

Abstract
The vascular and immune systems of mammals are closely intertwined: the individual components of the immune system must move between various body compartments to perform their function effectively. Sphingosine 1-phosphate (S1P), a bioactive lipid mediator, exerts effects on the two organ systems and influences the interaction between them. In the resting state, the vascular S1P gradient contributes to control of lymphocyte recirculation through the blood, lymphoid tissue and lymphatic vasculature. The high level of S1P in blood helps maintain endothelial barrier integrity. During the inflammatory process, both the level of S1P in different immune compartments and S1P receptor expression on lymphocytes and endothelial cells are modified, resulting in functionally important changes in endothelial cell and lymphocyte behaviour. These include transient arrest of lymphocytes in secondary lymphoid tissue, crucial for generation of adaptive immunity, and subsequent promotion of lymphocyte recruitment to sites of inflammation. This review begins with an outline of the basic biochemistry of S1P. S1P receptor signalling is then discussed, followed by an exploration of the roles of S1P in the vascular and immune systems, with particular focus on the interface between them. The latter part concerns crosstalk between S1P and other signalling pathways, and concludes with a look at therapies targeting the S1P-S1P receptor axis.
Keywords: sphingosine 1-phosphate, inflammation, endothelial cell, lymphocyte
Introduction
Under normal conditions, vascular capillary endothelial cells form tight junctions with each other ensuring that there is no net movement of plasma into peripheral organ tissues. Resting endothelial cells express adhesion molecules at low level and there is very limited presentation of chemoattractant cytokines (chemokines) on the luminal extracellular matrix. Few leucocytes are recruited into sub-endothelial tissues and normal tissue function is maintained by homeostatic processes.
Pathogens or other stimuli can trigger an inflammatory response in the periphery by induction of signalling by Toll-like receptors (TLRs) expressed on sentinel cells. Dendritic cells (DC) are perhaps the most important cells of this type: tissue resident DC, such as Langerhans cells, express a wide range of TLRs and are therefore able to respond to many different pro-inflammatory cues. These include pathogen-derived nucleic acids and lipids, and also endogeneous components – for example histones.
Production of inflammatory mediators, including tumour necrosis factor (TNF)-α, results in a reduction in endothelial barrier integrity and an increase in the amount of fluid (containing a number of non-cellular components which contribute to inflammation, including complement proteins) exuded into the local tissue. Adhesion molecules, including selectins and Ig-domain containing-molecules such as ICAM-1 and VCAM-1 become highly expressed on the surface of endothelial cells. Chemokines released within the tissue are also presented on the luminal surface of the endothelium. The result is that the fluid exudate is accompanied by an influx of captured leucocytes, which under healthy conditions would help resolve the inflammation (Fig. 1).
Fig 1.
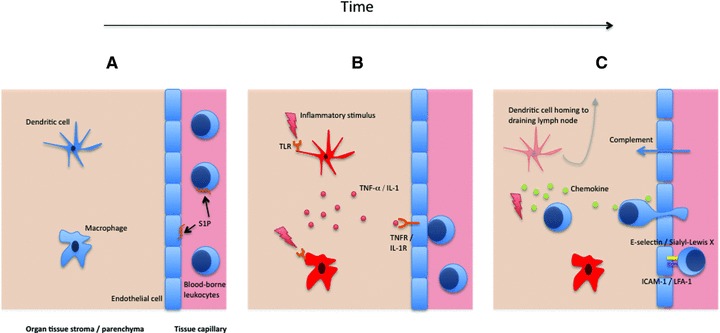
Progression of the inflammatory process. In the resting state, sentinel cells including DC and macrophages reside in peripheral tissue and are placed to respond to an inflammatory insult. Tissue capillary endothelial cells form tight junctions with each other (A). The constitutive signalling of S1P receptors plays an important role in maintenance of barrier integrity. Pathogen-associated molecular patterns or endogenous danger signals cause activation of the sentinel cells via specific receptors; the TLRs are an important class of these. Inflammatory cytokines, including TNF-α and interleukin-1 produced by stimulated macrophages and DC, bind cognate receptors expressed by the local endothelial cells (B). The endothelial cells become activated and the permeability of the vascular barrier decreases as cell–cell contacts are disrupted. Soluble components, for example complement which can opsonize and directly lyse pathogens, can thus move much more easily into the tissue. Adhesion molecules are also up-regulated on the surface of the endothelium, which facilitates capture of leucocytes from the blood and their ingress into the tissue. The carbohydrate-binding E-selectin and Ig-family protein ICAM-1 are particularly important for the rolling capture then firm adhesion, respectively, of passing leucocytes. Leucocyte entry into tissue and migration to the inflamed site is aided by a gradient of chemotactic cytokines – chemokines. DC migrate out of the tissue and pass via the afferent lymphatics into the local draining lymph node, where they can prime the adaptive immune response. Later, antigen-specific cells such as T lymphocytes are recruited to the tissue. Activated macrophages have an important role in the resolution of inflammation because of their ability to phagocytose dead cells, debris, and their ability to re-model the extracellular matrix (C).
The endogeneous compound sphingosine 1-phosphate (S1P) plays a central role in the regulation of the vascular and immune systems in the resting state and inflammation. There is an extensive literature on the involvement of S1P in the processes of angiogenesis and regulation of arterial vascular tone [1, 2]. The focus of this review lies at the interface of the vascular and immune systems: in this regard, two aspects of S1P biology are of key concern. First, S1P is effectively compartmentalized in different regions of the lymphatic and circulatory systems. Second, whilst the receptors for S1P are widely expressed, the functioning of particular receptor types is dependent on the immune state. This review explores these concepts in further detail.
Biochemistry of sphingosine 1-phosphate
S1P is a bioactive lipid and member of the lysophospholipid family. It is 379 Da in size and consists of a polar head group and a long saturated aliphatic tail (Fig. 2).
Fig 2.
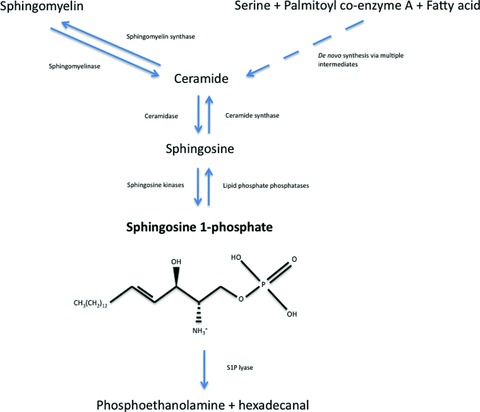
Synthesis, degradation and molecular structure of S1P. S1P is formed either de novo, from serine, palmitoyl coA and a fatty acid, or from the breakdown of membrane-resident sphingomyelin. S1P can be dephosphorylated, reversibly, to sphingosine, or degraded irreversibly to phosphoethanolamine and hexadecanal.
