant heart rate (600 bpm). The coronary flow and developed pressure were measured without or with adenosine (1 μmol/l). At the end, the hearts were fixed in distension with 10% formalin and cut into 5 μm serial slices.
Histomorphometry and assessment of myocardial infarction size
Serial sections (10–12 per mouse, 400 μm apart, up to the mitral valve) were stained with Gomori's 1-step trichrome stain. The infarcted area was determined in all sections using Diskus software (Hilgers) and expressed as percentage of total left ventricular volume.
Quantitative immunohistochemistry and immunofluorescence
Serial sections (three per mouse, 400 μm apart) were stained to analyse infarcted areas for the content of neutrophils (specific esterase, Sigma), macrophages (F4/80, Serotec), lymphocytes (CD3, Serotec), vessels (CD31, Santa Cruz) and myofibroblasts (α-smooth muscle actin, DAKO). Cells were numbered in six different fields pro-section and expressed as cells/mm2. Cardiomyocytes were stained with an anti-sarcomeric myosin antibody (Sigma). Collagen content was determined by Gomori's 1-step trichrome staining, and calculated as the positively stained area percentage of the infarcted area, using the AnalySIS software (Soft Imaging Systems). To determine apoptosis and proliferation in the infarcted area, serial sections (three per mouse, 400 μm apart) were stained with in situ cell death detection kit (Roche) or anti-Ki-67 (DAKO) respectively, and counter-stained with DAPI. Apoptotic cell or proliferation indexes were expressed as percentage of positive cells.
Flow cytometry
Blood samples were stained with anti-CD3ɛ-FITC (BD Pharmingen) for lymphocytes, anti-Gr-1-PerCP-Cy5.5 (BD Pharmingen) for neutrophils and CD115-PE (eBioscience) for monocytes and analysed by FACS to determine the distribution of blood cells at different time points after MI.
ELISA
ELISA was also performed from serum samples using DuoSet ELISA Development System kit for mouse MIP-1α and RANTES (R&D Systems).
mRNA isolation and RT-PCR
mRNA was also isolated from the infarcted area with TRIzol Reagent (Gibco) and RT-PCR was performed using Omniscript kit (Qiagen). Following oligonucleotide primers specific were used for murine RANTES cDNA (forward primer: 5′-GTGCCCACGTCAAGGAGTAT-3′ and reverse primer: 5′-GGGAAGCGTATACAGGGTCA-3′), murine MIP-1_ cDNA (forward primer: 5′-AGATTCCACGC-CAATTCATC-3′, reverse primer: 5′-CTCAAGCCCCT-GCTCTACAC-3′), murine MMP2 (forward primer: 5′-CACACCAGGTGAAGGATGTG-3′, reverse primer: 5′-AGGGCTGCATTGCAAATATC-3′), murine MMP9 (forward primer: 5′-CACCACCACAACTGAACCAC-3′, reverse primer: 5′-CTCAGAAGAGCCCGCAGTAG-3′) and murine TIMP-1 (forward primer: 5′-ATCAGT GCCTGCAGCTTCTT-3′, reverse primer: 5′-TGACGGCTCTGGTAGTCCTC -3′). Murine ß-actin cDNA served as housekeeping gene and was amplified in parallel with the gene of interest (forward primer: 5′-AGCCATGTACGTAGCCATCC-3′, reverse primer: 5′-CTCTCAGCTGTGGTGGTGAA-3′).
Transmigration assay
Neutrophils were isolated from wild-type and Ccr1−/− blood by gradient centrifugation using Lympholyte mammal (Cedarlane laboratory). After isolation, neutrophils were resuspended in assay medium (Hepes-buffered Hanks' balanced salt solution containing 0.5% bovine serum albumin and 1 μmol/l Mg2+ and Ca2+) and allowed to transmigrate across 5 μm transwell filters toward murine RANTES (100 ng/ml, PeproTech), MIP-1α (100 ng/ml, PeproTech) or buffer control in the lower chamber for 2 hrs at 37°C. Transmigrated neutrophils were counted by flow cytometry. The ratio of chemokine-induced versus spontaneous migration was defined as chemotactic index [24, 25].
Statistical analysis
Data represent mean±S. E. M. Statistical analysis was performed with Prism 4 software (Graph Pad) using unpaired Student's-t test or 1-way ANOVA followed by Newmann-Keuls post test. P-values <0.05 were considered significant.
