

Abstract
Many studies aim at improving therapeutic efficacy by combining strategies with oxidative stress-inducing drugs and histone deacetylase (HDAC) inhibitors in colorectal cancer. As p53 and p21WAF1 are essential in oxidative stress-induced DNA damage, we investigated epigenetic regulation of p21WAF1 promoter. Firstly, HCT116 p53+/+ and p53−/− colorectal cancer cells were treated with H2O2 for 6 hrs and 24 hrs (early/late response). Chromatin immunoprecipitation revealed transcriptional transactivation of p21WAF1 in HCT116 p53+/+ cells as shown by increased binding of p53 and acetylated H4 around two p21WAF1 promoter sites, the responsible element (RE) and the Sp1 site, while both proteins bound preferentially on the RE. Interestingly, H3 was not involved, suggesting H4-specific transactivation of the p21WAF1 promoter. H2O2 addition resulted in G2/M arrest of both HCT116 cell lines without significant cell death. To investigate whether a HDAC inhibitor strengthens G2/M arrest, we pretreated cells with Trichostatin A (TSA). In HCT116 p53+/+ cells, we found (i) remarkably increased acetylated H4 around both p21WAF1 promoter regions, especially at the Sp1 site; (ii) increased acetylation of p53 at lysines 320 and 382;(iii) displacement of HDAC1 from the Sp1 site, thus inhibiting its repression effect and increasing p53 binding.p53 seems to trigger H4-acetylation around the p21WAF1 promoter because there was nearly no H4 acetylation in HCT116 p53−/− cells. For the first time we show that there is a time-dependent TSA mode of action with increased p53-dependent histone H4 acetylation at the p21WAF1 promoter in early response, and decreased acetylation in late response. Reduced p53-triggered transactivation of p21WAF1 in late response allows cells to re-enter cell cycle, and TSA causes p53 to simultaneously induce apoptosis.
Keywords: colorectal cancer, HDAC inhibitor, oxidative stress, p53, p21WAF1, chromatin immunoprecipitation, cell cycle, apoptosis
Introduction
Oxidative stress-induced DNA damage is known to lead to cell cycle arrest, whereas p21WAF1 plays a crucial role [1]. Thus, reactive oxygen species (ROS)-generating anticancer drugs are of major clinical interest, because they induce DNA damage in tumor cells [2, 3]. While the overexpression of p21WAF1 results in G1, G2 or S phase arrest [4–7], p21WAF1-deficient cells fail to undergo cell cycle arrest in response to p53 activation after DNA damage [8]. In the process of gene expression regulation, chromatin re-modelling proved to be crucial because tightly bound DNA around the nucleosome core (histone residues H2A, H2B, H3 and H4) suppresses gene transcription. The activation proceeds by increasing the accessibility of transcription factors to DNA by histone modifications. A key function in this context is the acetylation of lysine residues in the N-terminal tails of core histone proteins, which results in an uncoiled, accessible DNA, thus regulating gene expression [9]. This is mediated by the counteracting activities of histone acetyltransferase (HAT) and histone deacetylase (HDAC). HDAC inhibitors, such as trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA) and sodium butyrate (NaB), can inhibit cancer cell growth in vitro and in vivo[10–12]. It has recently been shown that the HDAC inhibitor SAHA induces the accumulation of acetylated histones in the p21WAF1-associated chromatin, which could lead to an increase in p21WAF1 expression in T24 human bladder carcinoma cells [13]. Furthermore, it has been demonstrated that the HDAC inhibitor TSA inhibits the growth of cancer cells through induction of p21WAF1[14]. Histone acetylation induced by TSA executes alternate functions. On the one hand, TSA activates p21WAF1 transcription through down-regulation of c-Myc expression and release of c-Myc from the p21WAF1 promoter in cervical carcinoma cells in a p53-independent manner [15]. On the other hand, TSA can relieve the repression exerted by HDAC1 at p53-induced transcription from the p21WAF1 gene in human osteosarcoma cells [16]. In addition, it has been demonstrated that TSA induces apoptosis in human brain tumour cells [17] and hepatoma cells [18].
Cancer growth arrest and apoptosis are two main strategies to affect tumour cells in chemotherapy. The present work is part of our understanding of the linkage between HDAC inhibitors, which have led to promising anticancer therapies using TSA and oxidative stress-induced growth arrest in colorectal cancer cells. As oxidative stress-induced DNA damage and TSA lead to an up-regulation of p21WAF1, resulting in cell cycle arrest, we aimed to enhance the DNA-damaged transcriptional activation of p21WAF1 by a combined treatment. Due to the fact that p53-dependent histone acetylation following DNA damage in colorectal HCT116 cells was due to transcriptional activation of the p21WAF1 promoter [19], pretreatment of HCT116 cells with TSA could provide a great impact for stronger histone acetylation, thus increasing p21WAF1 transactivation. Combining this strategy with the known function of TSA to induce apoptosis [17, 18], we aimed to increase the effectiveness of ROS-based anticancer drugs. We simply chose H2O2 treatment, which mimics ROS-generating anticancer drugs, as a basic model for inducing DNA damage in HCT116 cells, because H2O2 itself displays a ROS in terms of a stable molecular oxidant. Our data suggest that TSA pre-treatment alters the chromatin structure, thus facilitating the accessibility of the transcription factor p53 to bind at p21WAF1 promoter as a consequence of oxidative stress. To the best of our knowledge, we are the first to show that there is a time-dependent TSA mode of action with an increased p53-dependent histone H4 acetylation at the p21WAF1 promoter in the early response, and decreased acetylation in the late response. Thus, the reduced p53-triggered transac-tivation of p21WAF1 in the late response allows cells to re-enter the cell cycle, and p53 simultaneously induces apoptosis, a finding that might be of therapeutical interest.
Materials and methods
Cell culture and treatment
HCT116 p53+/+ and p53−/− cells were maintained in RPMI with 10% foetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified 5% CO2 atmosphere at 37°C. Cells were pre-treated for 6hrs with the HDAC inhibitor trichostatin A (TSA, Sigma) at a final concentration of 200 ng/ml. Cells were treated with 30mM H2O2 for 3 min after pre-treatment with TSA or not and collected after 6 and 24 hrs following treatment.
Flow cytometric analysis of DNA content
One day before treatment cells were seeded in 12-well dishes at a density of 7.5 × 104 cells per well. After the indicated times, the supernatants were collected and combined with cells that were harvested by trypsin, washed twice with phosphate buffered saline (PBS), fixed with 70% ethanol, treated with 1% RNase and finally stained with a hypotonic propidium iodide solution (100 μg/ml). Distribution of cell cycle phases with different DNA contents was determined using a flow cytometer LSR1 (Becton-Dickinson, CA, USA). Cells whose DNA were less intensively stained than those of G1 cells (sub G1 cells) in flow cytometric histograms were considered apoptotic cells. Analysis of cell cycle distribution and the percentage of cells in the G0/G1, S, and G2/M phase of the cell cycle were determined using the software CellQuest Pro (BD).