Generation
S1P is generated inside cells through the action of sphingosine kinases 1 and 2 (SK1 and 2) on the primary hydroxyl group of sphingosine (which comprises a hydrophobic backbone and hydrophilic head group). Sphingosine is itself generated from the metabolism of sphingolipids, structural components of all eukaryotic cell membranes; this synthetic pathway is widespread among mammalian cells [3–5]. The primary mechanism by which S1P is produced involves the degradation of sphingomyelin (a membrane component enriched in lipid rafts) to ceramide, conversion to sphingosine and finally phosphorylation to produce S1P [6]. An important aspect of S1P biochemistry is the existence of the sphingolipid ‘rheostat’ inside cells. Sphingosine (and ceramide, with which it can be interconverted) promotes apoptosis, whereas S1P suppresses apoptosis and causes cell proliferation. Hence, the balance of these lipid metabolites determines cell survival, and the relative activity of the S1P synthetic and degradative enzymes is an important regulator of cell fate [5, 7]. Sphingosine kinases 1 and 2 share overall homology, both consisting of five conserved domains (C1–5). They do however display different catalytic properties, subcellular locations, tissue distribution and temporal expression patterns. The enzymes have specific and unique functions: SK1 prefers D-erythro sphingosine as a substrate, whereas SK2 phosphorylates a wider range of substrates. SK1 can be stimulated by growth factors, cytokines, hormones, various G protein-coupled receptor (GPCR) ligands and by lysophospholipids including S1P itself [3, 8, 9]. Activation of SK1 is essential for the pro-inflammatory effects on endothelium of cytokines such as TNF-α[2, 10]. Activation occurs through phosphorylation and translocation of the enzyme to the plasma membrane (into the vicinity of its substrate, where it causes localized production of S1P [11]), or via interactions with other proteins or acidic phospholipids.
Degradation
S1P can be dephosphorylated by enzymes of the lipid phosphate phosphatase family (three isoforms have been described – LPP1–3) to regenerate its immediate precursor sphingosine. In vitro experiments show that these enzymes have a broad substrate specificity [12]. They have six transmembrane domains and efficiently form oligomers in cell membranes [13]. LPP activity on the extracellular side of the plasma membrane (ecto-LPP) would be expected to reduce S1P receptor signalling activity through depletion of the agonist; intracellular LPP could influence second messenger signalling by S1P. S1P can also be depleted through irreversible degradation by its specific lyase to phosphoethanolamine and hexadecanal. S1P lyase is predominantly localized within the ER, with the active site facing the cytosol and appears to have a purely intracellular function [14, 15]. In this context, we are concerned with the critical role played by the enzyme in control of the steady state levels of S1P in different compartments of the immune system. S1P lyase expression is absent in platelets [16] and erythrocytes [17]; expression is higher in tissue, particularly, for example in the thymus [18]. This generates a vascular S1P gradient [5, 18, 19]. Inhibition of S1P lyase by 2-acetyl-4-tetrahydroxybutylimidazole caused a dramatic effect on the distribution of lymphocytes in mice through disruption of this gradient [18].
Transport
S1P can be transported across lipid bilayers by ABC transporters: in the case of astrocytes ABCA1 has a major role [20], in mast cells ABCC1 [21] and rat erythrocytes appear to utilize a novel ATP-dependent transporter [22]. It has been reported that S1P export by ABCC1 and autologous signalling via S1PR3 is necessary for the cytoprotective effect of dexamethasone on human fibroblasts [23]. Others have shown that the CFTR can regulate S1P uptake in a murine epithelial cell line [24]. Localized export of S1P and autocrine signalling is necessary for cell chemotaxis towards certain ligands – so-called inside-out signalling. Chemotaxis to platelet-derived growth factor (PDGF) showed dependence on SK and S1PR1 expression. These data are consistent with a mechanism in which PDGF signalling causes a membrane-proximal local increase in S1P production. The S1P is then exported and binds its specific GPCR to induce directional migration [7, 25].
S1P receptor biology
Extracellular S1P signals mainly through a family of five receptors denoted S1PR1–5. These have a high degree of sequence similarity and all belong to the heterotrimeric GPCR superfamily [26]. S1PR1 is expressed widely on immune cells, whereas the other receptor types have more limited expression, for example S1PR4 expression by T lymphocytes [27] and S1PR5 expression by NK cells [28]. Receptor expression can be either constitutive or modulated, depending on the cell type. The affinity of S1P for these receptors (Kd) is mostly in the sub-nanomolar range [29, 30]. FTY720-P, the phosphorylated form of the drug FTY720 and a structural analogue of S1P, is a potent agonist of S1PR1, 3, 4 and 5 [31]. SEW2871 by contrast does not structurally resemble S1P and is a selective agonist of S1PR1 [32].
The receptors couple to a variety of downstream signalling pathways (Fig. 3): some are common to a number of the receptors, others not. The signalling biology of the receptors is complicated by the fact that multiple receptor subtypes are often expressed on the same cell [29, 33] and certain individual receptor subtypes can be split into subpopulations based upon the coupled heterotrimeric G proteins –αi, αq or α12/13. There is abundant evidence for different signalling activities of individual receptor subtypes depending upon the level of receptor occupancy. For example, lymphocyte recirculation is turned off at sub-stoichiometric occupancies of S1PR1 [34], whereas at much higher concentrations of ligand S1PR1 and other S1P receptor subtypes can be internalized (and also degraded) [35].
Fig 3.
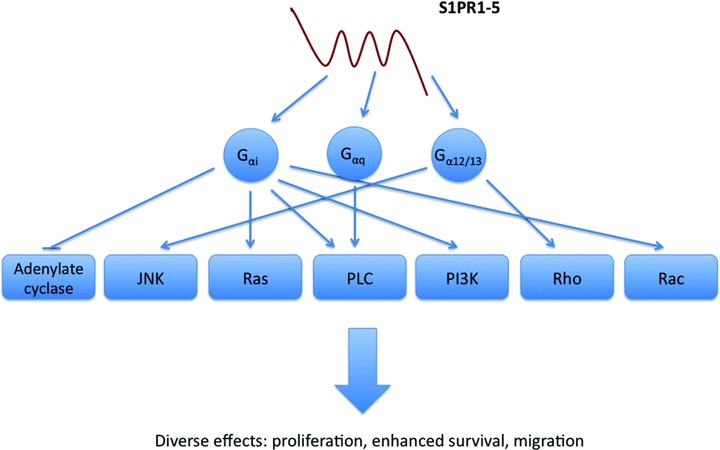
Signalling of S1P through its family of GPCRs. The receptors, denoted S1PR1–5, couple to various G proteins on binding of agonist and thereby trigger a multitude of signalling pathways. For example, whereas S1PR1 couples to Gαi and can induce cell chemotaxis in part via Rac activation, S1PR2 couples to Gα12/13 and can inhibit migration via activation of the Rho pathway.
S1PR1–5 are responsible for mediating many of the effects of S1P on cell biology including proliferation, cell migration, invasion, angiogenesis and vascular maturation, actin cytoskeleton rearrangement and adherens junction assembly [36, 37]. Interestingly, S1P is required for the transduction of signals by certain other GPCR (inside-out signalling) [3].