Results
Analysis of myocardial infarction
MI injury was induced in wild-type and Ccr1−/− mice. No differences were observed between survival rates between wild-type and Ccr1−/− mice (96% each). Initial infarct areas and areas at risk did not differ between the groups one day after MI (Fig. 1). Whereas the infarct size in wild-type mice reached 20.6±8.4% of the left ventricle (LV) at day 21 and following wound contraction decreased to 15.4±0.4% at day 28, a significant attenuation and prevention of this increase in infarct size was observed in Ccr1−/− mice, where infarct size remained at 11.2_1.2% of the LV at day 21 and only slightly further decreased to 10.3±1.7% after 28 days (P<0.05;Fig. 1). Concordantly, isolated heart studies according to Langendorff showed a decrease in LV function over time to a LV developed pressure (LVDP) of 49.0±19.7 mmHg in wild-type mice (Fig. 2A), whereas LV function significantly preserved in Ccr1−/− mice compared to wild-type mice (84.5±19.8 mmHg, P<0.01 versus wild-type). Coronary flow analysis revealed no differences in basal flow (Fig. 2B) but a significant increase and recovery in coronary flow after adenosine administration in Ccr1−/−mice versus wild-type mice (Fig. 2C). These data indicate a reduced infarct area and improved LV function following MI in Ccr1−/− mice. Analysis of cellular parameters and scar composition demonstrated that Ccr1−/−mice display a decrease in apoptosis rates in the scar area at intermediate time points (19.4 ± 2.9%versus 33.0±4.0% in wild-type mice at day 14, P<0.05, Fig. 3A and B). To further characterize the cell types undergoing apoptosis, double immunofluorescence staining revealed that predominantly cardiomyocytes became apoptotic at day 1–4 after MI and only few apoptotic neutrophils or myofibroblasts could be detected (Fig. 4B and C). No apoptosis in macrophages was observed at early time points (data not shown). Moreover, cell proliferation at earlier time points was increased in the infarct area of Ccr1−/− mice (17.0 ± 0.9%versus 8.3 ± 1.4% in wild-type mice, P<0.0001, Fig. 3C and D). Myofibroblasts were shown to proliferate at day 4 but began to undergo apoptosis at 1 week after MI (Fig. 4C and D). This was also associated with an earlier repopulation with α-actin+ myofibroblasts (3205 ± 251 cells/mm2versus 2523 ± 289 cells/mm2 in wild-type mice at day 4, Fig. 3E and F). In addition, a significant increase in the collagen content of the infarcted areas was observed in Ccr1−/− mice (3.6 ± 2.2%versus 10.4 ± 0.9% in wild-type mice at day 1, P<0.05 and 13.7 ± 0.8%versus 22.4 ± 1.0% in wild-type mice at day 7, P<0.01, Fig. 4A).
1.
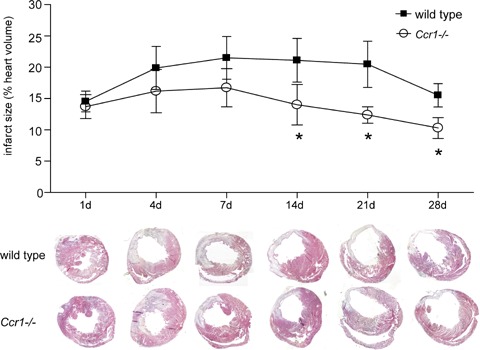
Histomorphometry of the infarcted myocardium. Paraffin-embedded hearts were cut in serial sections, stained with Gomori 1-step staining and heart wall and infracted area were measured by planimetry. Compared to wild-type mice, Ccr1−/− mice show a significantly smaller infarct size (mean ±S. E. M., n= 6–8 mice per group, *P<0.05 versus wild-type).
2.
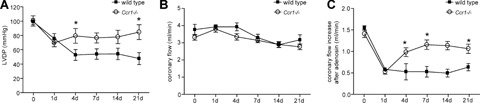
Functional assessment after myocardial infarction. Hearts collected at different time points after infarction were studied in a Langendorff perfusion system. (A) Left ventricular developed pressure (LVDP) shows a better-preserved basal function of the Ccr1−/− compared to wild-type hearts (n = 6–8 mice per group, mean ±S. E. M., *P<0.05 versus wild-type). While basal coronary flow did not differ (B), Ccr1−/− mice displayed a better vascular reserve, as shown after adenosine administration (C, mean ±S. E. M., *P<0.05 versus wild-type).
3.
