

Abstract
We previously showed that the human heart expresses all known P2X and P2Y receptors activated by extra-cellular adenine or uracil nucleotides. Despite evidence that, both in humans and rodents, plasma levels of ATP and UTP markedly increase during myocardial infarction, the differential effects mediated by the various adenine- and uracil-preferring myocardial P2 receptors are still largely unknown. Here, we studied the effects of adenine and uracil nucleotides on murine HL-1 cardiomyocytes. RT-PCR analysis showed that HL-1 cardiomyocytes express all known P2X receptors (except for P2X2), as well as the P2Y2,4,6,14 subtypes. Exposure of cardiomyocytes to adenine nucleotides (ATP, ADP or BzATP) induced apoptosis and necrosis, as determined by flow-cytometry. Cell death was exacerbated by tumour necrosis factor (TNF)-α, a cytokine implicated in chronic heart failure progression. Conversely, uracil nucleotides (UTP, UDP and UDPglucose) had no effect ‘per se’, but fully counteracted the deleterious effects induced by adenine nucleotides and TNF-α, even if added to cardiomyocytes after beginning exposure to these cell death-inducing agents. Thus, exposure of cardiomyocytes to elevated concentrations of ATP or ADP in the presence of TNF-α contributes to cell death, an effect which is counteracted by uracil-preferring P2 receptors. Cardiomyocytes do not need to be ‘primed’ by uracil nucleotides to become insensitive to adenine nucleotides-induced death, suggesting the existence of a possible ‘therapeutic’ window for uracil nucleotides-mediated protection. Thus, release of UTP during cardiac ischaemia and in chronic heart failure may protect against myocardial damage, setting the basis for developing novel cardioprotective agents that specifically target uracil-preferring P2Y receptors.
Keywords: apoptosis, heart failure, P2Y receptors, cytokines, signal transduction
Introduction
Extracellular purine and pyrimidine nucleotides are primordial molecules that serve multiple biological processes [1]. They are responsible for transmission of genetic information, provide currency units for biological energy transfer, and are also released from cells to provide integral elements of extracellular signalling. Within such ‘purinergic’ system, specificity and regulation is dictated by the source of extracellular nucleotides and by the expression of specific receptors on target cells: the P2 receptors.
Two distinct families of P2 receptors are currently recognized: the ligand-gated P2X receptors, and the G protein-coupled P2Y receptors [2–5]. So far, seven distinct P2X receptor subtypes (P2X1–7) and eight P2Y receptors (the P2Y1,2,4,6,11,12,13,14 subtypes) have been cloned and characterized (ibidem). Recently, a new putative member of the P2Y receptor family, previously known as the ‘orphan’ receptor GPR17, has been identified [6]. P2X receptors can form homomeric and heteromeric channels allowing Na+ and Ca2+ influxes, and have been implicated in fast excitatory transmission [4] and, under some circumstances, in apoptotic cell death [7]. Conversely, activation of metabotropic P2Y receptors leads to modulation of adenylyl cyclase and/or phospholipase C, depending upon the specific P2Y subtype [5, 8]. While P2X receptors primarily respond to adenine nucleotides (i.e. ATP and ADP), P2Y receptors can be further subdivided into (i) the adenine-nucleotide-preferring receptors (human and rodent P2Y1, P2Y12 and P2Y13, and human P2Y11), (ii) the uracil-nucleotide-preferring receptors, responding to either UTP or UDP (human P2Y4 and P2Y6), (iii) receptors of mixed selectivity, which respond to both adenine and uracil nucleotides (human and rodent P2Y2, rodent P2Y4) and, (iv) receptors responding solely to sugar nucleotides, such as UDPglucose and UDPgalactose (P2Y14) [5]. Interestingly, the newly identified P2Y-like receptor (GPR17) is solely activated by uracil nucleotides, with a pharmacological profile which is intermediate between P2Y6 and P2Y14[6].
Both P2X and P2Y receptors are widely distributed in human tissues and are known to mediate fundamental roles in the cardiovascular system [9]. At myocardial level, despite very early reports on the cardiac effects of adenine nucleotides and nucleo-sides [10], and demonstration that ATP is released from myocardial sympathetic terminals and hypoxic cardiomyocytes [9], the effects mediated by specific myocardial P2 receptors are just now beginning to be elucidated. Adenine nucleotides induce rat cardiac myocyte contraction possibly through P2X receptors, and/or by increasing L-type Ca2+ currents (summarized in [1]). Positive inotropism by ATP has been also attributed to P2Y receptors [11]. Besides adenine nucleotides, uracil nucleotides have been also recently reported to act as positive inotropic agents in rat and mouse cardiomyocytes [12,13]. The utilization of stable selective agonists has allowed the pharmacological characterization of the specific P2 receptors involved in the positive inotropic effects induced by uracil nucleotides. Most important, nucleotide plasma levels were measured to evaluate whether UTP is released in patients with coronary heart disease. Besides ATP, venous plasma levels of UTP were increased (57%) in patients with myocardial infarction, giving the first evidence for UTP release in man. Based on these results, authors concluded that extracellular pyrimidines (UTP and UDP) could be important inotropic factors involved in the development of chronic heart failure, a disease that still remains a major cause of ill health in industrial societies countries [14,15].
However, UTP may also play a significant role in protecting the ischaemic heart against injury [16,17]. Short-term activation of UTP receptors indeed protects newborn rat cardiomyocytes against hypoxic damage [16] and significantly reduces the cell death caused by hypoxia via activation of P2Y2 and P2Y4 receptors [17].
Thus, besides influencing cardiac contractility, P2 receptors may also directly regulate the viability of myocardial cells. In a recent study, we have shown that the full repertoire of known P2X and P2Y receptors is expressed in the human heart from both healthy donors and patients with chronic heart failure (CHF) undergoing cardiac transplantation [18]. We also showed that the ionotropic P2X6 receptor was up-regulated in CHF subjects, as confirmed by quantitative real time-PCR [18]. The potential significance of this change was studied in primary cardiac fibroblasts freshly isolated from young pigs. Exposure of cardiac fibroblasts to ATP or its hydrolysis-resistant-analogue benzoylATP (BzATP) induced apoptosis. Tumour necrosis factor (TNF)-α (a pro-inflammatory cytokine implicated in CHF progression) [19] exacerbated cell death. In cardiac fibroblasts, exposure to TNF-α inhibited the down-regulation of P2X6 mRNA induced by prolonged agonist exposure, suggesting that, by preventing ATP-induced P2X6 desensitization, TNF-α may abolish a defence mechanism meant at avoiding Ca2+ overload and, ultimately, Ca2+-dependent cell death. This may provide a basis for P2X6 up-regulation in CHF. Globally, these findings suggest that the interaction between TNF-α and the up-regulated P2X6 receptor may represent a novel pathogenic mechanism in CHF.
In the present study, based on our previous results and on the demonstration that, besides ATP, UTP is also released from the heart during cardiac ischaemia [12], we have investigated the effects of both adenine and uracil nucleotides on the viability of HL-1 cardiomyocytes, the only available cell line that spontaneously contracts in vitro and maintains a differentiated cardiac phenotype [12]. In line with our previous findings [20], we show that exposure of HL-1 cardiomyocytes to ATP or ADP induces cell death by both apoptosis and necrosis, an effect which was increased by pre-treatment of cells with TNF-α. On the contrary, uracil nucleotides (UTP, UDP and UDPglucose), utilized either alone or with TNF-α, did not induce apoptosis or necrosis ‘per se’, but significantly reduced cardiomyocyte death induced by adenine nucleotides. We conclude that activation of uracil-preferring P2Y receptors in cardiomyocytes counteracts the deleterious effects mediated by ATP and TNF-α on myocardial cells. We also suggest that release of UTP during cardiac ischaemia and during development of CHF may be protective against myocardial damage. These findings may have important therapeutic implications and set the basis for the development of novel cardioprotective agents that specifically target uracil-preferring P2Y receptors.