S1P can also act intracellularly and independently of S1PR1–5 to enhance cell proliferation and suppress apoptosis. This has been shown in the MCF-7 cell line, where depletion of SK2 attenuated the ability of phorbol myristate acetate to cause acetylation of histone H3 at the p21 and c-fos promoters. SK2 can physically associate with, and S1P can suppress the activity of, histone deacetylases 1 and 2 in the nucleus [38].
S1PR1 is the most highly expressed S1P receptor on lymphocytes and seems to be exclusively responsible for mediating the effects of S1P on cell trafficking. Similarly, S1PRs 1 and 3 are abundantly expressed by endothelial cells and seem largely responsible for transduction of the effects of their ligand on that cell type. These receptors are described in further detail below.
S1PR1
S1PR1 is expressed by a wide variety of tissues in mice and human beings. S1PR1-KO (knockout) mice die between E12.5 and E14.5 due to severe haemorrhage resulting from a defect in vascular stabilization [39]. Conditional gene deletion and experiments using selective receptor agonists show that the receptor has additional roles in angiogenesis, immune cell trafficking, regulation of vascular tone and endothelial barrier function [40–42].
S1PR1 couples to the G protein Gαi alone, as shown by the complete inhibition of S1PR1-mediated responses by pertussis toxin [43]. Through the activation of Gαi, receptor signalling results in an increase in mitogen-activated protein kinase activity, decrease in adenylate cyclase activity, increase in phospholipase C (PLC) activity, increase in Rac activation and increase in phosphatidyl inositol 3-kinase activity. Binding of S1P to S1PR1 transactivates receptor tyrosine kinases including the vascular endothelial growth factor (VEGF) and PDGF receptors. This occurs through intracellular receptor crosstalk, direct phosphorylation of receptor tyrosine kinases by protein tyrosine kinases, induced production or secretion of other receptor ligands and the formation of signalplexes [3, 33].
Plasma-membrane S1PR1 is internalized into endosomal compartments on receptor activation. The G protein receptor kinase GRK2 is activated on receptor signalling and phosphorylates the C-terminal tail of S1PR1 to initiate endocytosis [5]. Recently, the carboxyl terminal region of different GPCRs has been recognized as the interacting site of novel GPCR-regulatory proteins; in particular, complex PDZ domain containing proteins that help to localize the receptors to defined areas of the cell membrane, where signalling effectors are also concentrated. Proteins containing more than one PDZ domain often interact with the carboxyl terminus of different receptors thus helping to form functional aggregates. Using the HUVEC-derived immortalized cell line EA.hy926, our group has shown internalization of S1PR1 following treatment with the S1P analogue FTY720-P for 6 hrs (Fig. 4, unpublished data). Others have shown, in the HUVEC cell line, persistent signalling by S1PR1 after internalization by FTY720-P treatment [44]. This raises the intriguing possibility that the effects on endothelial cell phenotype, such as enhanced barrier function, mediated by FTY720-P might be due to persistent agonism, rather than functional antagonism, of S1PR1.
Fig 4.
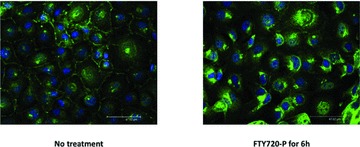
Internalization of surface S1PR1 on stimulation of endothelial cells (EA.hy 926) with FTY720-P. EA.hy 926 were grown to confluence on chamber slides and incubated overnight in serum-free medium. They were stimulated with vehicle or 1 μM FTY720-P for 6 hrs, then fixed and stained for examination by immunofluorescence confocal microscopy. S1PR1 protein in green; nucleus in blue.
Interestingly, it has been shown that S1P and phorbol myristate acetate (via protein kinase C) result in phosphorylation of residues on the tail of S1PR1 via distinct mechanisms and are associated with different pathways leading to loss of the receptor from the cell surface [45]. S1PR1 can undergo polyubiquitinylation upon internalization [46]. The degree of polyubiquitinylation is dependent on the type and concentration of agonist. The poorly ubiquitinylated receptor recycles from the late endosomal compartment back to the plasma membrane, the highly ubiquitinylated receptor is degraded in lysosomes [29]. Interestingly, activation of T cells through the T-cell receptor results in translocation of plasma membrane resident S1PR1 to the nuclear envelope, where the receptor affects changes in gene transcription (rather than, e.g. promoting cell migration) [47].
S1PR1 plays a central role in the S1P-mediated control of lymphocyte trafficking. It alone is sufficient to control lymphocyte recirculation, as shown by S1PR3 KO mice and selective receptor agonists [32]. Natural and functionally similar S1PR1 agonists are sufficient for the maintenance and enhancement of endothelial integrity [2, 48, 49].
S1PR3
Like S1PR1, S1PR3 is expressed widely in mice and human beings, particularly in the heart, lungs and kidneys. Deletion of the gene that codes for S1PR3 does not, however, result in any obvious abnormality [50]. The receptor couples to the Gαi, Gα12/13, Gαs and Gαq proteins, although the coupling to Gαq appears to predominate [33]. Through Gαq, receptor signalling causes activation of PLC and increased production of inositol trisphosphateand diacylglycerol. S1PR3 protein is expressed widely on vascular endothelial and smooth muscle cells and is probably important in the regulation of vascular tone. S1P signalling through S1PR3 contributes to stimulation of nitric oxide production by endothelial cells, which causes vasorelaxation; S1PR3 expressed on vascular smooth muscle cells mediates vasopressor effects [51]. Evidence suggests that S1P and its analogues can influence heart rate through S1PR3. Non-selective S1P receptor agonists in S1PR3 KO mice and S1PR1 selective agonists in wild-type mice do not induce bradycardia, implicating S1PR3 and not S1PR1 in bradycardia [32] in this species.
The vascular gradient of sphingosine 1-phosphate
Compartmentalization
The levels of S1P in the various compartments of the mammalian immune system are tightly controlled. S1P is present at single unit micromolar concentration in human serum, although reported values vary widely [17, 52], where it is mostly bound to lipoproteins, particularly HDL, and albumin [53] (Fig. 1A). Erythrocytes express SKs and lack both S1P lyase and LPPs and are the primary source of S1P in blood [54], and appear to be able to buffer its concentration there [17]. There also exists evidence for the transcellular transport of S1P from erythrocytes to other cell types including endothelial cells, and thus into tissues [55]. It has also been reported that vascular endothelium is a significant contributor to S1P in blood, and indeed endothelial cells respond to shear flow by modulating the level of S1P metabolic enzymes [56]. They are able to internalize and store S1P. HDL and serum albumin are the major triggers for erythrocyte S1P release into the blood [55].
S1P is present in lymph at around one quarter the level in blood. Experiments with tissue-specific conditional SK1 and constitutive SK2 KO mice have shown that lymphatic endothelial cells are the source of S1P in lymph, and that expression of the enzyme there is required for normal lymphocyte egress from lymph nodes and Peyer’s patches [57]. S1P is at a very low concentration in primary and secondary lymphoid tissue: the levels of S1P in thymus, lymph nodes and spleen are 20, 40 and 150 ng per g of tissue, respectively. A high level of S1P lyase activity in these tissues underlies this low concentration, as shown by the dramatic increase in tissue abundance of S1P on enzyme inhibition [18].