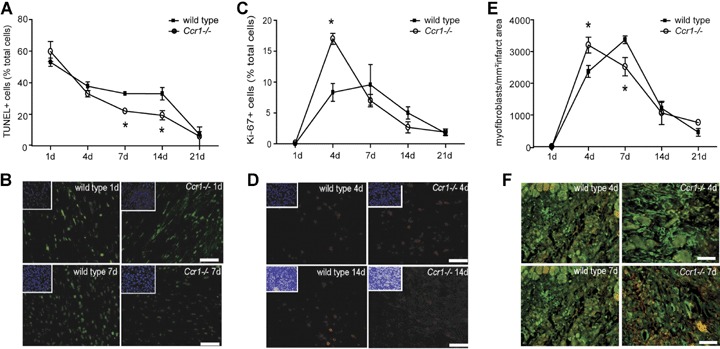
Scar remodelling after myocardial infarction. Apoptosis, as detected by TUNEL staining (A, B) and cell proliferation, as detected by Ki-67 staining (C, D) and myofibroblast content as determined by staining for a-actin (E, F) were determined and quantified in infarcted myocardium at different time points after injury. Ccr1−/− hearts showed lower apoptosis rates (A), earlier and higher cell proliferation rates (C) and an earlier myofibroblast population (E) in the infracted area (n= 6–8 mice per group, mean ±S. E. M., *P<0.05 versus wild-type). Insets in the representative images (B, D) show nuclear counter-staining of the same field with DAPI. Scale bar:50 μm.
4.
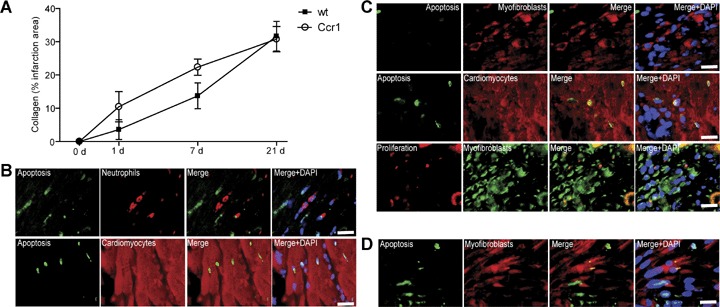
Scar remodelling after myocardial infarction. The collagen content of infarcted areas was determined in Ccr1−/−and wild-type mice after MI at the time points indicated (A). In wild-type mice, double immunofluorescence of apoptotic TUNEL+ or proliferating Ki-67+ cells with specific cell markers was performed (B–D). At 1 day after MI, cardiomyocytes (sarcomeric myosin) but not neutrophils (specific esterase) undergo apoptosis (B). After 4 days, cardiomyocytes but not myofibroblasts (α-smooth muscle actin) underwent apoptosis and myofibroblasts exhibited proliferation (C). At 7 days after MI, apoptosis can also be detected in myofibroblasts (D).
Staining for CD31+ endothelial cells giving rise to angiogenesis in the infarcted area by immunofluorescence revealed no differences between wild-type and Ccr1−/− mice (data not shown). Together, our data suggest an improved wound healing response in Ccr1−/− mice.
Inflammatory cell analysis after MI
We next analysed inflammatory cell recruitment into the infarcted area after MI. In wild-type mice, the neutrophil content in the infarcted area were transiently increased at 1 day after MI but subsequently reversed to baseline levels at day 4 (Fig. 5A). Notably, this increase was almost completely abrogated in Ccr1−/− mice (189 ± 33 cells/mm2versus 707 ± 244 cells/mm2 in wild-type, P<0.05, Fig. 5A). In contrast, the peak in the content of both monocytes and lymphocytes in the infarcted area appeared to occur earlier in Ccr1−/− mice (day 7) as compared to wild-type mice (day 14), in line with an accelerated healing response (Fig. 5B and C). Interestingly, the peripheral blood counts of these leukocyte subsets (neutrophils, monocytes, lymphocytes) did not differ between Ccr1−/− and wild-type mice (data not shown), indicating that changes in inflammatory cell content in the infarcted area were due to a defect in recruitment rather than mobilization of leukocytes.
5.