Annexin-V measurements
Direct fluorescence staining of apoptotic cells for flow cytometric analysis was performed with the Annexin-V-FLUOS staining Kit (Roche). After the indicated times, 5 × 105–1 × 106 cells were harvested by trypsin, washed twice with PBS, stained with both Annexin-V-FLUOS and propidium iodide and analysed in a flow cytometer.
Real-time RT-PCR
cDNA synthesis was done in a 20 μl reaction mix starting with 1 μg of total RNA using the reverse transcription system of Promega (Madison, WI; 42°C for 30 min; 99°C for 5 min, and 4°C for 5 min). Real-time RT-PCR was performed using a LightCycler (Roche Diagnostics, Mannheim, Germany), and threshold cycle numbers were determined using the LightCycler software, version 3.5. Sequences of primers and probes, PCR product length and annealing temperatures are given in Table 1. The real-time RT-PCR was performed in a final volume of 20 μl. The final reaction mixture contained the forward and reverse primer at 10 pmol each, the LC Red640 probe at 40 pmol, the FL probe at 20 pmol, 4 mM MgCl2, and 1x Master Amp hybridization mix. PCR was performed under the following conditions: 95°C for 600 s, followed by 45 cycles of 95°C for 10 sec, annealing temperature for 10 sec, and 72°C for 7 sec. We used serial dilutions of the positive control cDNA of HCT116 cells to create a standard curve. PCR was performed in triplicate, and the threshold cycle numbers were averaged. Fold induction was calculated according to the formula 2(Rt-Et)/2(Rn-En), where Rt is the threshold cycle number for the β2-Microglobulin gene in the treated cells, Et is the threshold cycle number for the experimental gene in treated cells, Rn is the threshold cycle number for the b2-Microglobulin gene in non-treated cells and En is the threshold cycle number for the experimental gene in non-treated cells.
1.
Sequences of primers, hybridization probes and lengths of PCR products
| Gene | Size (bp) | Sequence 5'-3' | TAnnealing (°C) | |
|---|---|---|---|---|
| p53 | Real-time RT-PCR | 149 | Fw: ATGAGCCGCCTGAGGTTG Rev: AGCTGTTCCGTCCCAGTAGATTA FL: GGCATGAACCGGAGGCCCA-FlLC: Red640-CCTCACCATCATCACACTGGAAGACTCC-p | 62 |
| p21WAF1 | Real-time RT-PCR | 211 | Fw: GGCAGACCAGCATGACAGATT Rev: GCGGCCAGGGTATGTACATGA FL: CCTGTGGGCGGATTAGGGCTTCC-FlLC: Red640-CTTGGAGAAGATCAGCCGGCGTTT-p | 62 |
| β2-Microglobulin | Real-time RT-PCR | 269 | Fw: CCAGCAGAGAATGGAAAGTC Rev: GATGCTGCTTACATGTCTCG FL: TTCTTCAGTAAGTCAACTTCAATGTCGGA-FlLC: Red640-ATGAAACCCAGACACATAGCAATTCAG-p | 53 |
| p21WAF1 promoter (RE) | ChIP | 169 | Fw: AGGCTGTGGCTCTGATTGG Rev: GGCTAAGGTTTACCTGGGG | 60 |
| p21WAF1 promoter ChIP | ChIP | 150 | Fw: GGGCGGGGCGGTTGTATATCAG Rev: GTCTGCCGCCGCTCTCTCACCT | 60 |
Fw, forward; Rev, reverse; Fl, fluorescein labelled probe; LC, LCRed640 labelled probe; p, phosphorylated.
Western Blotting
Whole cell lysates were prepared from HCT116 cells with or without treatment of TSA, H2O2. Protein concentration of lysates was determined with Bio-Rad Dc Protein Assay (BioRad Laboratories, Hercules, CA, USA), and equivalent amounts were loaded onto 10–13% SDS-PAGE. The gels were transferred to nitrocellulose membranes before immunodetection processing with anti-p53 (1:50; Oncogene, San Diego, CA), -acetyl-p53 (Lys320, 1:1000; and Lys382, 1:2000; Upstate, Lake Placid, NY), anti-p21 (1:25; DakoCytomation, Glostrup, Denmark), anti-acetyl- H3 (1:1000) and –H4 (1:3000; Upstate, Lake Placid, NY), anti-HDAC1 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), anti-caspase 3 (1:1000; Cell Signalling Technology), and with secondary antibodies (anti-mouse, 1:20000 or 1:30000; and anti-rabbit IgG peroxidase conjugated; 1:5000 or 1:10000; Pierce, Rockford, IL). Bound antibodies were detected by incubating the blots in West Pico chemiluminescent substrate (Pierce, Rockford, IL). The level of immunoreactivity was then measured as peak intensity using an image capture and analysis system (GeneGnome, Syngene, UK). Hybridization with anti-β-actin (1:30000; Sigma) was used to control equal loading and protein quality.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed using the ChIP Assay Kit according to the manufacturers protocol (Upstate, NY, USA). Briefly, approximately 1 × 106 cells were used per ChIP assay. Cells were cross-linked with 1% formaldehyde at 37°C for 10 min, rinsed with ice-cold PBS and harvested by brief centrifugation. Cell pellets were re-suspended in SDS-lysis buffer and sonicated to shear DNA to lengths between 200 and 1000 base pairs. After centrifugation, the supernatants were collected and diluted in ChIP dilution buffer. Two percent of the diluted cell supernatant was kept for DNA quantification and considered as inputs. Samples were incubated for 30 min at 4°C with salmon sperm DNA/protein A/protein G agarose 50% slurry before overnight immunoprecipitation with the appropriate antibody (anti-p53, anti-acetyl-H3 and -H4, anti-HDAC1). A portion of each sample was removed before immunoprecipitation and served as a negative control. After immunoprecipitation, the samples were incubated for 1 hr with salmon sperm DNA/protein A/protein G agarose 50% slurry and were centrifuged to collect the antibody/histone complex. After washing, the immune complexes and the negative controls (without antibody) were incubated in elu-tion buffer (1% SDS, 0.1 M NaHCO3). The elutes and inputes were then heated at 65°C for 4 hrs to reverse cross-link by addition of 5 M NaCl. Following proteinase K treatment, DNA was extracted by phenol/chloroform and precipitated with 96% ethanol. The recovered DNA was re-suspended in H2O for PCR. All the ChIP experiments and PCR were performed in triplicate. Sequences of primers, PCR product length and annealing temperatures are given in Table 1.