Materials and methods
HL-1 cardiomyocytes
HL-1 cardiomyocytes were a kind gift of Professor W.C. Claycomb, LSU Health Sciences Center, New Orleans, LA, USA [20]. Cells were cultured in Complete Claycomb Medium supplemented with 10% foetal calf serum (FCS) (JRH Biosciences, UK) and Norepinephrine (100 μM, Sigma-Aldrich), following Professor Claycomb's instructions. Cells were seeded in six-well plates at various densities (48–100–200 × 103); on the next day, medium was replaced and after 8 hrs, HL-1 cells were pre-treated overnight with TNF-α (10 ng/ml) followed by addition of ATP (25–500 μM) or ADP (250–500 μM) or 3′-benzoyl adenosine 5′-triphosphate (BzATP, 100 μM), adenosine 5′-O-(3-thiotriphosphate) (ATPγS, 500 μM), 2-methylthioadenosine 5′-triphosphate (2MeSATP, 500 μM) and 2-methylthioadenosine 5′-diphosphate (2MeSADP, 500 μM) (Sigma-Aldrich). Uracil nucleotides (UTP, UDP, UDPglucose, 100 nM-500 μM, Sigma-Aldrich) and all antagonists [8-cyclopentyl-1, 3-dipropylxanthine (CPX 10 μM, Sigma-Aldrich), 5-amino-7-(β-phenylethyl)-2-(8-furyl)pyrazolo(4,3-epsilon)-1,2, 4-triazolo(1,5-c)pyrimidine (SCH58261, 100 nM, from Schering-Plough), 9-chloro-2-(2-furyl)-5-phenylacety-lamino(1,2,4)-triazolo(1,5-c)quinazoline (MRS1220, 100 nM, Sigma-Aldrich), 2′ deoxy-N6-methyl adenosine 3′, 5′ diphosphate (MRS2179, 100 μM, Sigma-Aldrich), pyridoxal-phosphate-6-azophenyl-2′,4′ -disulfonate (PPADS, 100 μM, Sigma-Aldrich), suramin (100 μM, Sigma-Aldrich), reactive blue 2 (RB2, 100 μM, Sigma-Aldrich) and Cangrelor (10 μM, The Medicines Company, Parsippany, NJ, USA)] were all added to cultures 30 min before adenine nucleotides. In selected experiments, 24 hrs after seeding, medium was replaced with Claycomb medium without FCS and Norepinephrine, followed by addition of TNF-α and adenine/uracil nucleotides. In these experiments, after pharmacological treatments cells were maintained in Claycomb medium without serum and norepinephrine; under these conditions, cells are more sensitive to cell death and therefore ATP was used at lower concentration (100 μM).
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Extraction of total RNA and RT-PCR were performed as described [18], Control samples lacking reverse transcriptase were processed in parallel. All reagents were from Invitrogen (Milan, Italy). Amplifications were performed in a GeneAmp 9700 thermal cycler (Applied Biosystems) using primers designed with Oligo4 (see Table 1), as described [18].
1.
RT-PCR primers for murine P2X and P2Y receptors: sequences, annealing temperatures, accession number and expected amplification product lengths
| P2X and P2Y primers | TA (°C) | Accession number | cDNA (bp) | ||||
|---|---|---|---|---|---|---|---|
| P2X1 | S 5′-GTTTGGGATTCGCTTTGA-3′ | 55.8 | NM_008771 | 452 | |||
| A 5′-TCAGGAAGGGAAGTGTGG-3′ | |||||||
| P2X2 | S 5′-GGTGGAGGATGGGACTTC-3′ | 53.1 | Y10473, AB094664, | 498 | |||
| A 5′-ATGGTGGGAATGAGACTG-3′ | AB094663 | ||||||
| P2X3 | S 5′-ACTTTGTGGGGTGGGTTT-3′ | 55.6 | NM_145526 | 767 | |||
| A 5′-GCTGCCATTCTCCATCTT-3′ | |||||||
| P2X4 | S 5′-GGAATTGGGACTGGAAGG-3′ | 55.8 | NM_011026 | 774 | |||
| A 5′-GGAGTGGAGACCGAGTGA-3′ | |||||||
| P2X5 | S 5′-CCAATCTCTACTGCCCCATC-3′ | 54.2 | NM_033321 | 438 | |||
| A 5′-TGTCTCGGTAAAACTCGC-3′ | |||||||
| P2X6 | S 5′-GCTCAAGTCCAGGGCAGATG-3′ | 57.7 | NM_0011028 | 482 | |||
| A 5′-CAGTCAGAGCCTTTCGTGTCC-3′ | |||||||
| P2X7 | S 5′-GGCATCCGTTTTGACATC-3′ | 54 | NM_0011027 | 360 | |||
| A 5′-CTGGGGTCTTGGAACTTC-3′ | |||||||
| P2Y1 | S 5′-CCTGCGAAGTTATTTCATCTA-3′ | 51.6 | NM_008772 | 319 | |||
| A 5′-GTTGAGACTTGCTAGACCTCT-3 | |||||||
| P2Y2 | S 5′-GCAGCATCCTCTTCCTCACCT-3′ | 60.2 | NM_008773 | 503 | |||
| A 5′-CATGTTGATGGCGTTGAGGGT-3′ | |||||||
| P2Y4 | S 5′-CTTTGGCTTTCCCTTCTTGA-3′ | 56.5 | NM020621 | 427 | |||
| A 5′-GTCCGCCCACCTGCTGATGC-3′ | |||||||
| P2Y6 | S 5′-CGCTTCCTCTTCTATGCCAA-3′ | 59.6 | AF298899 | 480 | |||
| A 5′-GTAGGCTGTCTTGGTGATGTG-3′ | |||||||
| P2Y12 | S 5′-CCGCTACCTGAAGACCACCA-3′ | 55.1 | NM_027571 | 641 | |||
| A 5′-GTTCGCCACCTTCTTGTCCTT-3′ | |||||||
| P2Y13 | S 5′-CAGGGACACTCGGATGACA-3′ | 55.4 | NM_028808 | 577 | |||
| A 5′-CACCGCATAAAACAGAAGC-3′ | |||||||
| P2Y14 | S 5′-GTCTCTGCCGTCATCTTCT-3′ | 54.3 | NM_133200 | 591 | |||
| A 5′-GGGTCCAGACACACATTG-3′ | |||||||
For the expression profile of P2 receptors the following murine positive controls were used: trigeminal ganglia for P2X2 and P2X3, total brain for P2X1, P2X4, P2X5, P2X7, P2Y13 and P2Y14, heart for P2X6, liver for P2Y4, lung for P2Y2 and N9 cells for P2Y1, P2Y6 and P2Y12[8, 21, 22].
Nuclear staining of adhering cells
After 24 hrs in culture, cells were fixed in 4% paraformaldehyde in PBS and nuclear chromatin stained by using the fluorescent dye Hoechst 33258 (Società Italiana Chimici), as described [23].