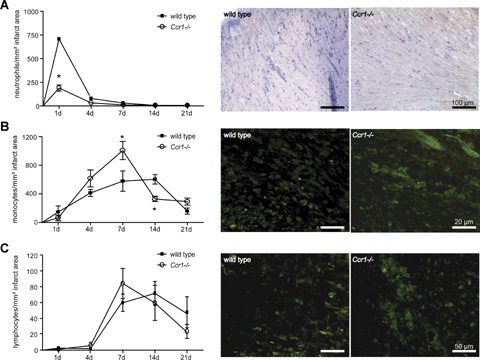
Myocardial infiltration with inflammatory blood cells after infarction. Neutrophils, monocytes and lymphocytes were analysed in infarcted myocardium by immunostaining for specific esterase, F4/80 or CD3. Compared to wild-type hearts, Ccr1−/− hearts displayed a significantly reduced infiltration with neutrophils (A), whereas infiltration with monocytes (B) and lymphocyte (C) occurred earlier and more prominent in Ccr1−/− myocardium (n= 6–8 mice per group, mean ±S. E. M., *P<0.05 versus wild-type).
Chemokine expression and function after MI
To analyse changes in the expression and function of chemokines accounting for the differences in inflammatory cell infiltration in Ccr1−/− mice, we determined serum levels and chemotactic activities of MIP-1α and RANTES as prominent Ccr1 ligands. As analysed by ELISA, both chemokines rapidly increased in serum 1 day after MI in wild-type mice (Fig. 6A). This increase was abrogated in Ccr1−/− mice for MIP-1α (27.0 ± 2.9 versus 69.5 ± 14.7 pg/ml in wild-type mice, P<0.05) and for RANTES (181.3 ± 50.7 versus 381.2 ± 84.1 in wild-type mice, P<0.05). To assess functional effects of these chemokines in neutrophils, we performed in vitro transmigration experiments using neutrophils isolated from Ccr1−/− and wild-type mice (Fig. 6B). Notably, MIP-1α rather than RANTES triggered substantial transmigration of wild-type neutrophils but not Ccr1−/− neutrophils, indicating that neutrophil transmi-gration was mediated by Ccr1 and that MIP-1α was more important than RANTES in this process (Fig. 6B). As analysed by RT-PCR, the expression of MIP-1α transcripts was up-regulated but did not differ between wild-type and Ccr1−/− hearts 1 day after MI (Fig. 6C). Notably, expression of RANTES mRNA was markedly enhanced in Ccr1−/− compared to wild-type mice 4 days after MI (Fig. 6C), whereas the expression of MCP-1 mRNA was unaltered (Fig. 6D), implying that RANTES rather than MCP-1 is responsible for the more prominent, that is accelerated monocyte and lymphocyte infiltration in Ccr1−/− mice at this time point. In contrast, the rapid decrease in monocyte recruitment might be related to the reduction in MCP-1 expression after 1 week in Ccr1−/− compared with wild-type mice (Fig. 6D). No differences in the expression levels of MMP2, MMP9 or TIMP-1 were observed after MI in Ccr1−/− compared with wild-type mice (Fig. 6D).
6.
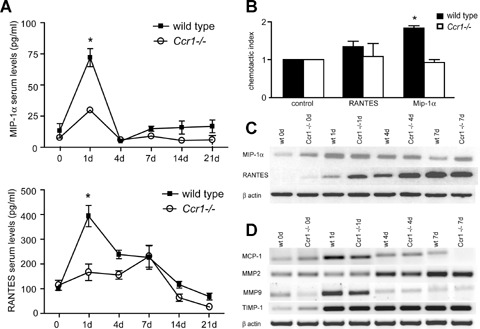
Expression and function of chemokines after myocardial infarction. Serum MIP-1α and RANTES were determined by ELISA at indicated time points after MI (A). MIP-1α and RANTES rapidly increase in serum after MI in wild-type but not in Ccr1−/− mice (n= 6–8 mice per group, mean ±S. E. M., *P<0.05 versus wild-type). Transmigration assays were performed using Ccr1−/− and wild-type neutrophils using transwell-filters (B). Wild-type, but not Ccr1−/− neutrophils migrated towards MIP-1α but not RANTES (mean ±S. E. M., n= 3, *P<0.05 versus control). Transcripts for MIP-1α and RANTES were analysed in myocardium by RT-PCR at day 1, 4 and 7 after MI (C, D). RANTES, but not MIP-1α mRNA expression was increased in Ccr1−/−mice compared with wild-type (C). mRNA expression levels of MMP2, MMP9 and TIMP-1 showed no differences between the two groups (D).