HDAC activity
HDAC activity in HCT116 cells was determined with a HDAC fluorimetric assay (Biomol, Plymouth Meeting, PA) according to the manufactures guidelines. Briefly, cells were plated in 96-well plates and, after 24 hrs treated with TSA and H2O2 as indicated previously. The medium was then replaced with the HDAC substrate for 20 min at 37°C. After incubation with the developer, deacetylation of the substrate was detected on a fluorimetric plate reader (excitation wavelength: 335 nm, emission: 460 nm).
Results
TSA pre-treatment switches H2O2 -damaged colorectal cancer cells from growth arrest into apoptosis
The H2O2- and TSA-associated specific alterations of cell cycle progression and apoptosis induction in HCT116 p53+/+ colorectal cancer cells were determined using flow cytometric analysis of DNA content by PI staining (FACS, Annexin-Assay). We could show that H2O2 treatment alone increases the number of cells in the G2 phase 1.3- and 2-fold after 6 hrs and 24 hrs, respectively (Fig. 1A and D). In parallel, cells underwent a low extent of apoptosis after 24 hrs, reflected by an increase of Pre-G1-cells from 3.1% to 5.9% (1.9-fold;Fig. 1A and D), which was confirmed by Annexin-V measurements and caspase 3 western blotting (Fig. 1B and C).
1.
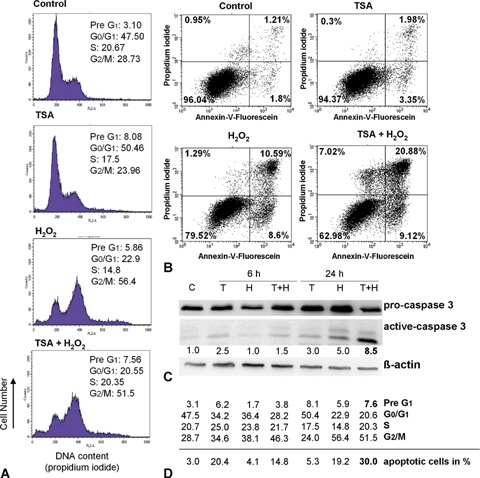
Effects of H2O2-treatment (30 mM, 3 min) on cell cycle profiles and cell viability of HCT116 p53+/+ cells after 6 hrs and 24 hrs, with and without Trichostatin A (TSA) (200 ng/ml, 6 hrs) pre-treatment.(A) H2O2 induces G2/M arrest in HCT116p53+/+ cells after 24 hrs, whereas TSA induces a switch from cell cycle arrest into apoptosis.(B) Annexin-V measurement shows that H2O2 induces apoptosis and TSA reinforces this effect.(C) Western Blotting of caspase 3 confirms the observed Annexin-V results.(D) Time-dependent effects (6 hrs and 24 hrs) of TSA, H2O2, and their combined treatment on cell cycle progression and apoptosis induction. Above the line, data of FACS analysis (in%) are shown and below the line apoptotic cells (in%) measured in the Annexin-V-Assay are given.
TSA treatment of HCT116 p53+/+ cells also resulted in G2/M arrest after 6 hrs (1.2-fold increase of cells in the G2 phase; Fig. 1D), while this growth arrest was completely reversed after 24 hrs (Fig. 1A and D). In addition, TSA treatment alone resulted in a low number of apoptotic cells and caspase 3 cleavage after 6 hrs and 24 hrs (Fig. 1B and C).
In order to induce stronger pro-apoptotic effects, we pre-treated H2O2-damaged HCT116 p53+/+ cells with TSA. While there was no further apoptosis induction after 6 hrs, we later found an increase of cells in the Pre-G1 phase from 3.1% to 7.6% (2.5-fold; Fig. 1A and D), an increase of apoptotic cells in the Annexin-Assay from 3.0% to 30.0% (10-fold, Fig. 1B and D), and an 8.5-fold increase of caspase 3 cleavage after 24 hrs (Fig. 1C). There seem to be time-dependent effects of TSA pre-treatment regarding growth arrest and apoptotic response: whereas efficient growth arrest was evident already after 6 hrs (Fig. 1D), cells continued to grow after 48 hrs (data not shown). In parallel, HCT116 p53+/+ cells underwent time-delayed apoptosis after 24 hrs (Figs. 1A–D), and apoptosis could even be increased after 48hrs (increase of PreG1 cells from 3.1% to 28.1% and increase of apoptotic cells in the Annexin-Assay from 3.0% to 43.0%, data not shown).
On the basis of these results, we showed that H2O2 addition alone resulted in a time-dependent inhibition of cancer cell growth paralleled by only minor apoptotic effects, while TSA pre-treatment of H2O2-damaged HCT116 p53+/+ cells induces a switch from this early growth arrest into efficient later apoptotic response.
H2O2 induces p53 and p21WAF1 expression
p53 and its target p21WAF1 play a major role in cell cycle control and apoptosis. To investigate H2O2-caused modulation of p53 and p21WAF1 expression in HCT116 p53+/+ cells, we determined their protein and mRNA level. There was a 2.2- and 2.4-fold increase in p53 protein expression 6 hrs and 24 hrs after H2O2 addition, respectively (Fig. 2A). p53 seemed to be regulated at the posttranscriptional level because its mRNA was not significantly up-regulated after H2O2 treatment (Fig. 2B). The higher amounts of p53 protein coincided with a significant increase in p21WAF1 protein expression (2-fold and 2.8-fold, respectively, Fig. 2A). As we could also observe a similar time-dependent increase in p21WAF1 mRNA (Fig. 2C), the induction of p21WAF1 protein expression seemed to be regulated transcriptionally.
2.