Flow cytometric evaluation of apoptosis
The percentage of apoptotic and necrotic cells in the total population (adhering+detached cells) was evaluated by means of hypotonic propidium iodide (PI) staining of DNA followed by flow cytometric analysis as described [18, 24, 25]. When utilized under hypotonic conditions, PI enters all cells and allows a direct measure of DNA fragmentation and of the ongoing cell death process. In selected experiments, induction of apoptosis was also confirmed by measuring Annexin V binding to the outer membrane of apoptotic cells; in these experiments, isotonic PI was used in parallel, as described [25]. When employed at isotonic conditions, PI can indeed only permeate cells with damaged membranes, thus providing a direct measure of earlier membrane events associated to necrosis (ibidem).
Statistical analysis
All results are expressed as mean ± S.E.M. of three independent experiments. Statistical significance between groups was derived from one-way-ANOVA followed by Scheffe's F-test. A P value less than 0.05 was considered significant.
Results
HL-1 cardiomyocytes express P2 receptors responding to both adenine and uracil nucleotides
As a first step to the characterization of the effects of P2 receptors on the viability of HL-1 cells, we assessed the presence of all rodent P2 receptors by RT-PCR. Amplification products with the expected molecular weight for all known P2X receptors (with the only exception of P2X2) and for P2Y2, P2Y4, P2Y6 and P2Y14 were obtained (Fig. 1). No specific signals for P2Y1, P2Y12 and P2Y13 were obtained; negative data were not due to technical problems, since specific amplification products for these receptors were detected in murine tissues or cell lines which express these receptors [8, 21, 22] here utilized as positive controls (Fig. 1).
1.
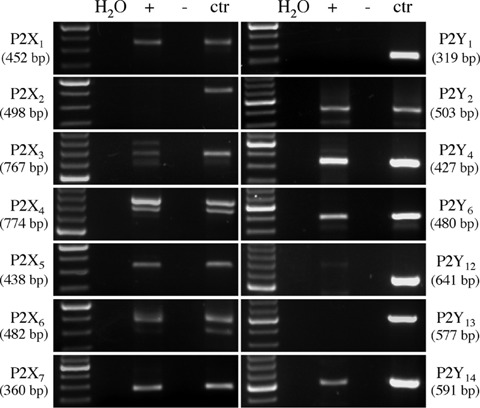
Murine HL-1 cardiomyocytes express P2X1,3,4,5,6,7 and P2Y2,4,6,14. The presence of P2 receptors was assessed using ad hoc designed RT-PCR primers for all cloned murine P2 receptors. For each receptor, amplification products in parallel with MW standards are reported. Various tissues or cell lines were analysed in parallel as a positive controls: trigeminal ganglia for P2X2 and P2X3, total brain for P2X1, P2X4, P2X5, P2X7, P2Y13 and P2Y14, heart for P2X6, liver for P2Y4, lung for P2Y2 and N9 cells for P2Y1, P2Y6 and P2Y12. No products were detected in samples that did not undergo retrotranscription (–).
Exposure of HL-1 cardiomyocytes to adenine nucleotides induces cell death, which is exacerbated by TNF-α
In line with our previous findings [18], a 24 hr-exposure of HL-1 cardiomyocytes to ATP and to the relatively-selective P2X agonist BzATP resulted in a dramatic reduction of the number of cells adhering to the culture substrate (Fig. 2A). Cell number reduction by ATP and BzATP was due to induction of cell death by both apoptosis and necrosis, as shown by flow cytometric analysis of PI-stained nuclei (Fig. 2B). In a similar way to cardiac fibroblasts [18], the pro-inflammatory cytokine TNF-α exerted little effect ‘per se’ on the viability of HL-1 cardiomyocytes, but dramatically potentiated the cell death induced by ATP, and, to a lesser extent, by BzATP (Fig. 2B). To further confirm that the cell death induced by adenine nucleotides is indeed apoptosis, we evaluated another established marker of apoptosis, that is, Annexin V binding to the outer membrane of apoptotic cells, as previously described [25]. Exposure to adenine nucleotides produced a significant increase in the percentage of Annexin V (apoptotic)-positive cells, from about 8.00% in Control cultures to more than 35.00% after exposure to ATP (Fig. 2C). With the latter method, in the ATP+TNF-α condition, apoptosis was 47.00%; thus, potentiation of ATP-induced effects by TNF-α was less evident if compared to that observed at the nuclear level with hypotonic PI (Fig. 2B). This could be explained by hypothesising that TNF-α is indeed accelerating the progression of the ATP-induced cell death program downstream the cell membrane. At a membrane level, where apoptosis-induced changes precede nuclear fragmentation, potentiation of ATP effects by TNF-α is less visible, since Annexin V-positive cells are still alive and will undergo cell death at later time points.
2.
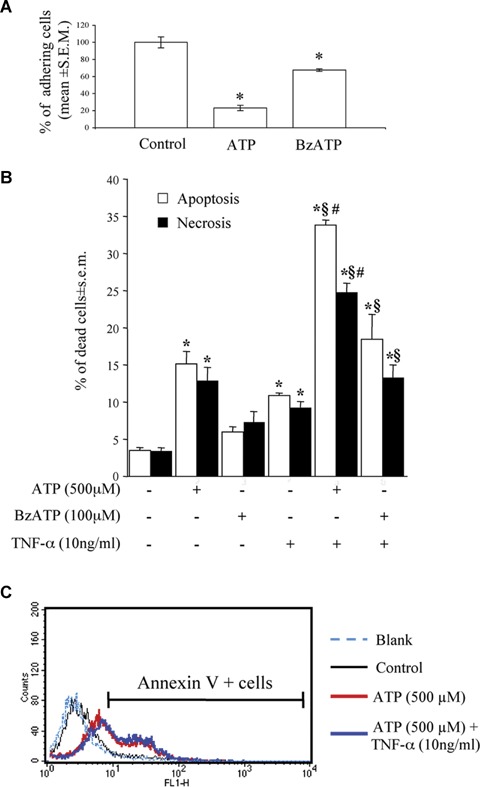
Cardiomyocyte death induced by ATP or BzATP alone or in combination with TNF-α. After 24hrs treatment with either ATP (500 M) or the selective P2X-agonist BzATP (100 μM), cardiomyocytes were fixed, stained with the Hoechst 33258 dye and the number of adhering cells evaluated by fluorescence microscopy. Data are reported as % of control cell number (A). For evaluation of apoptosis and necrosis (B), cells were exposed to ATP or BzATP, alone or in combination with TNF-α (10ng/ml), as indicated. After 24 hrs, cells were collected and the percentage of apoptotic and necrotic cells measured by flow cytometric analysis of DNA fragmentation with Propidium Iodide. Data represent the mean ± S.E.M. of three independent experiments. *P<0.05 versus control, (P<0.05 versus TNF-α alone, and §P<0.05 versus ATP or BzATP alone, by one way ANOVA (Scheffe's F test). (C) Annexin V staining confirmed the induction of apoptotic cell death. Cells were exposed to ATP alone or in combination with TNF-α, as previously described. At the end of the incubation period, cells were collected, and stained with Annexin V to detect membrane signs of apoptotic death. Blank refers to unstained negative control cells, used to calibrate the instrument. The graph shows a representative experiments (percentages of Annexin V-positive cells: Control, 8.61%; ATP alone, 38.57%; ATP+TNF-α, 47.12%). Comparable results were obtained in four independent experiments.
Of a number of other adenine nucleotides tested, only ADP and ATPγS (but not 2MeSATP or 2MeSADP) were able to induce cardiomyocyte apoptosis and necrosis; also in this case, induction of cell death was potentiated by TNF-α (Fig. 3A).
3.