T-cell trafficking
S1P plays a pivotal role in the immune system of mammals through its role in the control of lymphocyte egress from the thymus and secondary lymphoid tissue. This regulation is achieved through changes in the surface expression of S1PR1 on lymphocytes resulting from homologous desensitization of the receptor and by transduction of other cell signals [58].
T cells present in blood are exposed to a high concentration of S1P. This results in marked internalization of S1PR1 to endosomes and loss of sensitivity to extracellular S1P (Fig. 1A). This contrasts with the very low concentration found within lymphoid tissue in non-inflammatory conditions. So, S1PR1 that is sequestered in endosomes recycles onto the T-cell surface within a few hours of T-cell entry into such tissue [59]. Resting T cells patrol the paracortex of the lymph node and move towards exit structures under the influence of chemokines, most notably CCL19 which is produced by stromal cells there and binds surface CCR7. It has been proposed that T cells receive opposing retention and egress signals (via CCR7 and S1PR1, respectively) when adjacent to the cortical sinusoids. The signalling through S1PR1, which is strongest in the immediate vicinity of exit structures, overcomes the CCR7-mediated retention signal and cause T-cell egress [60, 61].
The situation is different under inflammatory conditions where the egress process is shut down locally and transiently in the course of an immune response. Activation of a T cell through the T-cell receptor (with co-stimulation), or simply through innate immune cytokines, causes up-regulation of CD69, which physically associates with S1PR1 and renders cells insensitive to signalling through this receptor [62]. The cell consequently does not receive exit signals and remains in the lymphoid tissue where it can proliferate and differentiate into other cell subtypes. The expression of CD69 is reduced following T-cell differentiation to an effector phenotype, allowing cells to exit [40].
There is also evidence for a role of lymphocyte intrinsic S1P signalling in control of T-cell movement from peripheral tissue into afferent lymphatics during inflammation (when the extracellular concentration of S1P concentration can increase). It has been shown that agonism of S1PR1 on T cells prevented movement of T cells across lymphatic endothelium. This was found to be dependent on the interaction of LFA-1 and VLA-4 on the T cells with their ligands ICAM-1 and VCAM-1 on the endothelium [63].
Role of S1P on vascular endothelium
Maintenance of endothelial barrier integrity in the resting state
Maintenance of vascular barrier integrity under normal conditions is necessary for proper organ function. Paracellular permeability, the passive movement of solutes with molecular radii less than 3 nm, is the main determinant of endothelial barrier permeability [64]. Barrier permeability is maintained by a complex balance of tethering forces at cell–cell and cell–matrix level as well as intracellular contractile forces mediated by actin and myosin.
Endothelial cells are connected to each other by three junctional complexes: adherens junctions, tight junctions and gap junctions. S1P enhances the integrity of vascular barrier through agonism of the receptor S1PR1 (Fig. 1A). Downstream signalling events result in enhanced generation of focal adhesion complexes and adherens junctions – mediated by activation of focal adhesion kinase and paxillin, and through VE-cadherin and β-catenin, respectively [65].
Rac activation, downstream of S1PR1, is important for S1P-induced adherens junction assembly and cytoskeletal rearrangement. Overexpression of dominant negative Rac in endothelial cells significantly reduced S1P-induced VE-cadherin and β-catenin accumulation at cell–cell junctions, whereas overexpression of active Rac produced changes similar to those produced following signalling by S1P [66]. S1P also causes an increase in intracellular Ca2+ concentration in endothelial cells via a Gαi-dependent pathway [67]. Inhibition of either Gαi, PLC or the inositol trisphosphate receptor in endothelial cells prevented the S1P-induced increase in intracellular Ca2+, Rac activation, adherens junction assembly and barrier enhancement. Interestingly, it has recently been shown that S1P signalling occurs within lipid microdomains and is associated with activation of a protein tyrosine kinase. The tyrosine phosphorylation of key cytoskeletal effectors including cortactin, non-muscle myosin light chain kinase and filamin potentially contribute to the regulation of endothelial barrier function by transiently stimulating endothelial cell adherence and focal junction assembly [68].
S1P also stimulates the formation of tight junctions, which are present on the outer leaflet of lateral membranes between endothelial cells. These junctions, composed of claudins, occludins and junctional adhesion molecules, are connected to the actin cytoskeleton via the zona occludens proteins (ZO-1 to 3). S1P stimulation, via S1PR1 and Rac activation, causes redistribution of ZO-1 to cell–cell junctions. The enhancement in barrier function mediated by S1P was inhibited by down-regulation of ZO-1 [69].
Disruption of barrier integrity during inflammation
During inflammation, thrombin antagonizes the effects of S1PR1 signalling to increase paracellular permeability. This occurs through activation of the Rho signalling pathway, causing cell contraction and disruption of the endothelial barrier [70] (Fig. 1C). Specifically, Rho signalling results in activation of the phosphatase PTEN, which inhibits the activity of Akt and Rac. S1PR2, which couples to Gα12/13, can also antagonize the barrier enhancement caused by S1PR1 signalling via the same mechanism. S1PR2 overexpression results in expression of a strong peripheral ring of stress fibres and complete inhibition of cortical actin formation; translocation of VE-cadherin to adherens junctions is also inhibited [71]. It might be that regulation of expression of S1PR1 and S1PR2 plays an important role in the change in the effect of S1P on vascular permeability.
Crosstalk between S1PR and other receptors
Crosstalk with growth factor receptors
Growth factors such as VEGF and transforming growth factor β (TGF-β) act alongside S1P in the progression of the inflammatory response. VEGF, acting through VEGFR1, stimulates recruitment of haematopoietic precursors, monocytes and macrophages to the site of inflammation. VEGFR2, expressed by vascular endothelial cells, is implicated in angiogenesis in the adult [72]. TGF-β signalling has been shown to be necessary in stabilization of the peripheral microvasculature, and TGF-β is a central player in the chronic inflammatory process [73, 74]. There is therefore a significant overlap between the biological effects of S1P, cytokines and growth factors. An early study suggested that the consequences of S1P stimulation of endothelial cells were mediated by activation of the VEGF receptor [75]. This S1P-mediated activation of VEGF was dependent on Gαi protein and Src family kinases. In addition, VEGF can stimulate an increase of S1P receptors, thereby enhancing the potential of S1P to activate VEGF receptors [76].
Recently, it has been demonstrated that S1P can transactivate the TGF-β pathway. TGF-β initiates the signal transduction pathway by stimulating the formation of a multimeric receptor complex consisting of two pairs of heterodimers. Each heterodimer consists of two serine-threonine kinases, a type I TGF-β receptor (TβRI) and type II (TβRII) receptor. The canonical Smad pathway is activated by the binding of TGF-β to TβRII, which then phosphorylates TβRI. Sauer et al. observed that S1P and TGF-β induced phosphorylation of Smad2 and Smad3, they also showed that S1P mediated activation of Smad3 was GPCR dependent [77]. The possibility that Smad3 activation was required for S1P induction by keratinocytes was further verified by the observation that neither S1P nor TGF-β could induce chemotaxis in keratinocytes derived from Smad3–/– mice [77].