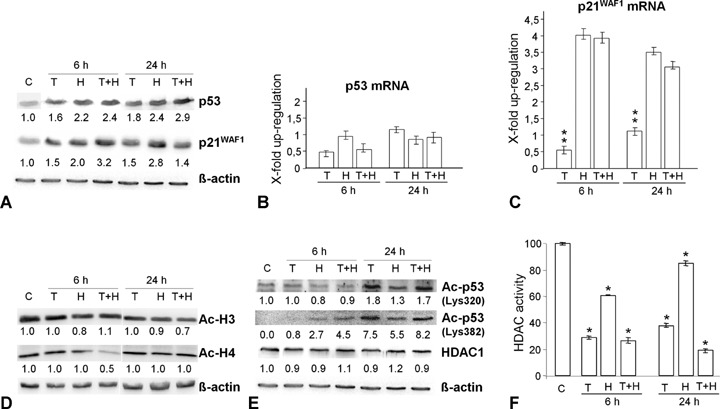
H2O2 and TSA modulate the expression of p53 and p21WAF1 and the acetylation status of core histones in HCT116 p53+/+ cells.(A) H2O2 and TSA induce p53 and p21WAF1 protein expression.(B–C) While H2O2 and TSA have no significant effect on p53 mRNA expression (real-time PCR, LightCycler), single H2O2 addition and TSA pre-treatment significantly increase p21WAF1 mRNA expression. (D) Western Blotting showing that neither H2O2 nor TSA had a significant effect on the acetylation form of H3 and H4. (E) Western Blotting showing that H2O2 modulates the acetylation levels of p53 (Lys320, Lys382) and TSA reinforces this effect. The protein amounts of HDAC1 remain unchanged during all treatments.(F) Histone deacetylase (HDAC) activity of HCT116 p53+/+ cells was measured with a HDAC fluorimetric assay. The control cell level (C) was adjusted to 100%. *P<0.05. As expected TSA decreases HDAC activity.
TSA pre-treatment modulates p53 and p21WAF1 expression in a time-dependent manner
To further elucidate the effects of TSA on p53 and p21WAF1 expression, we pre-treated HCT116 p53+/+ cells with TSA. After single application of TSA, as shown in Fig. 2A, the p53 and p21WAF1 expression levels were only slightly enhanced after 6 hrs and 24 hrs compared to H2O2 addition alone. By contrast, TSA pre-treatment induced continuous increase in p53 protein expression (Fig. 2A). Interestingly, after 6 hrs this p53 up-regulation was accompanied by a synergistic induction of p21WAF1 protein, where it was abolished after 24 hrs. Thus, TSA promotes p21WAF1 expression as an early response and represses the H2O2-induced p21WAF1 mRNA and protein expression at later time points.
H2O2 and TSA modulate acetylation
Since acetylation has been linked to activation of gene transcription [20], we investigated the effects of H2O2-associated DNA damage combined with TSA pre-treatment on the acetylation status of the core histones H3 and H4, as well as on p53 in HCT116 p53+/+ cells. While the total amounts of acetylated H3 did not significantly change after H2O2 and TSA treatment alone and in combination (Fig. 2D), the levels of acetylated p53 increased remarkably (lysine 382 > lysine 320; Fig. 2E). Interestingly, the protein level of acetylated H4 was decreased following TSA pre-treatment after 6 hrs (Fig. 2D). As expected, we observed a significant decrease in the HDAC activity (Fig. 2F). The protein level of HDAC1, however, remained constant irrespective of treatment (Fig. 2E).
H2O2 induces binding of p53 and acetylated H4 on the p21WAF1 promoter
As indicated above, H2O2 induced p21WAF1 expression through its transcriptional activation. In order to determine the involvement of acetylated histones in the transcriptional activation of p21WAF1 in DNA-damaged HCT116 p53+/+ cells, we performed ChIP analyses. To further investigate whether the increased p21WAF1 mRNA was due to direct transcriptional activation by p53 promoter binding, we also used a p53 antibody to immunoprecipitate chromatin from HCT116 p53+/+ cells 6 hrs and 24 hrs after H2O2 treatment. We observed both, an increased binding of p53 and acetylated H4 around the two p21WAF1 promoter sites, the responsible element (RE) and the Sp1 site (Figs. 3A–E). However, both proteins were present preferentially on the RE after H2O2 treatment. Interestingly, H3 was not involved in this process, giving evidence of a histone H4-specific acetylation as a consequence of H2O2-induced oxidative stress (Fig. 3F and G).
3.
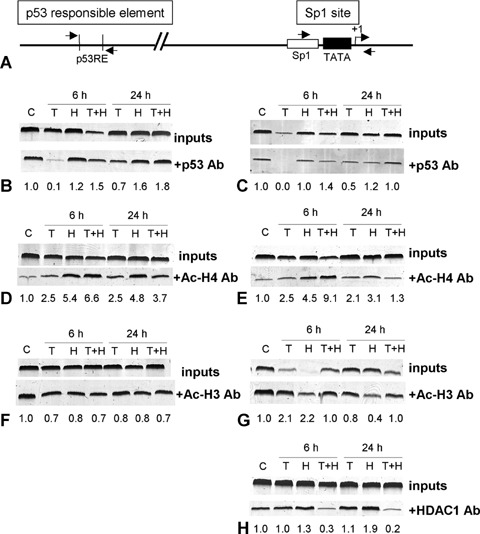
Effects of H2O2 and TSA on the promoter status of p21WAF1 in HCT116 p53+/+ cells. (A) Schematic drawing of the relative positions of PCR primers for the amplification of Sp1 site or p53 responsible element (RE) of the p21WAF1 promoter. (B–H) ChIP experiments using immunoprecipitation with a p53 (B, C), Ac-H4 (D, E), Ac-H3 (F, G) and HDAC1 antibody (H) in control cells (C), 6 hrs and 24 hrs after treatment with TSA for 6 hrs (200 ng/ml, T), and with H2O2 (30 mM, 3 min, H), and with TSA and H2O2 (T + H), respectively. (B, C) There is an increase in p53 binding only at the p21WAF1-p53 RE promoter region after H2O2. TSA pre-treatment enables p53 to bind also at the Sp1 site of the p21WAF1 promoter in the early response, while this binding was diminished in the late response. (D–G) Pre-treatment with TSA increases H2O2-induced accumulation of acH4 (D, E), but not acH3 (F, G) in the chromatin associated with both p21WAF1 promoter regions. (H) Combination of TSA and H2O2 treatment displaces HDAC1 from the Sp1 binding site.
TSA pre-treatment increases H2O2-induced accumulation of acetylated H4 in the early response
To investigate whether TSA-induced up-regulation of p21WAF1 was in fact due to a stronger histone acetylation at the p21WAF1 promoter, we performed ChIP analyses. Indeed, H2O2-damaged HCT116 p53+/+ cells pre-treated with TSA showed further enrichment of p21WAF1 promoter regions in the acetylated H4 chromatin pool 6 hrs after TSA pre-treatment (Fig. 3D and E). Moreover, the pattern of histone H4-associated chromatin at the p21WAF1 promoter seemed to be site-specific and time-dependent, showing more pronounced H4 acetylation at the Sp1 site after 6 hrs (9.1-fold vs. 6.6-fold, respectively, Fig. 3D and E) and lower H4 acetylation at the Sp1 site at a later time point (1.3-fold vs. 3.7-fold, respectively, Fig. 3D and E). Furthermore, acetylated histone H3 was not involved at either p21WAF1 promoter site (Fig. 3F and G). The increase in H4 acetylation at the p21WAF1 promoter after 6 hrs may cause the higher p21WAF1 mRNA and protein expression observed. However, comparing the 24 hrs with the 6 hrs time point, H4-acetylation, especially at the Sp1 site, was significantly reduced after 24 hrs (9.1-fold to 1.3-fold, Fig. 3E). Loss of H4 acetylation at the p21WAF1 promoter observed at a later time point seems to correlate with down-regulation of p21WAF1 expression and apoptosis induction.