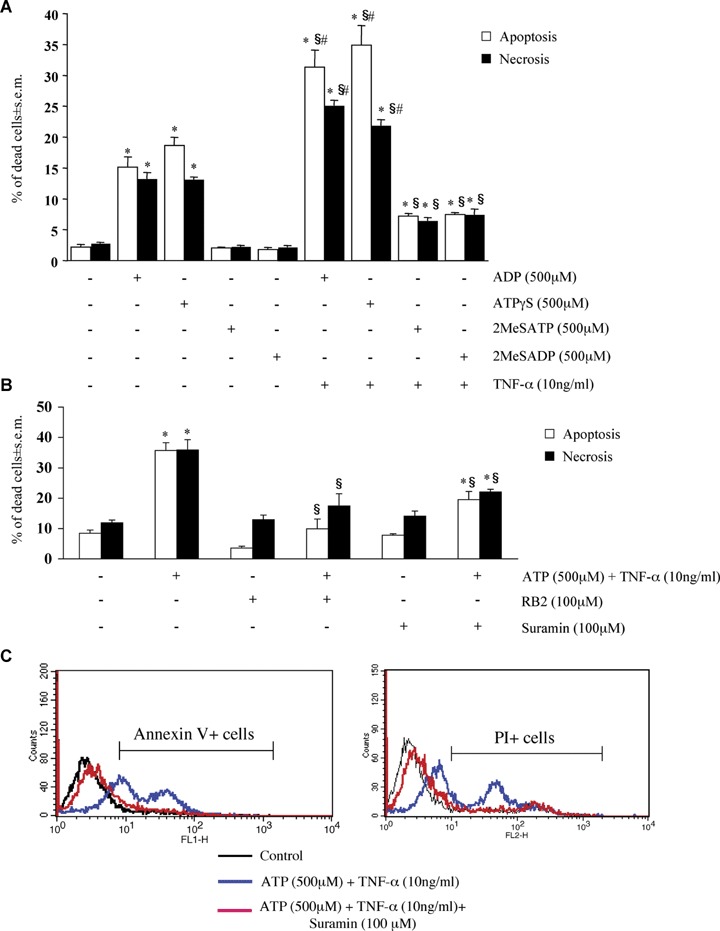
Pharmacological characterization of adenine nucleotides-induced cardiomyocyte death. HL-1 cardiomyocytes were exposed to either ADP, ATPγS, 2MeSATP or 2MeSADP (all 500 μM) for 24 hrs, alone or in combination with TNF-α (10 ng/ml), as indicated (A), or to ATP (500 μM) and TNF-α (10 ng/ml), either alone or in combination with the P2 receptor antagonists RB2 or suramin, all utilized at a 100 μM concentration (B). For suramin, the original traces of a representative experiment are shown in C. The percentages of apoptotic and necrotic cells measured by flow cytometric analysis with hypotonic PI (A) or by Annexin V/isotonic PI (B) (see Materials and methods) are shown. In A and B, data represent the mean ± S.E.M.of three and four independent experiments, respectively.*P<0.05 versus control. In A, #P<0.05 versus TNF-α and §P<0.05 versus corresponding adenine nucleotide alone, and in B §P<0.05 versus ATP + TNF-α, by one way ANOVA (Scheffe's F test).
Adenine nucleotides-induced cardiomyocyte death is mediated by activation of P2 receptors
To verify if the cell death effect reported above is actually due to activation of P2 receptors, HL-1 cardiomyocytes were exposed to ATPand TNF-α in the presence of various P2 receptor antagonists such as Reactive Blue 2 (RB2), PPADS and suramin [8, 26, 27]. Suramin was the most potent of the three tested antagonists in inhibiting the nuclear signs of apoptotic death induced by ATP and TNF-α; a lower, but statistically significant inhibition was observed with PPADS and RB2 (Table 2). Also in this case, the specificity of these effects was confirmed by the Annexin V method. In these experiments, isotonic PI (see Materials and methods) was used to measure necrotic cell death in the same samples. Figure 3B shows the results from four independent Annexin V/isotonic PI experiments confirming the protective effects of RB2 and suramin on both the apoptosis (white bars) and the necrosis (black bars) induced by ATP and TNF-α (for suramin, the original traces of one representative experiment are shown in Fig. 3C). Basically, suramin and RB2 were equivalent in reducing cell death at the membrane level, with RB2 showing a little more pronounced effect. Conversely, RB2 did not modify the cell death effects mediated by the P2X agonist BzATP (data not shown), suggesting that different P2 receptor subtypes may be involved in the cell death induced by different adenine nucleotides (see also below). We also tested MRS2179 and cangrelor which act as selective P2Y1 and P2Y12,13 receptor antagonists, respectively [26]. In line with the lack of expression of these three receptor subtypes (Fig. 1), none of these antagonists had any effect on the cell death induced by ATP and TNF-α (data not shown). Upon addition to cells, ATP is quickly degraded to adenosine, which can then activate the P1 receptors (subdivided into the A1, A2A, A2B and A3 subtypes [28]). Based on previous data suggesting important functional effects of these receptors in the cardiovascular system [29, 30], we next verified if P1 receptors could contribute to the detected effects by utilizing specific P1 receptor antagonists. In our experimental model, ATP-induced cytotoxicity was not counteracted by either 8-cyclopentyl-1,3-dipropylxanthine (CPX, which primarily antagonizes the A1 and A2B receptors [31–33]), or by the selective A2A antagonist SCH58261, or by the selective A3 antagonist MRS1220 [28] (data not shown), thus ruling out a role for these receptors in the detected effects.
2.
Reduction of ATP+TNF-α-induced apoptotic cell death by P2 receptor antagonists
| Apoptosis (% of ATP+TNF-α set to 100%± S.E.M.) | |
|---|---|
| ATP+TNF-α | 100.00±5.64 |
| + Suramin (100 μM) | 33.52±6.85* |
| + PPADS (100 μM) | 49.3±2.1*§ |
| + RB2 (100 μM) | 42.11±7.69*§ |
Murine cardiomyocytes were exposed to ATP (500 μM) in combination with TNF-α (10 ng/ml), together with the various P2 receptor antagonists (which were added 30 min before). At the end of the incubation period, the percentage of apoptotic death was evaluated by isotonic PI staining of nuclei (see Materials and methods), followed by flow cytometric analysis. ATP+TNF-α-induced apoptosis was set to 100%. The three antagonists had no effect on cell survival when utilized alone (data not shown). Data represent the mean ± S.E.M of three independent experiments.*P<0.05 versus ATP+TNF-α, and §P<0.05 versus control, by one-way ANOVA (Scheffe's F test).
As mentioned above, BzATP primarily behaves as a P2X receptor-agonist [4], whereas ATP activates both P2X and adenine-sensitive P2Y receptors [4]. Of the utilized P2 receptor antagonists, RB2 is relatively ineffective at P2X receptors and primarily acts as a P2Y4,6,11 antagonist, PPADS antagonises P2X1,2,3,5 and P2Y4,6, whereas suramin effectively blocks P2X1,2,3,5 and P2Y2,6,11[4, 34]. On the basis of the results reported above, we suggest that both P2X and P2Y receptors mediate cardiomyocytic cell death by adenine nucleotides. Based on the expression profile of P2 receptors in HL-1 cells (Fig. 1), we also suggest that, among P2Y receptors, P2Y2/P2Y4, are the most likely candidates for induction of cell death. Among P2X receptors, P2X1 and P2X5 which are antagonised by both suramin and PPADS, may be involved in cell death induction; however, since P2X receptors can form heteromers characterized by different and unpredictable pharmacological profiles [4], in this case, a definite conclusion regarding the specific receptor subtypes involved cannot be drawn.