Discussion
Here, we show that genetic deficiency in Ccr1 protects against myocardial re-modelling and inflammatory changes and improves myocardial function after MI. This appeared to be due to an attenuation of neutrophil infiltration and associated with reduced apoptosis in Ccr1−/− compared with control mice. Similarly, treatment with a blocking anti-IL8 antibody targeting neutrophil transmigration or genetic deletion of Cxcr2 in neutrophils impairing their recruitment inhibited myocardial ischaemia-reperfusion injury [11, 26]. Since CXCR2 has been described to mainly act as an arrest receptor [27, 28], it is not inconceivable that Ccr1 can complement CXCR2 functions during neu-trophil transmigration. Indeed, Ccr1 can exert non-redundant functions in haematopoiesis, host defence and inflammation [29], and blocking Ccr1 has been found to significantly reduce the inflammation in models of sepsis [30] or lung injury [31]. Moreover, a function of Ccr1 in neutrophil recruitment has recently been supported by a study employing a model of ischaemia-reperfusion, where Ccr1−/− mice display an impairment in early neutrophil arrest and transmigration in the cremasteric microcirculation [32]. On the other hand, the reduction in apoptosis encountered in the scar area of Ccr1−/− mice was mainly evident between day 7 and 14 and not at day 1 and 4, when cardiomyocytes apoptosis is most abundant, and thus appears to be attributable to effects on recruited leukocytes and fibroblasts, rather than early effects on cardiomyocytes.
In accordance with our findings, a rapid and transient up-regulation of CCR1 and MIP-1α transcripts has been observed after myocardial ischaemia-reperfusion injury in a study by Dewald et al., which differed from a more prolonged up-regulation of CCR2. Moreover, the genetic deficiency in CCR2 or its ligand CCL2 attenuated LV remodelling after experimental MI, resulting in decreased and/or delayed macrophage infiltration, reduced gelatinolytic activity, delayed phagocytic clearance of injured cardiomyocytes and diminished granulation and myofibroblast accumulation, but did not affect neutrophil recruitment [16, 17]. However, in contrast to our findings in Ccr1-deficient mice, these mice did not display a reduction in infarct size, suggesting that an abrogation of Ccr1 effects on neutrophil recruitment may more markedly limit infarct size with subsequent benefits for LV function after MI.
Unlike in mice, MIP1-α appears to produce only negligible levels of calcium flux and chemotaxis in human neutrophils [33]. However, CCR1 expression in these cells can be increased by granulocyte-macrophage colony stimulating factor [34], which is elevated in serum after MI [14, 35, 36], or by interferon-γ[37], which is up-regulated in myocardium after MI [38]. This may result in relevant effects of CCR1 in human disease. Indeed, blocking human CCR1 by a specific antagonist CP-481715 decreased cell infiltration and inflammatory responses in human CCR1-transgenic mice [39]. Moreover, the human CCR1 antagonist BX471 reduced heart and renal transplant rejection in rats, which may also involve a component of initial reperfusion damage [40, 41]. It is further notable that Ccr1 deficiency resulted in a prevention of acute chemokine secretion as evident by reduced serum levels after MI. This may be related to local attenuation inflammatory cell recruitment and their interactions with platelets and microvascular endothelium.
Another important mechanism contributing to the preserved LV function and the improved tissue healing with higher content of collagen and myofibroblasts could be the earlier monocyte and lymphocyte infiltration of the infarcted area, likely due to the increased RANTES expression in infarcted myocardium. The exact role of monocyte and lymphocytes in the healing scar has not been fully investigated; however they might have a pivotal role in the transition between inflammation and repair. They create an appropriate environment to promote neoves-sel formation, fibroblast proliferation and extracellular matrix metabolism and serve as a major source of cytokines and growth factors [42]. In particular, a Gr-1low (or in the human system CD14+CD16+) monocyte subset with high expression of Ccr5 [43] has recently been found to display angiogenic properties [44]. Therefore, an earlier recruitment of a distinct mono-cyte subset in the infarcted area could be one mechanism for an improved cardiac function in Ccr1−/− compared to wild-type mice. This accumulation could be favoured by the marked up-regulation of the Ccr5 ligand in the infarcted myocardium of Ccr1−/− mice.