TSA pre-treatment induces time-dependent binding of p53 at the p21WAF1 promoter Sp1 site
As we have shown, H2O2 induced a preferential enrichment of p53 binding at the RE site of the p21WAF1 promoter (Fig. 3B and C). Further, ChIP analyses manifested that 6 hrs after TSA pre-treatment, p53 is able to bind not only to the RE site but also to the Sp1 site of the p21WAF1 promoter in HCT116 p53+/+ cells (Fig. 3C). However, p53 lost its association with the Sp1 site after 24 hrs. p53 release from the promoter, which is in accordance with the H4 acetylation pattern, seems to be a further reason for the p21WAF1 down-regulation observed at a later time point.
p53 and HDAC1 compete for binding at the p21WAF1 promoter in a time-dependent manner
Using the ChIP assay with a HDAC1 antibody, we examined the competition between HDAC1 and p53 binding at the Sp1 site of the p21WAF1 promoter (Fig. 3C–H) as described previously for osteosarcoma cells [16]. We could show that after TSA pre-treatment H2O2 damage induced a significant decrease in HDAC1 binding in HCT116 p53+/+ cells after 6 hrs and 24 hrs (Fig. 3H). By contrast, p53 shows increased binding after 6 hrs as mentioned above, whereas it is again released at a later time point (Fig. 3C). We therefore suggest that the DNA damage signal reveals a replacement of HDAC1 by p53 at the Sp1 site after TSA pre-treatment as an early response. At a later time point, the competition between both proteins seems to disappear.
Effects of H2O2 and TSA on p53-deficient HCT116 cells
Because colorectal cancer therapy often fails due to drug resistance of colon cancer cells mostly due to p53 mutations, we investigated the effectiveness of our developed pro-apoptotic combination strategy for HCT116 p53−/− cells. In contrast to the p53+/+ cells, we observed no significant increase in the Pre-G1 cell population (Fig. 4A and D), reflecting the fact that apoptosis was not efficiently induced. Additionally, this result was confirmed by Annexin-V measurements and caspase 3 western blotting (Fig. 4B and C), suggesting that HCT116 p53−/− cells lost their apoptotic competence if p53 is lost. However, we observed H2O2-induced G2/M arrest, which could even be reinforced after TSA pre-treatment and was associated with significantly increased p21WAF1 expression (Figs. 4 and 5B). Nevertheless, ChIP experiments and RT-PCR gave no evidence of a tran-scriptionally caused up-regulation of p21WAF1 protein after TSA treatment alone (9-fold) at an early time point (Figs. 5A, E and F), suggesting posttranscriptional processes for p21WAF1 regulation. By contrast, the p21WAF1 up-regulation after H2O2 treatment or in combination with TSA at a later time point seems to be regulated transcriptionally (Fig. 5A), but was obviously not caused by HDAC1 release or acetylated H4 recruitment at the p21WAF1 promoter (Fig. 5E and F). Furthermore, HDAC1 activity was enhanced following single H2O2 treatment after 6 hrs and 24 hrs in both HCT cell lines (Figs. 3C and 5C). However, only in p53-deficient cells this rise in activity was accompanied by an increase in the HDAC1 protein amounts (Figs. 5B, C, 3B and C). The higher HDAC1 binding on the Sp1 site (Fig. 5F), compared to the p53+/+ cells (Fig. 3F), supports our idea that p53 actively displaces HDAC1 from the p21WAF1 promoter in H2O2-damaged, TSA pre-treated colorectal cancer cells.
4.
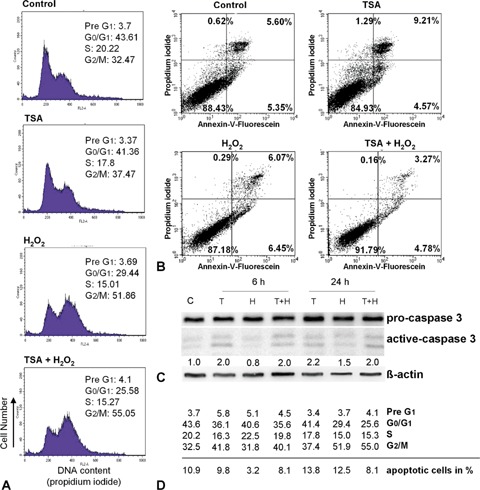
Effects of H2O2-treatment (30 mM, 3 min) on cell cycle profiles and cell viability of HCT116 p53−/− cells after 6 hrs and 24 hrs, with and without TSA (200 ng/ml, 6 hrs) pre-treatment. (A) H2O2 induces G2/M arrest in HCT116 p53−/− cells after 24 hrs while TSA induces a slight G2/M stop.(B) Annexin-V measurement shows that neither H2O2 nor TSA induce a significant apoptosis. (C) Western Blotting of caspase 3 confirms the observed Annexin-V results. (D) Time-dependent effects (6 hrs and 24 hrs) of TSA, H2O2, and their combined treatment on cell cycle progression and apoptosis induction. Above the line, data of FACS analysis (in%) are shown and below the line apoptotic cells (in%) measured in the Annexin-V-Assay are given.
5.