Uracil nucleotides do not affect cardiomyocytic viability and protect against cell death induced by adenine nucleotides
Since several of the P2Y receptors expressed by HL-1 cardiomyocytes also respond to uracil nucleotides (i.e. P2Y2, P2Y4, P2Y6 and P2Y14, Fig. 1), the effects of uracil agonists on HL-1 cardiomyocytes were also assessed. At variance from ATP, neither UTP (which activates P2Y2 and P2Y4) nor UDP nor UDPglucose (which activate the P2Y6 and P2Y14 receptor subtypes, respectively [34]), had any effect ‘per se’ on the viability of HL-1 cardiomyocytes, not even when assayed in presence of TNF-α (Fig. 4A). We then tested the possible effect of these ligands on adenine nucleotides-induced death. In a first set of experiments, uracil nucleotides were added to cells 30 min before addition of either ATP alone or ATP+TNF-α. As expected, exposure of HL-1 cardiomyocytes to ATP and TNF-α for 24 hrs resulted in dramatic cytotoxicity (see micrograph in Fig. 4C) and in highly significant induction of both necrosis and apoptosis (see cytogram in Fig. 4C) with respect to control healthy cells (Fig. 4B). Addition of either UDP (Fig. 4D), or UTP or UDPglucose (Fig. 4E) fully reversed cell death induced by ATP+TNF-α. As an example, in the experiment reported in Figure 4E, the number of necrotic and apoptotic cells in the ATP+TNF-α group was reduced from 22.8% and 28.8% to 6.5% and 6.2%, respectively, upon addition of UDPglucose. Results were fully confirmed also by evaluating Annexin V staining of apoptotic cells (Fig. 4F). Here again, Annexin V binding induced by ATP+TNF-α was prevented by UDP. A similar protective effect was observed against ADP-induced cell death. UDP significantly reduced both the apoptosis and necrosis induced by either ADP alone, or by ADP+TNF-α (Fig. 4G).
4.
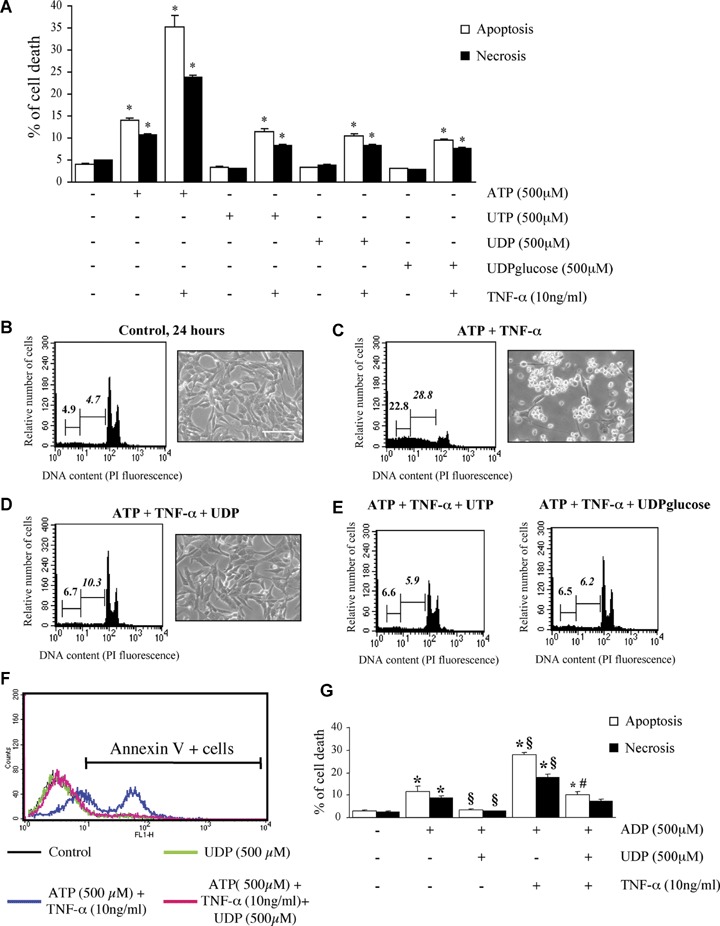
Uracil nucleotides have no effect ‘per se’ on cardiomyocytic viability, but protect cells from ATP and ADP-induced cytotoxicity. To assess the effect of uracil nucleotides on HL-1 cardiomyocytic viability, cells were exposed to UTP, UDP or UDPglucose (500 μM) alone or in combination with TNF-α, and apoptosis and necrosis determined after 24 hrs by flow-cytometry. Parallel cultures were treated with ATP, here utilized as a positive control (A). The effect of TNF-α alone is shown in Figure 2B.*P <0.05 versus corresponding Control, by one-way ANOVA (Scheffe's F test). To assess the ability of uracil nucleotides to counteract cardiomyocyte death induced by ATP+TNF-α, cells were treated with either vehicle (B), or ATP+TNF-α (500 μM and 10 ng/ml, respectively) in the absence (C) or presence of UDP (100 μM, D), UTP (25 μM) or UDPglucose (10 μM) (E). In these experiments, uracil nucleotides were added to cells 30 min before adenine nucleotides. After 24 hrs, micrographs of adhering cells were taken (scale bar: 50 μm) and flow cytometric analysis was performed on the total cell population. In cytograms, first and second numbers on the left represent the percentage of necrotic and apoptotic cells, respectively. Results from a typical experiment are shown in (B–E). Similar data were obtained in five independent experiments. UDP similarly protected cells from the apoptosis and necrosis induced by either ADP alone (500 μM) or ADP and TNF-α (10 ng/ml) (F). Annexin V staining of cells confirmed the complete protection exerted by uracil nucleotides. The percentages of Annexin V-positive cells in this representative experiment (out of three) were: Control, 8.1%; UDP alone, 8.4%; ATP+TNF-α, 56.4%; ATP+TNF-α+UDP, 11.3%. (G) *P <0.05 versus corresponding Control, §P<0.05 versus ADP alone, #P <0.05 versus ADP+TNF-α, by one-way ANOVA (Scheffe's F test); the mean values ± S.E.M.of four independent experiments are shown.
Both adenine nucleotides-induced death (sum of apoptosis and necrosis) and uracil nucleotides-mediated cytoprotection are concentration-dependent events. In particular, ATP displayed an EC50 value (i.e. concentration eliciting 50% of maximal effect) of 99.68 μM and 91.41 μM in the absence and presence of TNF-α, respectively (Fig. 5A; Table 3); thus, potentiation of cell death by TNF-α seems mainly due to increased effectiveness rather than to changes in the potency of adenine nucleotides (see also Table 3). Also the protective effects mediated by uracil nucleotides were strictly concentration-dependent, with IC50 values (i.e. concentrations needed to inhibit 50% of ATP-induced cell death) of 7.29 μM, 10.64 μM and 2.76 μM for UTP, UDP and UDPglucose, respectively (Figs 5B–D; Table 3). This suggests a rank order of potency of UDPglucose ≥ UTP ≥ UDP. A comparable rank order of potency for uracil nucleotides was obtained when the effects of UTP, UDP and UDPglucose were analysed separately against ATP-induced apoptosis or necrosis (data not shown). All experiments above have been performed by adding uracil nucleotides to cells 30 min before adenine nucleotides. We next tested the time-dependence of uracil nucleotides-mediated protection. Addition of UDP together with (Fig. 6C) or 30 min after ATP (Fig. 6D) still fully protected cells against adenine nucleotide-induced death, suggesting the existence of a possible ‘therapeutic’ window for uracil nucleotides-mediated protection, and that cardiomyocytes do not need to be ‘primed’ by uracil nucleotides to become insensitive to ATP-induced death.