Monocytes/macrophages also produce transforming growth factor-β1 (TGF-β1), a major fibrogenic cytokine, which can mediate the differentiation of fibroblasts into myofibroblasts [45]. As a versatile cell type, myofibroblasts can assume different functions in extracellular matrix metabolism and contractile activity [3, 46, 47] and contribute to scar formation after MI. Our results indicate that the appearance and proliferation of myofibroblasts occurs earlier in Ccr1−/− than in wild-type mice. This might be related to the early RANTES-induced recruitment of specific monocyte subsets releasing TGF-β1 or of fibroblast progenitors and their differentiation into myofibroblasts in Ccr1−/− mice. In addition, the attenuation of neutrophil recruitment in Ccr1−/− mice associated with a reduced release of proteolytic enzymes or reactive oxygen species might contribute to an auspicious and less inflammatory environment permitting other cells, such as fibroblast to proliferate and further differentiate in response to ischaemic injury. The exact functions of these cell types, however, warrants further investigation and refinement.
Angiogenesis, that is the formation of new vessels in the area of ischaemia is also important to preserve and improve cardiac function [1, 3, 5, 48] and Ccr1 has been reported to mediate endothelial cell migration and to promote angiogenesis through CCL23, which besides MIP-1α and RANTES is another ligand for Ccr1 [20, 21]. However, we did not find differences in CD31 staining between Ccr1−/− and wild-type mice. This mechanism is therefore unlikely to explain the improvement of tissue healing and LV function after MI in our model.
In summary, our study provides evidence that Ccr1 deficiency preserves cardiac function and precipitates wound healing processes by attenuating the neutrophil-induced myocardial injury and necrosis and by supporting the early infiltration with mono-cytes, collagen synthesis and myofibroblast accumulation. Given the availability of effective small molecular antagonists [30, 31, 42, 43], we therefore propose that targeting CCR1 may represent a promising and accessible strategy to optimize cardiac repair and to prevent ventricular remodelling after myocardial injury.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (FOR809-TP7, 5FB542-C12), the Interdisciplinary Centre for Clinical Research ‘BIOMAT’(NTV 113-c, TVB 113-d) within the faculty of Medicine at RWTH Aachen University. We want to thank Dr. M. Hristov for excellent technical advice.
References
- 1.Blankesteijn WM, Creemers E, Lutgens E, Cleutjens JP, Daemen MJ, Smits JF. Dynamics of cardiac wound healing following myocardial infarction: observations in genetically altered mice. Acta Physiol Scand. 2001;173:75–82. doi: 10.1046/j.1365-201X.2001.00887.x. [DOI] [PubMed] [Google Scholar]
- 2.Yang F, Liu YH, Yang XP, Xu J, Kapke A, Carretero OA. Myocardial infarction and cardiac remodelling in mice. Exp Physiol. 2002;87:547–55. doi: 10.1113/eph8702385. [DOI] [PubMed] [Google Scholar]
- 3.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovascular research. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 4.Filippatos G, Parissis JT, Adamopoulos S, Kardaras F. Chemokines in cardiovascular remodeling: clinical and therapeutic implications. Currt Mol Med. 2003;3:139–47. doi: 10.2174/1566524033361546. [DOI] [PubMed] [Google Scholar]
- 5.Lu L, Zhang JQ, Ramires FJ, Sun Y. Molecular and cellular events at the site of myocardial infarction: from the perspective of rebuilding myocardial tissue. Biochem Biophys Res Commun. 2004;320:907–13. doi: 10.1016/j.bbrc.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 6.Marx N, Neumann FJ, Ott I, Gawaz M, Koch W, Pinkau T, Schomig A. Induction of cytokine expression in leukocytes in acute myocardial infarction. J Am Coll Cardiol. 1997;30:165–70. doi: 10.1016/s0735-1097(97)00116-2. [DOI] [PubMed] [Google Scholar]
- 7.Weber C, Schober A, Zernecke A. Chemokines:key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 8.Weber C. Chemokines take centre stage in vascular biology. Thromb Haemost. 2007;97:685–7. [PubMed] [Google Scholar]
- 9.Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med. 2005;15:163–9. doi: 10.1016/j.tcm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–47. [PubMed] [Google Scholar]