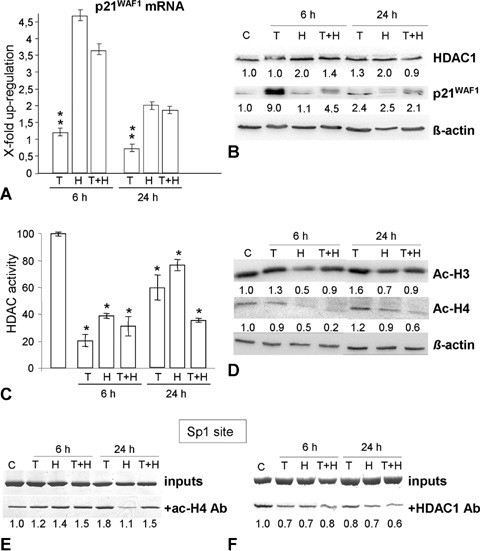
Effects of H2O2 and TSA on p21WAF1, acH3, acH4 and HDAC1 expression and on the p21WAF1 promoter status in HCT116 p53−/− cells. (A) H2O2 and TSA pre-treatment induce p21WAF1 mRNA expression after 6 hrs. (B) Whereas H2O2 induces both, p21WAF1 and HDAC1 protein expression, TSA alone and H2O2+ TSA have no effect on HDAC1 expression. (C) There was a significant decrease (*P<0.05) in the activity of HDACs 6 hrs after H2O2 or TSA addition, but TSA pre-treatment resulted in prolonged reduction of the HDACs activity. (D) Western Blotting showed that there was a decrease in the total acH3 and acH4 levels after TSA pre-treatment. (E, F) ChIP experiment using immunoprecipitation with Ac-H4 (E) and HDAC1 (F) antibodies in control cells (C), and 6 hrs and 24 hrs after treatment with TSA (200 ng/ml, 6 hrs, T), and with H2O2 (30 mM, 3 min, H), and TSA and H2O2 (T + H), respectively, showing that there is only a slight increase in acH4 around the p21WAF1-Sp1 promoter region after H2O2 treatment. Pre-treatment with TSA slightly reinforces the acH4 on this region. Furthermore, the pre-treatment do not significantly affect HDAC1 binding at the Sp1 promoter site.
Discussion
Many efforts have been made to improve the therapeutic efficacy by using combination strategies with oxidative stress-inducing drugs and HDAC inhibitors in colorectal cancer. The tumour suppressor p53 and its target gene p21WAF1 play a major role in drug response to growth arrest and apoptosis. How p53 and p21WAF1 decide between cell cycle arrest and apoptosis is only poorly understood. Here we show for the first time, that the most critical determinant of the function of the HDAC inhibitor TSA in DNA-damaged HCT116 p53+/+ colorectal cancer cells is p53 with activation of p21WAF1 in the early response and diminishing the transcriptional transactivation of p21WAF1 in the late response with simultaneous switching on the apoptotic machinery.The major conclusion is that TSA enhances a p53-dependent switch from G2/M arrest into apoptosis through site-specific and time-dependent binding of acetylated H4 on the p21WAF1 promoter. Therefore, p53 executes a dual function, whereas it can be considered as a bridge between cell cycle arrest and apoptosis.
The effects of H2O2 on colorectal cancer cells
In a recent study, Kaeser and Iggo [19] reported for the first time that histone acetylation of the p21WAF1 promoter is p53-dependent and H4-specific using 5-Fluorouracil (5-FU) DNA-damaged HCT116 colorectal cancer cells. Our study focussed on the acetylation-dependent regulation of the p53/p21WAF1 pathway after setting HCT116 p53+/+ cells under H2O2-induced oxidative stress because it is known that oxidative stress contributes to early modifications of histone proteins and associated gene expressions [21].In our ChIP experiments, we could show that the p21WAF1 transcriptional activation after H2O2 treatment was at least partly due to an increase in p53 recruitment to the p21WAF1 promoter.The increase in p53 binding to the p21WAF1 promoter was accompanied by a rise in acetylated H4, but not in acetylated H3 in the chromatin associated with the p21WAF1 gene.We therefore suggest that p53-mediated trans-activation of p21WAF1 transcription correlates with increased H4 acetylation. Such an increase in H4 acetylation conferred an open chromatin structure around the p21WAF1 promoter and thus permitted the accessibility to p53. Furthermore, p53 is able to recruit HATs, such as p300/CBP and PCAF/hGen5, thus inducing H4 acetylation at the p21WAF1 promoter [22–24]. Indeed, we found that H2O2-damaged HCT116 p53+/+ cells exhibit p53-dependent and histone H4-specific acetylation of the p21WAF1 promoter as 5-FU-treated cells as well. As a consequence, induced p21WAF1 expression leads to cell cycle arrest at the G2/M transition but only to a low extent of apoptosis. Thus, we suggest that the observed up-regulation of p21WAF1 after DNA damage by p53-dependent histone H4 specific acetylation is directly linked to cell cycle arrest, which might be a more common principle of tumour cells.
Nevertheless, besides p53-dependent transcriptional transactivation of p21WAF1, we found that p21WAF1 can also be activated via p53-independent pathways reflected by cell cycle arrest in HCT116 p53−/− cells. As loss of p53 gene function, which occurs in most colon cancer cells, often leads to resistance to anticancer drugs, the presented finding of a p53-independent action of H2O2 might overcome resistance problems at least at the growth arrest level.Definitely, targeted binding of acetylated H4 on the p21WAF1 promoter does not proceed in p53-deficient cells which substantiate p53-dependent H4 acetylation and p53-directed recruitment of HATs which catalyse the acetylation of histone proteins. Taken these results together, we conclude that transcriptional activation of p21WAF1 is not essentially p53-dependent, but the binding of acetylated H4 on the p21WAF1 gene.We could further show that binding of p53 to the p21WAF1 promoter was site-specific, which also caused site-specific binding of acetylated H4 to the promoter regions. p53 is able to bind to different sites in the p21WAF1 promoter.We studied two DNA sequences of the p21WAF1 promoter showing different affinity for p53 binding. One of these sequences contained a p53-RE [25], and the other one was located between positions –60 and +90 relative to the transcriptional starting point, and contained a Sp1 site [26].In our study, both sites showed similar patterns of H4 acetylation and p53 binding after H2O2 treatment.However, the level of acetylated H4 and p53 at the RE was globally higher than that at the Sp1 site.Otherwise, the increased H4 acetyla-tion localized at the Sp1 site of the p21WAF1 gene after H2O2 treatment was transient and correlated directly with the level of transcriptional activation of the gene. In accordance with Berthiaume et al. [27], this effect seemed to be selective for only a limited number of genes because the level of histone acetylation in the β-actin gene remained unchanged after H2O2 treatment (data not shown). Despite the fact that we observed a high amount of acetylated H4 in the p21WAF1 promoter, H2O2 had no significant effect on the overall acetylation level of H4.This might further support the idea that histone acetylation can be specifically targeted to the p21WAF1 promoter by transcription factors, such as p53 [13, 27].DNA damage signals are passed to p53 activity through posttrans-lational modifications such as acetylation of lysine residues [28, 29]. In our studies, H2O2 caused an increase in p53 acetylated at Lys320 and 382.It has been shown that acetylation in the C-terminal region of p53 by PCAF and p300 enhances the ability of p53 to bind target DNA [30], therefore stimulating its activity and binding efficiency at the p21WAF1 promoter, and promoting histone acetylation around the p21WAF1 promoter through the recruitment of HATs [22].