5.
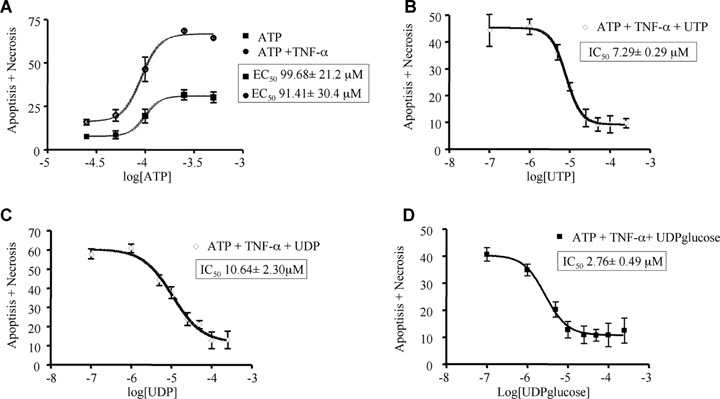
Concentration dependence of ATP-induced cardiomyocytic death (apoptosis+necrosis) and of uracil nucleotides-mediated protection. HL-1 cardiomyocytes were exposed to graded concentrations of ATP (25–250 μM) alone or in combination with TNF-α (10 ng/ml, A). Cultures pre-treated with TNF-α and incubated with 250 μM ATP were exposed to graded (100 nM to 250 μM) concentrations of UTP (B), UDP (C), or UDPglucose (D). After 24 hrs, the percentage of cell death was evaluated by cytofluorimetric analysis of PI stained nuclei. Data represent the mean ± S.E.M.of three independent experiments. Curves were calculated using GRAPH PAD Prism 4.0 software. EC50 values for induction of cell death by ATP or ATP+TNF-α and IC50 values for uracil nucleotides protection are also shown in individual panels and summarized in Table 3.
3.
Potency of adenine and uracil nucleotides in regulation of HL-1 cardiomyocytes viability
| Nucleotide | Type of effect | EC50 or IC50± S.E.M. |
|---|---|---|
| ATP | Induction of apoptosis and necrosis | 99.68±21.2 μM |
| ATP + TNF-α | Induction of apoptosis and necrosis | 91.41±30.4 μM |
| UTP | Protection against cell death induced by ATP+TNF-α | 7.29±0.29 μM |
| UDP | Protection against cell death induced by ATP+TNF-α | 10.64±2.30 μM |
| UDPglucose | Protection against cell death induced by ATP+TNF-α | 2.76±0.49 μM |
Murine cardiomyocytes were exposed to graded concentrations of either ATP alone (25–500 μM), or ATP in the presence of the cytokine TNF-α (10 ng/ml), or UTP, UDP and UDP-glucose (100 μM to 250 μM). In experiments where uracil nucleotides were challenged for their ability to inhibit the effects induced by adenine nucleotides, an ATP concentration of 250 μM in the presence of 10 ng/ml TNF-α was utilized. In all experiments, effects were detected after 24 hrs. At the end of treatments, the percentage of cell death (apoptosis+necrosis) was evaluated by cytofluorimetric analysis of PI stained nuclei. Reported EC50 and IC50 values refer to experiments shown in Fig. 5 (for more details, see Fig. 5 legend and text).
6.
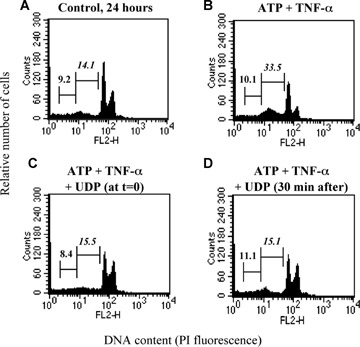
Characterization of UDP-induced protection. HL-1 cardiomyocytes were exposed to either vehicle (Control, A), ATP (100 μM) and TNF-α (10 ng/ml) (B), as indicated, for 24 hrs. UDP (100 μM) was added to cultures either together with ATP (C) or 30 min after its addition (D). At the end of treatment, flow cytometric analysis was performed on the total cell population. In cytograms, first and second numbers on the left represent the percentage of necrotic and apoptotic cells, respectively.
Discussion
Here we demonstrate that adenine and uracil nucleotides, which both in rodents [9, 35, 36], pig [37] and man [9, 12] are released in the blood stream during heart hypoxia, play strikingly different effects on cardiomyocyte viability. The novelty of the present results is twofold: first, we show a role for P2 receptors (both P2X and adenine-preferring P2Y receptors) in the cardiomyocyte death induced by adenine nucleotides (an effect which is exacerbated by TNF-α, a well-established pathogenetic factor in cardiac disease development), and, second, we demonstrate that uracil nucleotides fully protect against the deleterious effects mediated by adenine nucleotides and TNF-α. A combination of pharmacological and RT-PCR studies suggests that several uracil-preferring P2Y receptors expressed on cardiomyocytes likely contribute to these pro-survival effects. These receptors may thus represent new appropriate targets for innovative drug therapies aimed at cardiac protection during myocardial infarction and in CHF.
In the heart, hypoxic conditions induce ATP release [35, 36]. Functionally, released ATP has been involved in inotropy and hypertrophy of cardiomyocytes [38], and, as more recently demonstrated, in myocardial cell death (our previous data [18] and the present results). All these effects are likely mediated by the activation of specific P2 receptors on either cardiomyocytes or fibroblasts. Here we show that murine cardiomyocytes express a wide panel of P2X and P2Y receptors known to either exclusively respond to adenine nucleotides (P2X receptors), to both adenine and uracil nucleotides (P2Y2, P2Y4, P2Y6) or to sugar nucleotides (P2Y14 receptor). Such a large heterogeneity of P2 receptor expression is consistent with previous studies [18, 39–42], and suggests involvement of these receptors in multiple functional effects. Specifically, our data suggest that activation of P2 receptors by adenine nucleotides in the presence of TNF-α results in apoptosis and necrosis. In a similar way to pig cardiac fibroblasts [18], only modest effects on HL-1 cardiomyocytes viability were detected when adenine nucleotides and TNF-α were utilized alone ([18] and the present data), suggesting that a synergistic interaction between ATP/ADP and TNF-α is needed to induce cell death. Effects induced by ATP and ADP are concentration-dependent, with EC50 values in the μMolar range, which are consistent with the estimated nucleotide concentrations at the cell surface during cardiac ischaemia [12, 37], further confirming that the effects reported here may bear a pathophysiological significance. EC50 values for ATP and ADP in induction of cell death were not significantly altered by TNF-α, suggesting that the higher degree of cell death observed in the presence of the cytokine is likely due to increased effectiveness rather than increased affinity of nucleotides towards their receptors. Induction of cardiomyocyte apoptosis by adenine nucleotides is a receptor-mediated event, as also confirmed by data with the P2 receptor antagonists RB2, PPADS and suramin. Based on pharmacological data, we suggest that both P2X and P2Y receptors may contribute to induction of cell death in murine cardiomyocytes.
At variance from ATP, it is only recently that UTP has been shown to be released during heart ischaemia. The first demonstration was provided by Erlinge and coworkers in a pig cardiac ischaemia in vivo model [37]. UTP levels in the venous blood from the cardiac vein increased according to a biphasic pattern, with an increase 1 min after onset of cardiac ischaemia, and a second increase 1 min after reperfusion.