- 11.Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW, Kitsis RN. Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia-reperfusion injury:recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 2003;108:2387–92. doi: 10.1161/01.CIR.0000093192.72099.9A. [DOI] [PubMed] [Google Scholar]
- 12.Ono K, Matsumori A, Furukawa Y, Igata H, Shioi T, Matsushima K, Sasayama S. Prevention of myocardial reperfusion injury in rats by an antibody against monocyte chemotactic and activating factor/monocyte chemoattractant protein-1. Lab Invest. 1999;79:195–203. [PubMed] [Google Scholar]
- 13.Koyanagi M, Egashira K, Kitamoto S, Ni W, Shimokawa H, Takeya M, Yoshimura T, Takeshita A. Role of monocyte chemoattractant protein-1 in cardiovascular remodeling induced by chronic blockade of nitric oxide synthesis. Circulation. 2000;102:2243–8. doi: 10.1161/01.cir.102.18.2243. [DOI] [PubMed] [Google Scholar]
- 14.Parissis JT, Adamopoulos S, Venetsanou KF, Mentzikof DG, Karas SM, Kremastinos DT. Serum profiles of C-C chemokines in acute myocardial infarction: possible implication in postinfarction left ventricular remodeling. J Interferon Cytokine Res. 2002;22:223–9. doi: 10.1089/107999002753536194. [DOI] [PubMed] [Google Scholar]
- 15.Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N, Wen J, Egashira K, Takeshita A. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:2134–40. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- 16.Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165:439–47. doi: 10.1016/S0002-9440(10)63309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 18.Hayasaki T, Kaikita K, Okuma T, Yamamoto E, Kuziel WA, Ogawa H, Takeya M. CC chemokine receptor-2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia-reperfusion in mice. Circ J. 2006;70:342–51. doi: 10.1253/circj.70.342. [DOI] [PubMed] [Google Scholar]
- 19.Von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 20.Hwang J, Son KN, Kim CW, Ko J, Na DS, Kwon BS, Gho YS, Kim J. Human CC chemokine CCL23, a ligand for CCR1, induces endothelial cell migration and promotes angiogenesis. Cytokine. 2005;30:254–63. doi: 10.1016/j.cyto.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Son KN, Hwang J, Kwon BS, Kim J. Human CC chemokine CCL23 enhances expression of matrix metalloproteinase-2 and invasion of vascular endothelial cells. Biochem Biophys Res Commun. 2006;340:498–504. doi: 10.1016/j.bbrc.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 22.Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, Carthy PM, Penn MS. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem cells. 2007;25:245–51. doi: 10.1634/stemcells.2006-0293. [DOI] [PubMed] [Google Scholar]
- 23.Zernecke A, Liehn EA, Gao JL, Kuziel WA, Murphy PM, Weber C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: involvement of IL-10. Blood. 2006;107:4240–3. doi: 10.1182/blood-2005-09-3922. [DOI] [PubMed] [Google Scholar]
- 24.Weber C, Lu CF, Casasnovas JM, Springer TA. Role of alpha L beta 2 integrin avidity in transendothelial chemotaxis of mononuclear cells. J Immunol. 1997;159:3968–75. [PubMed] [Google Scholar]
- 25.Baltus T, Weber KS, Johnson Z, Proudfoot AE, Weber C. Oligomerization of RANTES is required for CCR1-mediated arrest but not CCR5-mediated transmigration of leukocytes on inflamed endothelium. Blood. 2003;102:1985–8. doi: 10.1182/blood-2003-04-1175. [DOI] [PubMed] [Google Scholar]
- 26.Boyle EM, Jr, Kovacich JC, Hebert CA, Canty TG, Jr, Chi E, Morgan EN, Pohlman TH, Verrier ED. Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 1998;116:114–21. doi: 10.1016/S0022-5223(98)70249-1. [DOI] [PubMed] [Google Scholar]
- 27.Weber KS, Von Hundelshausen P, Clark-Lewis I, Weber PC, Weber C. Differential immobilization and hierarchical involvement of chemokines in monocyte arrest and transmigration on inflamed endothelium in shear flow. Eur J Immunol. 1999;29:700–12. doi: 10.1002/(SICI)1521-4141(199902)29:02<700::AID-IMMU700>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, Mack M, Jung S, Littman DR, Ley K. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 2001;108:1307–14. doi: 10.1172/JCI12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao JL, Wynn TA, Chang Y, Lee EJ, Broxmeyer HE, Cooper S, Tiffany HL, Westphal H, Kwon-Chung J, Murphy PM. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J Exp Med. 1997;185:1959–68. doi: 10.1084/jem.185.11.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He M, Horuk R, Moochhala SM, Bhatia M. Treatment with BX471, a CC chemokine receptor 1 antagonist, attenuates systemic inflammatory response during sepsis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1173–80. doi: 10.1152/ajpgi.00420.2006. [DOI] [PubMed] [Google Scholar]