Cross talk between S1P and the TGF-β signalling pathway has also been observed in renal mesengial cells [78] in which TβRII was required for S1P-mediated activation of Smads and S1P induced TGF-β oligomerization. Though the exact mechanism for S1P activation of Smad signalling is not known, this cross-talk is of significance as it impacts on the understanding of various disease processes.
Cross-talk with other GPCR
Leucocytes typically express a range of G-protein coupled chemokine receptors as well as a wide range of other chemotactic and non-chemotactic GPCR. As these receptors may be engaged simultaneously or sequentially by multiple mediators at inflammatory sites, the cellular response is likely to be cross-regulated. Indeed, GPCR are known to be regulated by multiple processes which include down-regulation of both receptor function and cell-surface expression. Receptor desensitization is a process by which receptors become refractory to continued stimulation within minutes of initial agonist exposure.
At the point of exit from lymphoid tissue, resting T cells receive signals through both CCR7 and S1PR1. It is suggested that these signals are integrated inside the cells and that the result determines whether they exit or remain inside the tissue. This hypothesis is supported by a number of experiments examining lymphocyte egress in vivo. After FTY720 treatment to down-modulate surface S1PR1, CCR7-deficient T cells egress more efficiently than wild-type T cells. Treatment with pertussin toxin to inactivate Gαi-dependent signalling of CCR7 restores egress of S1PR1-deficient T cells [79]. It seems that T cells at the boundaries of the cortical sinusoids are desensitized to the CCR7-mediated retention signal by S1P, thus permitting the cells to leave.
Recent evidence has shown that S1P can modulate T-cell chemotaxis in response to certain chemokines. Our group has shown that 1 hr pre-treatment with the S1PR1-specific agonist SEW2871 potently reduced the chemotactic response of resting T cells to CXCL12 (Fig. 5).
Fig 5.
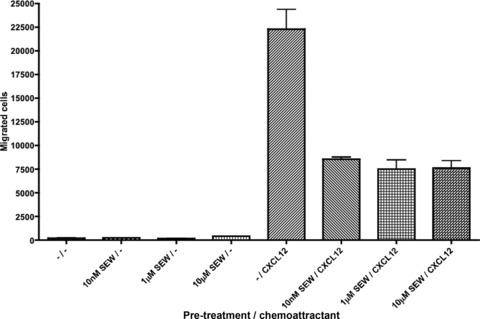
Inhibition of CXCR4-mediated chemotaxis by S1PR1 signalling. Primary resting human T cells were incubated overnight in serum-free medium. The cells were pre-treated for 1 hr with vehicle, 10 nM, 1 μM or 10 μM of the selective S1PR1 agonist SEW2871, then 100,000 cells left to migrate towards 6.25 nM CXCL12 for 2 hrs.
Others have shown that pre-treatment of CD4+ T cells with 10–100 nM S1P resulted in increased chemotaxis of CD4+ T cells towards CCL5 and CCL21, whereas pre-treatment with 300 nM to 3 μM S1P strongly suppressed migration towards those chemokines [80]. Strategies to modulate chemotaxis for possible therapeutic benefit are being devised [81].
CXCL12 is an important homeostatic chemokine that regulates the trafficking of lymphocytes and haematopoietic stem cells. S1PR1 overexpression in Jurkat T cells caused a reduction in surface CXCR4 and potent inhibition of ligand-induced chemotaxis and signalling responses [82]. Others have shown that S1P stimulation can support CXCR4-dependent migration, suggesting that the signalling pathways may synergize [83]. One group has identified phosphatidyl inositol 3-kinase as a possible mechanistic link between S1P and enhanced migration towards CXCL12.
There is also evidence for S1P and chemokine receptor crosstalk in neutrophils. S1P can reduce the chemotactic response of neutrophils towards CXCL8 or FMLP; it also antagonizes neutrophil apoptosis and induces pertussis toxin sensitive calcium signals.
Therapies targeting the S1P signalling axis
FTY720
Early in vitro experiments using the sphingosine analogue FTY720 did not show any effect until concentrations were increased into the millimolar range (whereupon it induces programmed cell death [84]), far in excess of the active in vivo concentrations. This has now been explained by the fact that FTY720 is rapidly phosphorylated in vivo by SK2, mostly inside platelets. FTY720-P is structurally similar to S1P and binds all S1P receptors except S1PR2. It binds with similar affinity to S1P at S1PR3, S1PR4 and S1PR5 [30]. Although it is an agonist of S1PR1, it behaves as a functional antagonist. FTY720-P is much more potent than S1P at causing ubiquitination and degradation of S1PR1, even if the concentration of S1P is raised to concentrations sufficient to cause internalization of almost all of the receptor [35, 46, 85]. It is likely that this effect is critical to its ability to render lymphocytes insensitive to egress signals in lymphoid tissue [85]. After treatment of mice with FTY720, lymphocytes can be observed packed next to sinus-lining endothelium but are unable to exit [30]. The efferent lymphatics are emptied of lymphocytes, with this reduction preceding a drop in blood lymphocyte numbers. It is believed that the block in egress is primarily lymphocyte intrinsic, but an additional role for FTY720-mediated enhancement of sinus-endothelial barrier function at lymphoid tissue exit structures has not been entirely ruled out [86]. Interestingly, it has also been shown that FTY720 can suppress a VEGF-induced increase in vascular permeability in mice [87]. This points to potential utility of S1P analogues in the treatment of disorders associated with aberrant angiogenesis and increased vascular permeability.
FTY720 has potent immunosuppressive activity and was found to prolong the survival of skin allografts in mice. Dosing in the 0.05 mg/kg range in human beings results in depletion of almost all CD4+ and CD8+ lymphocytes in blood, whereas granulocytes, monocytes and NK cells in blood are relatively unaffected [88, 89]. Some protective immunity is retained, certainly to a much greater extent than following treatment with the calcineurin inhibitors tacrolimus and cyclosporine [90]. FTY720 has been tested in transplantation, but was no better than mycophenolate mofetil in combination with cyclosporine in multiple phase II [91, 92] and phase III [93, 94] renal transplantation trials (its efficacy was comparable, but adverse events exceeded those caused by the comparator). Side effects included transient bradycardia (dose-dependent and dose-limiting), dyspnea and macular oedema. Initial studies indicated a role for S1PR3 signalling in the drug-induced effect on heart rate [32], but further studies support a role for S1PR1-dependent activation of the G protein gated potassium channel IKAch in cardiac myocytes [95, 96].