The effects of TSA pre-treatment on colorectal cancer cells
The effects of H2O2 were strengthened by the HDAC inhibitor TSA by inducing increased acetylation of H4 in the chromatin associated with the p21WAF1 gene, especially at the Sp1 site in the early response.We could show that, as expected, a significantly increased histone acetylation is the consequence of TSA pre-treatment caused by inhibition of HDAC1 reflected by decreased HDAC activity. It is worth mentioning that although the protein level of total acetylated H4 was decreased following TSA pretreatment after 6 hrs, we observed an enhanced specific binding of ac-H4 on the p21WAF1 promoter.This suggests that the total protein amount must not necessarily be correlated with the binding efficiency of a protein to the promoter region of a specific gene. Interestingly, histone acetylation induced by TSA promotes p21WAF1 expression at early time points and represses the H2O2-induced induction of p21WAF1 mRNA and protein expression at a later time point which is accompanied by decreased histone H4 acetylation. Here, we suggest that after 24 hrs decreased acetylation at the p21WAF1 promoters Sp1 site causes lower p21WAF1 mRNA and protein expression in TSA pre-treated HCT116 p53+/+ cells, thus switching cells from cell cycle arrest into remarkably p53-dependent apoptosis. Furthermore, according to Liu et al. [31], the extent of acetylated histones at the Sp1 site, and not the region containing the p53 RE, directly correlates with the activity of p53 to induce endogenous p21WAF1, while TSA pretreatment leads to stronger transcriptional activation in the early response. Moreover, HDAC1 has been shown to repress p21WAF1 by competing with p53 for DNA binding to the p21WAF1 promoter at the Sp1 binding site using actinomycin D-stimulated osteosarcoma cells [16]. In our study, H2O2-induced DNA damage in HCT116 p53+/+ cells led to a recruitment of p53 and hyperacetylation of histones at the p21WAF1 promoters Sp1 site, without any decrease in HDAC1 binding. However, pre-treatment with TSA before H2O2 damage caused the dissociation of HDAC1 from the p21WAF1 promoter accompanied by further recruitment of p53.The fact that HDAC1 is still bound at the Sp1 site of the p21WAF1 promoter in p53-deficient cells supports this idea. Furthermore, as expected, the increase in acetylated p53 was strengthened by pre-treatment with TSA.One explanation could be that TSA inhibited MDM2-HDAC1-mediated deacetylation of p53 [32, 33].
Interestingly, as for the p21WAF1 protein expression, we observed a switch from activation at an early time point as a consequence of H2O2 and TSA treatment to a repression at a later time point. In addition, TSA induces apoptosis by activating caspase 3. This is in accordance with Zhao et al.[34], who postulated that p21WAF1 down-regulation seems to be necessary for the activation of apoptosis. Furthermore, increased apoptotic sensitivity to drugs has been reported in colon cancer cells lacking p21WAF1[35]. However, in our study, p21WAF1 is also decreased in p53-deficient cells but without significant apoptosis, suggesting that p21WAF1 down-regulation alone does not provide efficient cell death but in combination with the presence of p53.The minor increase in activated caspase 3 in HCT p53−/− cells after TSA pre-treatment could be explained by the fact that HDAC inhibitors also exert their pro-apop-totic effects through p53-independent, but death receptor-dependent (TRAIL, Fas) pathways [36].
Proposed model
The results presented here support the following model (Fig. 6): when exposed to agents which cause DNA damage, HCT116 p53+/+ colorectal cancer cells are stimulated to produce p53. This induced p53 transcriptionally activates p21WAF1 expression in association with acetylated H4 by directly interacting especially with the p53 RE. Thus, p21WAF1 inhibits cyclin-dependent kinases, which results in a failure of cells to exit G2. This G2/M arrest, following induction of p53, prevents cancer cells from growing. Simultaneously, loss of wild-type p53 function does not prevent the activation of this growth control pathway because cell cycle arrest can also occur without activation of p53. These observations suggest a rationale for the design of drugs against colon cancer based on oxidative stress-induced DNA damage.
6.
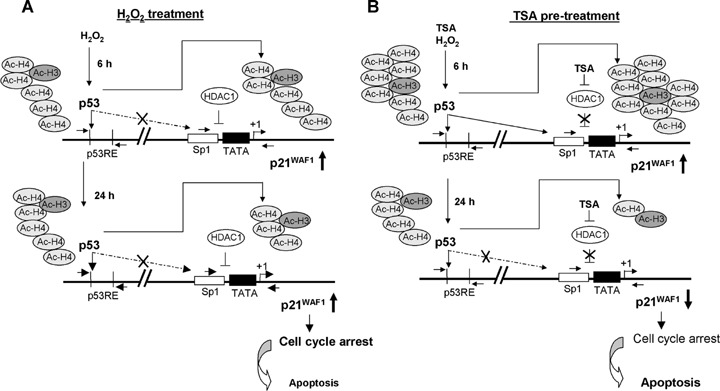
Mechanistic model to the action of H2O2 and TSA in oxidative-damaged HCT116 p53+/+ colorectal cancer cells. (A) H2O2 exposure causes a p53-induced transcriptional activation of p21WAF1 expression in association with acH4 by directly interacting especially with the p53 responsible element.(B) The combination of this DNA-damage pathway with TSA pre-treatment resulted in time-dependent effects on cell cycle arrest and apoptosis characterized as early and late responses.Firstly, TSA pre-treatment enables p53 to bind not only on the p53 RE but similarly on the Sp1 site of the p21WAF1 promoter by simultaneous displacement of HDAC1 and strengthened binding of acetylated H4, especially at the Sp1 site.Secondly, binding of p53 and therefore of acetylated H4 on the Sp1 site is significantly decreased in the late response as a consequence of TSA pre-treatment. This allows the cells to enter the cell cycle again and simultaneously to switch cells into apoptosis.
Moreover, the combination of this DNA-damaged pathway with TSA pre-treatment resulted in time-dependent effects characterized as early and late responses.Firstly, TSA pre-treatment enables p53 to bind not only to the p53-RE but also to the Sp1 site of the p21WAF1 promoter by simultaneous displacement of HDAC1 and strengthened binding of acetylated H4 to both promoter regions, but especially to the Sp1 site. In this manner, induced p21WAF1 leads to stronger G2/M arrest than oxidative damage alone. However, secondly, binding of p53 and therefore of acetylated H4 to the Sp1 site is significantly decreased in the late response as a consequence of TSA addition.Hence, p53 time-dependently transactivates p21WAF1 and, simultaneously, the activation of apoptosis. Reduced p53-triggered transactivation of p21WAF1 in late response allows cells to re-enter the cell cycle. In this context, the re-entry into cell cycle prior to repair of DNA damage causes endoredupli-cation, which may subsequently result in cell death, further enhancing apoptosis caused by TSA.