- 31.He M, Horuk R, Bhatia M. Treatment with BX471, a nonpeptide CCR1 antagonist, protects mice against acute pancreatitis-associated lung injury by modulating neutrophil recruitment. Pancreas. 2007;34:233–41. doi: 10.1097/mpa.0b013e31802e7598. [DOI] [PubMed] [Google Scholar]
- 32.Reichel CA, Khandoga A, Anders HJ, Schlondorff D, Luckow B, Krombach F. Chemokine receptors Ccr1, Ccr2, and Ccr5 mediate neutrophil migration to postischemic tissue. J Leukoc Biol. 2006;79:114–22. doi: 10.1189/jlb.0605337. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Youn BS, Gao JL, Murphy PM, Kwon BS. Differential effects of leukotactin-1 and macrophage inflammatory protein-1 alpha on neutrophils mediated by CCR1. J Immunol. 1999;162:4938–42. [PubMed] [Google Scholar]
- 34.Cheng SS, Lai JJ, Lukacs NW, Kunkel SL. Granulocyte-macrophage colony stimulating factor up-regulates CCR1 in human neutrophils. J Immunol. 2001;166:1178–84. doi: 10.4049/jimmunol.166.2.1178. [DOI] [PubMed] [Google Scholar]
- 35.Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J. 1994;72:561–6. doi: 10.1136/hrt.72.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parissis JT, Adamopoulos S, Venetsanou K, Kostakis G, Rigas A, Karas SM, Kremastinos D. Plasma profiles of circulating granulocyte-macrophage colony-stimulating factor and soluble cellular adhesion molecules in acute myocardial infarction. Contribution to post-infarction left ventricular dysfunction. Eur Cytokine Netw. 2004;15:139–44. [PubMed] [Google Scholar]
- 37.Bonecchi R, Polentarutti N, Luini W, Borsatti A, Bernasconi S, Locati M, Power C, Proudfoot A, Wells TN, Mackay C, Mantovani A, Sozzani S. Up-regulation of CCR1 and CCR3 and induction of chemotaxis to CC chemokines by IFN-gamma in human neutrophils. J Immunol. 1999;162:474–9. [PubMed] [Google Scholar]
- 38.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reper-fused myocardium. Am J Pathol. 1995;146:419–28. [PMC free article] [PubMed] [Google Scholar]
- 39.Gladue RP, Cole SH, Roach ML, Tylaska LA, Nelson RT, Shepard RM, McNeish JD, Ogborne KT, Neote KS. The human specific CCR1 antagonist CP-481715 inhibits cell infiltration and inflammatory responses in human CCR1 transgenic mice. J Immunol. 2006;176:3141–8. doi: 10.4049/jimmunol.176.5.3141. [DOI] [PubMed] [Google Scholar]
- 40.Grone HJ, Weber C, Weber KS, Grone EF, Rabelink T, Klier CM, Wells TN, Proudfood AE, Schlondorff D, Nelson PJ. Met-RANTES reduces vascular and tubular damage during acute renal transplant rejection: blocking monocyte arrest and recruitment. FASEB J. 1999;13:1371–83. [PubMed] [Google Scholar]
- 41.Horuk R, Clayberger C, Krensky AM, Wang Z, Grone HJ, Weber C, Weber KS, Nelson PJ, May K, Rosser M, Dunning L, Liang M, Buckman B, Ghannam A, Ng HP, Islam I, Bauman JG, Wei GP, Monahan S, Xu W, Snider RM, Morrissey MM, Hesselgesser J, Perez HD. A non-peptide functional antagonist of the CCR1 chemokine receptor is effective in rat heart transplant rejection. J Biol Chem. 2001;276:4199–204. doi: 10.1074/jbc.M007457200. [DOI] [PubMed] [Google Scholar]
- 42.Weihrauch D, Arras M, Zimmermann R, Schaper J. Importance of monocytes/macrophages and fibroblasts for healing of micronecroses in porcine myocardium. Mol Cell Biochem. 1995;147:13–9. doi: 10.1007/BF00944778. [DOI] [PubMed] [Google Scholar]
- 43.Weber C, Belge KU, Von Hundelshausen P, Draude G, Steppich B, Mack M, Frankenberger M, Weber KS, Ziegler-Heitbrock HW. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 44.Venneri MA, Palma MD, Ponzoni M, Pucci F, Scielzo C, Zonari E, Mazzieri R, Doglioni C, Naldini L. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109:5276–85. doi: 10.1182/blood-2006-10-053504. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–6. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 46.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–38. [PMC free article] [PubMed] [Google Scholar]
- 47.Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–83. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- 48.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–9. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]