FTY720 shows promise for the treatment of multiple sclerosis, an autoimmune disorder in which autoreactive T cells cross the blood–brain barrier and attack myelin sheaths leading to axonal damage [97]. FTY720 can itself efficiently cross the blood–brain barrier and both FTY720 and FTY720-P accumulate at rather higher concentrations in the central nervous system (CNS) than in the blood [98, 99]. FTY720 has already been through successful phase II trials [100, 101] for the treatment of multiple sclerosis. The latest report from a 2-year double-blind placebo-controlled randomized trial of 1033/1272 (completed trial/enrolled) shows a relapse rate of 0.18 with 0.5 mg/day FTY720 and 0.16 with 1.25 mg/day FTY720 compared with 0.4 in the placebo group [102]. The effectiveness of FTY720 in this condition may be due to broader mechanisms of action than inhibition of migration of autoantigen-specific (particularly Th17 cells [103, 104]) lymphocytes to the CNS [105]. S1P receptors (S1PRs 1, 2, 3 and 5) are expressed at significant levels in the CNS and are believed to play a number of roles in brain cell function, such as controlling astrocyte proliferation and migration, oligodendrocyte differentiation and survival, and neurite outgrowth and neurogenesis [106, 107].
Monoclonal antibody therapy
A monoclonal antibody of very high specificity and affinity for S1P has been developed. It has proven to be an effective inhibitor of tumour-associated angiogenesis in several murine models [108]. This is likely due to efficient depletion of tumour- associated S1P – which would otherwise drive endothelial cell migration, proliferation and ultimately tumour-supportive neovascularization. This antibody is now undergoing clinical trials for the treatment of cancer and age-related macular degeneration.
Summary and future directions
The endothelium is of crucial importance during health and disease by regulating the passage of solutes and immune cells between the blood or lymph and body tissues. It is now clear that S1P occupies a central position in this biology by maintenance of normal endothelial barrier functions and control of potentially destructive immune responses during inflammation. This review has integrated current data which indicate how both of these processes are regulated by variable concentrations of the S1P ligand in different compartments, and by differential expression, modulation by other cell-surface proteins, internalization and possible degradation of the various S1P receptors. It is now imperative to explore further crosstalk between the S1P and other immune signalling pathways. Here we have touched upon some initial evidence for an interaction between the S1P and chemokine signalling axes, raising the prospect that it may be possible to design strategies to modulate globally trafficking of pathogenic immune cells.
This knowledge has already led to clinical trials of S1P analogues in the fields of transplantation and CNS autoimmunity. A number of additional compounds, many of which are selective agonists of S1P receptors (primarily S1PR1), are already in the drug development stream. The results of clinical trials of these molecules, which are being applied to a wide spectrum of inflammatory disorders, including rheumatoid arthritis, multiple sclerosis, age-related macular oedema and various cancers, will likely be reported over the next few years. There is also an ongoing effort to find highly specific inhibitors of sphingosine kinase, with appropriate pharmacokinetic profiles, for clinical therapy. SK1 may play a critical role in PAR1 signalling [109] and targeting this kinase might prove effective for treatment of atherosclerosis. Finally, it has been speculated that targeting the S1P signalling system through inhibition of the degradative enzyme S1P lyase might prove an effective strategy for impairment of damaging adaptive immune responses, whilst avoiding the side – effects associated with S1P receptor agonism.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Takuwa Y, Okamoto Y, Yoshioka K, et al. Sphingosine-1-phosphate signaling and biological activities in the cardiovascular system. Biochim Biophys Acta. 2008;1781:483–8. doi: 10.1016/j.bbalip.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Limaye V. The role of sphingosine kinase and sphingosine-1-phosphate in the regulation of endothelial cell biology. Endothelium. 2008;15:101–12. doi: 10.1080/10623320802125342. [DOI] [PubMed] [Google Scholar]
- 3.Takabe K, Paugh SW, Milstien S, et al. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–95. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynch KR, Macdonald TL. Sphingosine 1-phosphate chemical biology. Biochim Biophys Acta. 2008;1781:508–12. doi: 10.1016/j.bbalip.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kihara A, Mitsutake S, Mizutani Y, et al. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res. 2007;46:126–44. doi: 10.1016/j.plipres.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18:300–7. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Hait NC, Oskeritzian CA, Paugh SW, et al. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006;1758:2016–26. doi: 10.1016/j.bbamem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006;39:113–31. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- 10.Xia P, Wang L, Moretti PA, et al. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem. 2002;277:7996–8003. doi: 10.1074/jbc.M111423200. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar S, Maceyka M, Hait NC, et al. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005;579:5313–7. doi: 10.1016/j.febslet.2005.08.055. [DOI] [PubMed] [Google Scholar]
- 12.Brindley DN, English D, Pilquil C, et al. Lipid phosphate phosphatases regulate signal transduction through glycerolipids and sphingolipids. Biochim Biophys Acta. 2002;1582:33–44. doi: 10.1016/s1388-1981(02)00135-x. [DOI] [PubMed] [Google Scholar]
- 13.Sigal YJ, McDermott MI, Morris AJ. Integral membrane lipid phosphatases/phosphotransferases: common structure and diverse functions. Biochem J. 2005;387:281–93. doi: 10.1042/BJ20041771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda M, Kihara A, Igarashi Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5’-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun. 2004;325:338–43. doi: 10.1016/j.bbrc.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 15.Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends Mol Med. 2007;13:210–7. doi: 10.1016/j.molmed.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Yatomi Y, Yamamura S, Ruan F, et al. Sphingosine 1-phosphate induces platelet activation through an extracellular action and shares a platelet surface receptor with lysophosphatidic acid. J Biol Chem. 1997;272:5291–7. doi: 10.1074/jbc.272.8.5291. [DOI] [PubMed] [Google Scholar]
- 17.Hanel P, Andreani P, Graler MH. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 2007;21:1202–9. doi: 10.1096/fj.06-7433com. [DOI] [PubMed] [Google Scholar]
- 18.Schwab SR, Pereira JP, Matloubian M, et al. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–9. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 19.Sciorra VA, Morris AJ. Roles for lipid phosphate phosphatases in regulation of cellular signaling. Biochim Biophys Acta. 2002;1582:45–51. doi: 10.1016/s1388-1981(02)00136-1. [DOI] [PubMed] [Google Scholar]
- 20.Sato K, Malchinkhuu E, Horiuchi Y, et al. Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J Neurochem. 2007;103:2610–9. doi: 10.1111/j.1471-4159.2007.04958.x. [DOI] [PubMed] [Google Scholar]