In the same animals, total venous blood flow from the heart and episodes of ventricular fibrillation or tachycardia peaked at the same time points, suggesting that nucleotide release positively correlated with both cardiac blood flow and ventricular arrhythmia. Importantly, release of UTP during myocardial infarction has been more recently confirmed also in man [12]. There still is little information regarding the source of UTP release during heart ischaemia. Although both myocytes, endothelial cells and platelets may theoretically release uridine nucleotides into the blood stream, the only published evidence for significant release of UTP is from endothelial cells during changes in flow (shear stress) [43]. Released UTP may then act on myocardial P2 receptors; in line with our previous findings [18], several uracil-preferring P2Y receptors were indeed found by Wihlborg and coworkers [12] in the human heart (P2Y2, P2Y4, P2Y6). These receptors are also expressed in the murine cardiomyocyte model utilized here, thus supporting its validity for unveiling the functional roles of these receptors in mammalian heart. In the Wihlborg et al. study, UTP and UDP were also demonstrated to mediate inotropic effects likely via P2Y6. On this basis, and based on previous findings demonstrating induction of vasoconstriction by UDP and UTP [44], these authors proposed that uracil nucleotides may play a similar role as angiotensin II in the development of cardiac disease, and that, in a similar way to angiotensin II receptor antagonists and angiotensin converting enzyme inhibitors, selective antagonists of UDP and UTP receptors may represent novel agents in the treatment of hypertension and heart failure via inhibition of peripheral resistance, inotropy and cardiac hypertrophy. Our results instead suggest that uracil nucleotides may also induce beneficial effects on cardiomyocytes. Both UTP and UDP and the sugar-nucleotide UDPglucose effectively counteracted cell death induced by adenine nucleotides and TNF-α on HL-1 cardiomyocytes. Protection by uracil nucleotides is concentration-dependent, with strikingly low IC50 values (in the low μMolar range) against ATP and ADP-induced cell death. Pharmacological and expression data suggest that cardiomyocyte protection is mediated by multiple uracil-sensitive P2Y receptors (P2Y2, P2Y4, P2Y6 and P2Y14; in this respect, see also below).
The protective effects described here are consistent with the previous demonstration that UTP protects neonatal rat cardiomyocytes against injury induced by 120 min of hypoxia [16]. However, the possible functional antagonism between uracil and adenine nucleotides was not addressed in this study, and only UTP, but not UDP, effectively protected cardiomyocytes. P2Y2 and P2Y4 receptors, whose presence in neonatal cardiomyocytes was confirmed by immunofluorescence staining and western blot, are likely to mediate protection in this study, whereas a role for P2Y6 was ruled out, based on the lack of effect of UDP. This protection profile seems different from that reported here, where all tested uracil nucleotides were found to be protective. Moreover, the effects described above [16] seems related to a preconditioning mechanism, since cells needed to be pre-exposed to UTP before hypoxia to show protection. Here, both UTP and other uracil nucleotides were able to effectively protect cells even when added after adenine nucleotides and TNF-α. Strikingly, our data suggest that the same receptors may even be involved in both cell death and cell protection. This applies in particular to the HL-1 P2Y2 and P2Y4 receptors, that in rodents respond to both ATP and UTP [8, 32]. It may be hypothesised that, depending upon the identity of the nucleotide (adenine versus uracil) binding to the receptor, different intracellular pathways are activated, eventually leading to either cell death (in the case of ATP) or cell protection (in the case of UTP). Alternatively, when the receptor is activated by one of its endogenous agonists (e.g. ATP) another agonist (UTP) can act as an antagonist. This would not be unexpected for P2Y receptors: for example, under specific conditions, the agonist ATP can act as an antagonist at the ADP-activated P2Y12 receptor subtype [8]. This interesting issue will be investigated in HL-1 cardyomiocytes future studies.
The protective effects of UTP have been also very recently confirmed in rats upon induction of myocardial infarction [17]. In animals injected with UTP 30 min before infarction, the ratio of infarct size to area at risk was significantly smaller with respect to control untreated rats.
In conclusion, therapeutic targeting of pyrimidinergic receptors may represent a novel promising strategy for myocardial protection that certainly deserves further exploration.
Acknowledgments
Authors warmly thank Professor William Claycomb, LSU Health Sciences Center, New Orleans, LA, USA, for the kind gift of HL-1 cells and Drs Alessandro Parolari and Cristina Banfi, Monzino Cardiologic Center IRCCS, Milan, for useful discussion. Cangrelor was a kind gift of the Medicine Company, Parsippany, NJ, USA.
References
- 1.Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- 2.Abbracchio MP, Burnstock G. Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther. 1994;64:445–75. doi: 10.1016/0163-7258(94)00048-4. [DOI] [PubMed] [Google Scholar]
- 3.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–92. [PubMed] [Google Scholar]
- 4.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 5.Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G. Characterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol Sci. 2003;24:52–5. doi: 10.1016/S0165-6147(02)00038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, Guerrini U, Belcredito S, Cimino M, Sironi L, Tremoli E, Rovati GE, Martini C, Abbracchio MP. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006;25:4615–27. doi: 10.1038/sj.emboj.7601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egan TM, Khakh BS. Contribution of calcium ions to P2X channel responses. J Neurosci. 2004;24:3413–20. doi: 10.1523/JNEUROSCI.5429-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelleg A, Vassort G. P2 receptors in the cardiovascular system. In: Abbracchio MP, Williams M, editors. Purinergic and pyrimidergic signalling II. Berlin Heidelberg: Springer-Verlag; 2001. pp. 73–100. [Google Scholar]
- 10.Drury A, Szent-Gyorgyi A. The physiological activity of adenine compounds with special reference to their action upon the mammalian heart. J Physiol. 1929;68:213–37. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mantelli L, Amerini S, Filippi S, Ledda F. Blockade of adenosine receptors unmasks a stimulatory effect of ATP on cardiac contractility. Br J Pharmacol. 1993;109:1268–71. doi: 10.1111/j.1476-5381.1993.tb13759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wihlborg AK, Balogh J, Wang L, Borna C, Dou Y, Joshi BV, Lazarowski E, Jacobson KA, Arner A, Erlinge D. Positive inotropic effects by uridine triphosphate (UTP) and uridine diphosphate (UDP) via P2Y2 and P2Y6 receptors on cardiomyocytes and release of UTP in man during myocardial infarction. Circ Res. 2006;98:970–6. doi: 10.1161/01.RES.0000217402.73402.cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Froldi G, Galzignato G, Zanetti M, Montopoli M, Dorigo P, Caparrotta L. Are prostanoids related to positive inotropism by UTP and ATP? Pharmacology. 2005;73:140–5. doi: 10.1159/000082315. [DOI] [PubMed] [Google Scholar]
- 14.Magner JJ, Royston D. Heart failure. Br J Anaesth. 2004;93:74–85. doi: 10.1093/bja/aeh167. [DOI] [PubMed] [Google Scholar]