In summary, our results suggest that TSA in combination with an ROS-based anticancer drug might have remarkable pro-apoptotic effects on p53-wt col-orectal cancer cells, while this strategy may have mainly growth arrest effects in cancer cells with nonfunctional p53.Therefore, TSA can be considered as a novel therapeutic strategy for colon cancer based on the possibility of sensitization to ROS-based antitumour agents, suggesting that combined TSA and H2O2 treatment merits further investigations for their role as cell cycle blocker and cancer chemopreventive agents.
Acknowledgments
We thank Simone Staeck for her excellent technical assistance.This work was supported by a DAAD-fellowship (C.H.).
References
- 1.Chen J-H, Stoeber K, Kingsbury S, Ozanne SE, Williams GH, Hales N. Loss of proliferative capacity and induction of senescence in oxidatively stressed human fibroblasts. J Biol Chem. 2004;279:49439–46. doi: 10.1074/jbc.M409153200. [DOI] [PubMed] [Google Scholar]
- 2.Schmitt E, Paquet C, Beauchemin M, Bertrand R. DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J Zhejiang Univ Sci B. 2007;8:377–97. doi: 10.1631/jzus.2007.B0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozben T. Oxidative stress and apoptosis: Impact on cancer therapy. J Pharm Sci. 2007;96:2181–96. doi: 10.1002/jps.20874. [DOI] [PubMed] [Google Scholar]
- 4.Ogryzko VV, Wong P, Howard BH. WAF1 retards S-phase progression primarily by inhibition of cyclin-dependent kinases. Mol Cell Biol. 1997;17:4877–82. doi: 10.1128/mcb.17.8.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niculescu AB, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/WAF1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–43. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radhakrishnan SK, Feliciano CS, Najmabadi F, Haegebarth A, Kandel ES, Tyner AL, Gartel AL. Constitutive expression of E2F-1 leads to p21-dependent cell cycle arrest in S phase of the cell cycle. Oncogene. 2004;23:4173–6. doi: 10.1038/sj.onc.1207571. [DOI] [PubMed] [Google Scholar]
- 7.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 8.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90. [PubMed] [Google Scholar]
- 9.Wu C. Chromatin remodelling and the control of gene expression. J Biol Chem. 1997;272:28171–4. doi: 10.1074/jbc.272.45.28171. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–9. [PubMed] [Google Scholar]
- 11.Wharton W, Savell J, Cress WD, Seto E, Pledger WJ. Inhibition of mitogenesis in Balb/c-3T3 cells by trichostatin A. J Biol Chem. 2000;275:33981–7. doi: 10.1074/jbc.M005600200. [DOI] [PubMed] [Google Scholar]
- 12.Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, Coombes RC. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res. 2001;7:971–6. [PubMed] [Google Scholar]
- 13.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li GC, Zhang X, Pan TJ, Chen Z, Ye ZQ. Histone deacetylase inhibitor trichostatin A inhibits the growth of bladder cancer cells through induction of p21WAF1 and G1 cell cycle arrest. Int J Urol. 2006;13:581–6. doi: 10.1111/j.1442-2042.2006.01344.x. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Wu X. Histone deacetylase inhibitor, Trichostatin A, activates p21WAF1/CIP1 expression through downregulation of c-myc and release of the repression of c-myc from the promoter in human cervical cancer cells. Biochem Biophys Res Commun. 2004;324:860–7. doi: 10.1016/j.bbrc.2004.09.130. [DOI] [PubMed] [Google Scholar]
- 16.Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, Tischler J, Chiocca S, Suske G, Rotheneder H, Wintersberger E, Seiser C. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol. 2003;23:2669–79. doi: 10.1128/MCB.23.8.2669-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang ZM, Hu J, Zhou D, Xu ZY, Panasci LC, Chen ZP. Trichostatin A inhibits proliferation and induces expression of p21WAF and p27 in human brain tumor cell lines. Ai Zheng. 2002;21:1100–5. [PubMed] [Google Scholar]
- 18.Herold C, Ganslmayer M, Ocker M, Hermann M, Geerts A, Hahn EG, Schuppan D. The histone-deacetylase inhibitor Trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J.Hepatol. 2002;36:233–40. doi: 10.1016/s0168-8278(01)00257-4. [DOI] [PubMed] [Google Scholar]
- 19.Kaeser MD, Iggo RD. Promoter-specific p53-dependent histone acetylation following DNA damage. Oncogene. 2004;23:4007–13. doi: 10.1038/sj.onc.1207536. [DOI] [PubMed] [Google Scholar]
- 20.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 21.Ullrich O, Grune T. Proteasomal degradation of oxidatively damaged endogenous histones in K562 human leukemic cells. Free Radic Biol Med. 2001;31:887–93. doi: 10.1016/s0891-5849(01)00672-4. [DOI] [PubMed] [Google Scholar]
- 22.Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–54. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 23.Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 24.Espinosa JM, Verdun RE, Emerson BM. P53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–27. doi: 10.1016/s1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- 25.Wei G, Li AG, Liu X. Insights into selective activation of p53 DNA binding by c-Abl. J Biol Chem. 2005;280:12271–8. doi: 10.1074/jbc.M409522200. [DOI] [PubMed] [Google Scholar]
- 26.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 27.Berthiaume M, Boufaied N, Moisan A, Gaudreau L. High levels of oxidative stress globally inhibit gene transcription and histone acetylation. DNA Cell Biol. 2006;25:124–34. doi: 10.1089/dna.2006.25.124. [DOI] [PubMed] [Google Scholar]
- 28.Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–55. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 29.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 30.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 31.Liu G, Xia T, Chen X. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J Biol Chem. 2003;278:17557–65. doi: 10.1074/jbc.M210696200. [DOI] [PubMed] [Google Scholar]
- 32.Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Apella E, Yao TP. P300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20:1331–40. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Apella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–45. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao LY, Niu Y, Santiago A, Liu J, Albert SH, Robertson KD, Liao D. An EBF3-mediated transcriptional program that induces cell cycle arrest and apoptosis. Cancer Res. 2006;66:9445–52. doi: 10.1158/0008-5472.CAN-06-1713. [DOI] [PubMed] [Google Scholar]
- 35.Mahyar-Roemer M, Roemer K. p21WAF1/Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene. 2001;20:3387–98. doi: 10.1038/sj.onc.1204440. [DOI] [PubMed] [Google Scholar]
- 36.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG. Inhibitors of histone deacetylases induce tumour-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]