- 21.Mitra P, Oskeritzian CA, Payne SG, et al. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci USA. 2006;103:16394–9. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi N, Yamaguchi A, Nishi T. Characterization of the ATP-dependent sphingosine 1-phosphate transporter in rat erythrocytes. J Biol Chem. 2009;284:21192–200. doi: 10.1074/jbc.M109.006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nieuwenhuis B, Luth A, Chun J, et al. Involvement of the ABC-transporter ABCC1 and the sphingosine 1-phosphate receptor subtype S1P(3) in the cytoprotection of human fibroblasts by the glucocorticoid dexamethasone. J Mol Med. 2009;87:645–57. doi: 10.1007/s00109-009-0468-x. [DOI] [PubMed] [Google Scholar]
- 24.Boujaoude LC, Bradshaw-Wilder C, Mao C, et al. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J Biol Chem. 2001;276:35258–64. doi: 10.1074/jbc.M105442200. [DOI] [PubMed] [Google Scholar]
- 25.Hobson JP, Rosenfeldt HM, Barak LS, et al. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science. 2001;291:1800–3. doi: 10.1126/science.1057559. [DOI] [PubMed] [Google Scholar]
- 26.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–70. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Graeler MH, Goetzl EJ. Type 4 sphingosine 1-phosphate G protein-coupled receptor (S1P4) transduces S1P effects on T cell proliferation and cytokine secretion without signaling migration. FASEB J. 2005;19:1731–3. doi: 10.1096/fj.05-3730fje. [DOI] [PubMed] [Google Scholar]
- 28.Jenne CN, Enders A, Rivera R, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469–81. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosen H, Gonzalez-Cabrera P, Marsolais D, et al. Modulating tone: the overture of S1P receptor immunotherapeutics. Immunol Rev. 2008;223:221–35. doi: 10.1111/j.1600-065X.2008.00645.x. [DOI] [PubMed] [Google Scholar]
- 30.Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 31.Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 32.Sanna MG, Liao J, Jo E, et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–48. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 33.Ishii I, Fukushima N, Ye X, et al. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–54. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 34.Rosen H, Liao J. Sphingosine 1-phosphate pathway therapeutics: a lipid ligand-receptor paradigm. Curr Opin Chem Biol. 2003;7:461–8. doi: 10.1016/s1367-5931(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Cabrera PJ, Hla T, et al. Mapping pathways downstream of sphingosine 1-phosphate subtype 1 by differential chemical perturbation and proteomics. J Biol Chem. 2007;282:7254–64. doi: 10.1074/jbc.M610581200. [DOI] [PubMed] [Google Scholar]
- 36.Spiegel S, Milstien S. Functions of a new family of sphingosine-1-phosphate receptors. Biochim Biophys Acta. 2000;1484:107–16. doi: 10.1016/s1388-1981(00)00010-x. [DOI] [PubMed] [Google Scholar]
- 37.Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hait NC, Allegood J, Maceyka M, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–7. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Wada R, Yamashita T, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–61. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 41.Singleton PA, Dudek SM, Chiang ET, et al. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005;19:1646–56. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 42.Sanna MG, Wang SK, Gonzalez-Cabrera PJ, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2:434–41. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 43.Lee MJ, Evans M, Hla T. The inducible G protein-coupled receptor edg-1 signals via the G(i)/mitogen-activated protein kinase pathway. J Biol Chem. 1996;271:11272–9. doi: 10.1074/jbc.271.19.11272. [DOI] [PubMed] [Google Scholar]
- 44.Mullershausen F, Zecri F, Cetin C, et al. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–34. doi: 10.1038/nchembio.173. [DOI] [PubMed] [Google Scholar]
- 45.Watterson KR, Johnston E, Chalmers C, et al. Dual regulation of EDG1/S1P(1) receptor phosphorylation and internalization by protein kinase C and G-protein-coupled receptor kinase 2. J Biol Chem. 2002;277:5767–77. doi: 10.1074/jbc.M110647200. [DOI] [PubMed] [Google Scholar]
- 46.Oo ML, Thangada S, Wu MT, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- 47.Liao JJ, Huang MC, Graler M, et al. Distinctive T cell-suppressive signals from nuclearized type 1 sphingosine 1-phosphate G protein-coupled receptors. J Biol Chem. 2007;282:1964–72. doi: 10.1074/jbc.M608597200. [DOI] [PubMed] [Google Scholar]
- 48.Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4:1019–25. doi: 10.1111/j.1600-6143.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 49.Rosen H, Sanna MG, Cahalan SM, et al. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–7. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Ishii I, Friedman B, Ye X, et al. Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J Biol Chem. 2001;276:33697–704. doi: 10.1074/jbc.M104441200. [DOI] [PubMed] [Google Scholar]
- 51.Takuwa Y, Okamoto Y, Yoshioka K, et al. Sphingosine-1-phosphate signaling and biological activities in the cardiovascular system. Biochim Biophys Acta. 2008;1781:483–8. doi: 10.1016/j.bbalip.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Hla T, Venkataraman K, Michaud J. The vascular S1P gradient-Cellular sources and biological significance. Biochim Biophys Acta. 2008;1781:477–82. doi: 10.1016/j.bbalip.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murata N, Sato K, Kon J, et al. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352:809–15. [PMC free article] [PubMed] [Google Scholar]
- 54.Pappu R, Schwab SR, Cornelissen I, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–8. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 55.Bode C, Sensken SC, Peest U, et al. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem. 2010;109:1232–43. doi: 10.1002/jcb.22507. [DOI] [PubMed] [Google Scholar]
- 56.Venkataraman K, Lee YM, Michaud J, et al. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102:669–76. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pham TH, Baluk P, Xu Y, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. 2010;207:17–27. doi: 10.1084/jem.20091619. , S1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 59.Lo CG, Xu Y, Proia RL, et al. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J Exp Med. 2005;201:291–301. doi: 10.1084/jem.20041509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dustin ML, Chakraborty AK. Tug of war at the exit door. Immunity. 2008;28:15–7. doi: 10.1016/j.immuni.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grigorova IL, Schwab SR, Phan TG, et al. Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat Immunol. 2009;10:58–65. doi: 10.1038/ni.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shiow LR, Rosen DB, Brdickova N, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–4. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 63.Ledgerwood LG, Lal G, Zhang N, et al. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat Immunol. 2008;9:42–53. doi: 10.1038/ni1534. [DOI] [PubMed] [Google Scholar]
- 64.Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2007;293:L823–42. doi: 10.1152/ajplung.00436.2006. [DOI] [PubMed] [Google Scholar]
- 65.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 66.Garcia JG, Liu F, Verin AD, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mehta D, Konstantoulaki M, Ahmmed GU, et al. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–8. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- 68.Sun X, Shikata Y, Wang L, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res. 2009;77:304–13. doi: 10.1016/j.mvr.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee JF, Zeng Q, Ozaki H, et al. Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J Biol Chem. 2006;281:29190–200. doi: 10.1074/jbc.M604310200. [DOI] [PubMed] [Google Scholar]