- 15.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415:227–33. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 16.Yitzhaki S, Shneyvays V, Jacobson KA, Shainberg A. Involvement of uracil nucleotides in protection of cardiomyocytes from hypoxic stress. Biochem Pharmacol. 2005;69:1215–23. doi: 10.1016/j.bcp.2005.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yitzhaki S, Shainberg A, Cheporko Y, Vidne BA, Sagie A, Jacobson KA, Hochhauser E. Uridine-5’-triphosphate (UTP) reduces infarct size and improves rat heart function after myocardial infarct. Biochem Pharmacol. 2006;72:949–55. doi: 10.1016/j.bcp.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banfi C, Ferrario S, De Vincenti O, Ceruti S, Fumagalli M, Mazzola A, D’Ambrosi N, Volonte C, Fratto P, Vitali E, Burnstock G, Beltrami E, Parolari A, Polvani G, Biglioli P, Tremoli E, Abbracchio MP. P2 receptors in human heart: upregulation of P2X6 in patients undergoing heart transplantation, interaction with TNFalpha and potential role in myocardial cell death. J Mol Cell Cardiol. 2005;39:929–39. doi: 10.1016/j.yjmcc.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Batista ML, Jr, Santos RV, Cunha LM, Mattos K, Oliveira EM, Seelaender MC, Costa Rosa LF. Changes in the pro-inflammatory cytokine production and peritoneal macrophage function in rats with chronic heart failure. Cytokine. 2006;34:284–90. doi: 10.1016/j.cyto.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 20.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–9. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 21.Simonetti M, Fabbro A, D’Arco M, Zweyer M, Nistri A, Giniatullin R, Fabbretti E. Comparison of P2X and TRPV1 receptors in ganglia or primary culture of trigeminal neurons and their modulation by NGF or serotonin. Mol Pain. 2006;2:11. doi: 10.1186/1744-8069-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bianco F, Fumagalli M, Pravettoni E, D’Ambrosi N, Volonte C, Matteoli M, Abbracchio MP, Verderio C. Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res Brain Res Rev. 2005;48:144–56. doi: 10.1016/j.brainresrev.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Ceruti S, Franceschi C, Barbieri D, Malorni W, Camurri A, Giammarioli AM, Ambrosini A, Racagni G, Cattabeni F, Abbracchio MP. Apoptosis induced by 2-chloro-adenosine and 2-chloro-2’-deoxy-adenosine in a human astrocytoma cell line: differential mechanisms and possible clinical relevance. J Neurosci Res. 2000;60:388–400. doi: 10.1002/(SICI)1097-4547(20000501)60:3<388::AID-JNR14>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 24.Ceruti S, Beltrami E, Matarrese P, Mazzola A, Cattabeni F, Malorni W, Abbracchio MP. A key role for caspase-2 and caspase-3 in the apoptosis induced by 2-chloro-2’-deoxy-adenosine (cladribine) and 2-chloro-adenosine in human astrocytoma cells. Mol Pharmacol. 2003;63:1437–47. doi: 10.1124/mol.63.6.1437. [DOI] [PubMed] [Google Scholar]
- 25.Ceruti S, Mazzola A, Abbracchio MP. Resistance of human astrocytoma cells to apoptosis induced by mitochondria-damaging agents: possible implications for anticancer therapy. J Pharmacol Exp Ther. 2005;314:825–37. doi: 10.1124/jpet.105.085340. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson KA, Costanzi S, Joshi BV, Besada P, Shin DH, Ko H, Ivanov AA, Mamedova L. Agonists and antagonists for P2 receptors. Novartis Found Symp. 2006;276:58–68. doi: 10.1002/9780470032244.ch6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wildman SS, Unwin RJ, King BF. Extended pharmacological profiles of rat P2Y2 and rat P2Y4 receptors and their sensitivity to extracellular H+ and Zn2+ ions. Br J Pharmacol. 2003;140:1177–86. doi: 10.1038/sj.bjp.0705544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–64. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Picano E, Abbracchio MP. Adenosine, the imperfect endogenous anti-ischemic cardio-neuroprotector. Brain Res Bull. 2000;52:75–82. doi: 10.1016/s0361-9230(00)00249-5. [DOI] [PubMed] [Google Scholar]
- 30.Sexl V, Mancusi G, Holler C, Gloria-Maercker E, Schutz W, Freissmuth M. Stimulation of the mitogen-activated protein kinase via the A2A-adenosine receptor in primary human endothelial cells. J Biol Chem. 1997;272:5792–9. doi: 10.1074/jbc.272.9.5792. [DOI] [PubMed] [Google Scholar]
- 31.Golembiowska K, Dziubina A. Striatal adenosine A(2A) receptor blockade increases extracellular dopamine release following l-DOPA administration in intact and dopamine-denervated rats. Neuropharmacology. 2004;47:414–26. doi: 10.1016/j.neuropharm.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 32.Goldenberg I, Shainberg A, Jacobson KA, Shneyvays V, Grossman E. Adenosine protects against angiotensin II-induced apoptosis in rat cardiocyte cultures. Mol Cell Biochem. 2003;252:133–9. doi: 10.1023/a:1025551229566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji X, Kim YC, Ahern DG, Linden J, Jacobson KA. [3H]MRS 1754, a selective antagonist radioligand for A(2B) adenosine receptors. Biochem Pharmacol. 2001;61:657–63. doi: 10.1016/s0006-2952(01)00531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abbracchio MP, Verderio C. Pathophysiological roles of P2 receptors in glial cells. Novartis Found Symp. 2006;276:91–103. [PubMed] [Google Scholar]
- 35.Clemens MG, Forrester T. Appearance of adenosine triphosphate in the coronary sinus effluent from isolated working rat heart in response to hypoxia. J Physiol. 1981;312:143–58. doi: 10.1113/jphysiol.1981.sp013621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vial C, Owen P, Opie LH, Posel D. Significance of release of adenosine triphosphate and adenosine induced by hypoxia or adrenaline in perfused rat heart. J Mol Cell Cardiol. 1987;19:187–97. doi: 10.1016/s0022-2828(87)80561-8. [DOI] [PubMed] [Google Scholar]
- 37.Erlinge D, Harnek J, Van Heusden C, Olivecrona G, Jern S, Lazarowski E. Uridine triphosphate (UTP) is released during cardiac ischemia. Int J Cardiol. 2005;100:427–33. doi: 10.1016/j.ijcard.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Aartsen WM, Schuijt MP, Danser AH, Daemen MJ, Smits JF. The role of locally expressed angiotensin converting enzyme in cardiac remodeling after myocardial infarction in mice. Cardiovasc Res. 2002;56:205–13. doi: 10.1016/s0008-6363(02)00516-3. [DOI] [PubMed] [Google Scholar]
- 39.Webb TE, Boluyt MO, Barnard EA. Molecular biology of P2Y purinoceptors: expression in rat heart. J Auton Pharmacol. 1996;16:303–7. doi: 10.1111/j.1474-8673.1996.tb00040.x. [DOI] [PubMed] [Google Scholar]
- 40.Bogdanov Y, Rubino A, Burnstock G. Characterisation of subtypes of the P2X and P2Y families of ATP receptors in the foetal human heart. Life Sci. 1998;62:697–703. doi: 10.1016/s0024-3205(97)01168-5. [DOI] [PubMed] [Google Scholar]
- 41.Balogh J, Wihlborg AK, Isackson H, Joshi BV, Jacobson KA, Arner A, Erlinge D. Phospholipase C and cAMP-dependent positive inotropic effects of ATP in mouse cardiomyocytes via P2Y11-like receptors. J Mol Cell Cardiol. 2005;39:223–30. doi: 10.1016/j.yjmcc.2005.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang L, Bardini M, Keogh A, Dos Remedios CG, Burnstock G. P2X1 receptors are closely associated with connexin 43 in human ventricular myocardium. Int J Cardiol. 2005;98:291–7. doi: 10.1016/j.ijcard.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 43.Saiag B, Bodin P, Shacoori V, Catheline M, Rault B, Burnstock G. Uptake and flow-induced release of uridine nucleotides from isolated vascular endothelial cells. Endothelium. 1995;2:279–85. [Google Scholar]
- 44.Hrafnkelsdottir T, Erlinge D, Jern S. Extracellular nucleotides ATP and UTP induce a marked acute release of tissue-type plasminogen activator in vivo in man. Thromb Haemost. 2001;85:875–81. [PubMed] [Google Scholar]