- 70.Van Nieuw Amerongen GP, Natarajan K, Yin G, et al. GIT1 mediates thrombin signaling in endothelial cells: role in turnover of RhoA-type focal adhesions. Circ Res. 2004;94:1041–9. doi: 10.1161/01.RES.0000125627.77235.0C. [DOI] [PubMed] [Google Scholar]
- 71.Sanchez T, Skoura A, Wu MT, et al. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27:1312–8. doi: 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- 72.Olsson AK, Dimberg A, Kreuger J, et al. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 73.Walshe TE, Saint-Geniez M, Maharaj AS, et al. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS One. 2009;4:e5149. doi: 10.1371/journal.pone.0005149. doi: 10.1371/journal.pone.0005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–61. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 75.Tanimoto T, Jin ZG, Berk BC. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) J Biol Chem. 2002;277:42997–3001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- 76.Igarashi J, Erwin PA, Dantas AP, et al. VEGF induces S1P1 receptors in endothelial cells: implications for cross-talk between sphingolipid and growth factor receptors. Proc Natl Acad Sci USA. 2003;100:10664–9. doi: 10.1073/pnas.1934494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sauer B, Vogler R, Von Wenckstern H, et al. Involvement of Smad signaling in sphingosine 1-phosphate-mediated biological responses of keratinocytes. J Biol Chem. 2004;279:38471–9. doi: 10.1074/jbc.M313557200. [DOI] [PubMed] [Google Scholar]
- 78.Xin C, Ren S, Kleuser B, et al. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem. 2004;279:35255–62. doi: 10.1074/jbc.M312091200. [DOI] [PubMed] [Google Scholar]
- 79.Pham TH, Okada T, Matloubian M, et al. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008;28:122–33. doi: 10.1016/j.immuni.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Graeler M, Shankar G, Goetzl EJ. Cutting edge: suppression of T cell chemotaxis by sphingosine 1-phosphate. J Immunol. 2002;169:4084–7. doi: 10.4049/jimmunol.169.8.4084. [DOI] [PubMed] [Google Scholar]
- 81.Ali S, Robertson H, Wain JH, et al. A non-glycosaminoglycan-binding variant of CC chemokine ligand 7 (monocyte chemoattractant protein-3) antagonizes chemokine-mediated inflammation. J Immunol. 2005;175:1257–66. doi: 10.4049/jimmunol.175.2.1257. [DOI] [PubMed] [Google Scholar]
- 82.Ryser MF, Ugarte F, Lehmann R, et al. S1P(1) overexpression stimulates S1P-dependent chemotaxis of human CD34+ hematopoietic progenitor cells but strongly inhibits SDF-1/CXCR4-dependent migration and in vivo homing. Mol Immunol. 2008;46:166–71. doi: 10.1016/j.molimm.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 83.Whetton AD, Lu Y, Pierce A, et al. Lysophospholipids synergistically promote primitive hematopoietic cell chemotaxis via a mechanism involving Vav 1. Blood. 2003;102:2798–802. doi: 10.1182/blood-2002-12-3635. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki S, Li XK, Enosawa S, Shinomiya T. A new immunosuppressant, FTY720, induces bcl-2-associated apoptotic cell death in human lymphocytes. Immunology. 1996;89:518–23. doi: 10.1046/j.1365-2567.1996.d01-777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Graler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004;18:551–3. doi: 10.1096/fj.03-0910fje. [DOI] [PubMed] [Google Scholar]
- 86.Wei SH, Rosen H, Matheu MP, et al. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6:1228–35. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- 87.Sanchez T, Estrada-Hernandez T, Paik JH, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278:47281–90. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- 88.Budde K, R LS, Nashan B, et al. Pharmacodynamics of single doses of the novel immunosuppressant FTY720 in stable renal transplant patients. Am J Transplant. 2003;3:846–54. doi: 10.1034/j.1600-6143.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- 89.Kahan BD, Karlix JL, Ferguson RM, et al. Pharmacodynamics, pharmacokinetics, and safety of multiple doses of FTY720 in stable renal transplant patients: a multicenter, randomized, placebo-controlled, phase I study. Transplantation. 2003;76:1079–84. doi: 10.1097/01.TP.0000084822.01372.AC. [DOI] [PubMed] [Google Scholar]
- 90.Brinkmann V, Lynch KR. FTY720: targeting G-protein-coupled receptors for sphingosine 1-phosphate in transplantation and autoimmunity. Curr Opin Immunol. 2002;14:569–75. doi: 10.1016/s0952-7915(02)00374-6. [DOI] [PubMed] [Google Scholar]
- 91.Mulgaonkar S, Tedesco H, Oppenheimer F, et al. FTY720/cyclosporine regimens in de novo renal transplantation: a 1-year dose-finding study. Am J Transplant. 2006;6:1848–57. doi: 10.1111/j.1600-6143.2006.01404.x. [DOI] [PubMed] [Google Scholar]
- 92.Tedesco-Silva H, Mourad G, Kahan BD, et al. FTY720, a novel immunomodulator: efficacy and safety results from the first phase 2A study in de novo renal transplantation. Transplantation. 2005;79:1553–60. [PubMed] [Google Scholar]
- 93.Salvadori M, Budde K, Charpentier B, et al. FTY720 versus MMF with cyclosporine in de novo renal transplantation: a 1-year, randomized controlled trial in Europe and Australasia. Am J Transplant. 2006;6:2912–21. doi: 10.1111/j.1600-6143.2006.01552.x. [DOI] [PubMed] [Google Scholar]
- 94.Tedesco-Silva H, Pescovitz MD, Cibrik D, et al. Randomized controlled trial of FTY720 versus MMF in de novo renal transplantation. Transplantation. 2006;82:1689–97. doi: 10.1097/01.tp.0000251718.95622.b3. [DOI] [PubMed] [Google Scholar]
- 95.Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 96.Koyrakh L, Roman MI, Brinkmann V, et al. The heart rate decrease caused by acute FTY720 administration is mediated by the G protein-gated potassium channel I. Am J Transplant. 2005;5:529–36. doi: 10.1111/j.1600-6143.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- 97.DeAngelis T, Lublin F. Multiple sclerosis: new treatment trials and emerging therapeutic targets. Curr Opin Neurol. 2008;21:261–71. doi: 10.1097/WCO.0b013e328300c70d. [DOI] [PubMed] [Google Scholar]
- 98.Miron VE, Schubart A, Antel JP. Central nervous system-directed effects of FTY720 (fingolimod) J Neurol Sci. 2008;274:13–7. doi: 10.1016/j.jns.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 99.Foster CA, Howard LM, Schweitzer A, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–75. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- 100.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–40. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 101.O’Connor P, Comi G, Montalban X, et al. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology. 2009;72:73–9. doi: 10.1212/01.wnl.0000338569.32367.3d. [DOI] [PubMed] [Google Scholar]
- 102.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 103.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Massberg S, Von Andrian UH. Fingolimod and sphingosine-1-phosphate–modifiers of lymphocyte migration. N Engl J Med. 2006;355:1088–91. doi: 10.1056/NEJMp068159. [DOI] [PubMed] [Google Scholar]
- 106.Dev KK, Mullershausen F, Mattes H, et al. Brain sphingosine-1-phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol Ther. 2008;117:77–93. doi: 10.1016/j.pharmthera.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 107.Brinkmann V. FTY720 (fingolimod) in Multiple Sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158:1173–82. doi: 10.1111/j.1476-5381.2009.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Visentin B, Vekich JA, Sibbald BJ, et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. 2006;9:225–38. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 109.Billich A, Urtz N, Reuschel R, et al. Sphingosine kinase 1 is essential for proteinase-activated receptor-1 signalling in epithelial and endothelial cells. Int J Biochem Cell Biol. 2009;41:1547–55. doi: 10.1016/j.biocel.2009.01.001. [DOI] [PubMed] [Google Scholar]