

Abstract
DNA lesions trigger the DNA damage response (DDR) machinery, which protects genomic integrity and sustains cellular survival. Increasing data underline the significance of the integrity of the DDR pathway in chemotherapy response. According to a recent work, persistent exposure of A549 lung carcinoma cells to doxorubicin induces an initial DDR-dependent checkpoint response, followed by a later DDR-independent, but p27Kip1-dependent one. Prompted by the above report and to better understand the involvement of the DDR signaling after chemotherapeutic stress, we examined the potential role of the canonical DDR pathway in A549 cells treated with doxorubicin. Exposure of A549 cells, prior to doxorubicin treatment, to ATM, ATR and DNA-PKcs inhibitors either alone or in various combinations, revealed that the earlier documented two-step response was DDR-dependent in both steps. Notably, inhibition of both ATM and ATR or selective inhibition of ATM or DNA-PKcs resulted in cell-cycle re-entry despite the increased levels of p27Kip1 at all time points analyzed. We further investigated the regulation of p27Kip1 protein levels in the particular setting. Our results showed that the protein status of p27Kip1 is mainly determined by p38-MAPK, whereas the role of SKP2 is less significant in the doxoroubicin-treated A549 cells. Cumulatively, we provide evidence that the DNA damage signaling is responsible for the prolonged cell cycle arrest observed after persistent chemotherapy-induced genotoxic stress. In conclusion, precise identification of the molecular mechanisms that are activated during the chemotherapeutic cycles could potentially increase the sensitization to the therapy applied.
Keywords: DNA damage response, cell cycle arrest, chemotherapy, ATM, ATR, DNA-PKcs, p27Kip1, p38-MAPK, SKP2
Maintenance of genome integrity is critical for proper development of the organism and prevention of genetic diseases including cancer. To guard against genomic alterations due to intrinsic or extrinsic genotoxic insults, eukaryotic cells harbour surveillance mechanisms collectively known as the DNA damage response (DDR). The DDR machinery is a network of pathways orchestrated by the phosphatidylinositol-3-OH-kinase-like (PIKKs) family of protein kinases that in mammals encompasses ATM, ATR and DNA-PKcs [1]. Within minutes upon genotoxic insults, the PIKKs become activated and through signalling cascades trigger checkpoint pathways that delay cell cycle progression to allow time for cells to repair the DNA damage. In case of the damage being too severe or difficult to repair, the DDR activation can induce premature cellular senescence or even cell death [1, 2], properties that inspired the concept of DDR as an intrinsic barrier against activated oncogenes and tumour progression [2].
The DDR machinery is also fundamental in cellular responses to major standard-of-care non-surgical treatment modalities in oncology, namely radiotherapy and genotoxic chemotherapy. Therefore, better understanding of the DDR-mediated checkpoint responses to genotoxic insults is important for both, insights into cancer biology and efforts to optimize cancer therapy. In this context, a two-step checkpoint response to persistent exposure of human A549 lung carcinoma cells to doxorubicin has recently been reported [3]. Although the initial checkpoint response (the ‘first step’ within several hours of treatment) was dependent on the DDR kinases ATM and ATR and the p53-p21Cip1 cascade as expected, the delayed cell-cycle arrest approximately between 24 and 72 hrs of drug treatment (the ‘second step’) was mediated by protein stabilization of the CDK inhibitor p27Kip1 in a mechanism that required the p38-MAPK kinase, but was apparently completely independent of DDR signalling by the canonical kinases ATM and ATR [3]. The main conclusion was that the DDR machinery was responsible only for the early cell-cycle arrest in that particular setting. This categorical conclusion that implied no contribution of the major DDR kinases to the p27-related chemotherapy-induced checkpoint was very surprising. Therefore, we decided to re-evaluate the notion of the requirement for DDR kinases in such delayed response to doxorubicin, particularly because the three major upstream DDR kinases ATM, ATR and DNA-PKcs function in at least partly redundant manner [1, 4], and this feature of DNA damage signalling was not explored by Cuadrado et al.[3] who did not consider potential involvement of DNA-PKcs. The redundancy in DDR signalling is conceptually important, because for example ATM-depleted cells respond to doxorubicin by compensatory hyperactivation of DNA-PKcs [4]. In addition, although Cuadrado et al. examined the effect of blocking ATM and ATR activity by caffeine on the abundance of p27Kip1, the critical issue of whether or not ATM/ATR inhibition might abrogate the delayed cell cycle arrest was not addressed [3].
To address any potential role of the canonical DDR signalling in the doxorubicin-triggered changes of p27Kip1 and the delayed cell cycle arrest we used a set of small molecule inhibitors of the three DDR kinases. Specifically, we exposed A549 cells, prior to doxorubicin treatment, to caffeine (ATM/ATR inhibitor), Ku55933 (ATM inhibitor) and Nu7441 (DNA-PKcs inhibitor) either individually or in various combinations. Notably, the combined treatment with caffeine and Nu7441 allowed us to restrain the activities of all three apical DDR kinases, a scenario not explored by Cuadrado et al.[3] Our analysis revealed that administration of the dual ATM/ATR inhibitor caffeine or even selective inhibition of ATM by Ku55933 and DNA-PKcs by Nu7441 released the doxorubicin-treated cells from the delayed G2 arrest and allowed cell-cycle re-entry, as documented by the induced mitotic marker MPM-2 both at 24 hrs (first step) and 48 hrs (second step) after doxorubicin addition. Importantly, this escape from the doxorubicin-evoked arrest occurred despite persistent high levels of p27Kip1 (Fig. 1A–C). Concurrent treatment with caffeine and Nu7441, i.e. combination that inhibits all three apical DDR kinases, not only released the cells from the G2 arrest, but in this case the p27Kip1 protein levels dropped significantly (P < 0.001) below those observed in any of the other doxorubicin treatment scenarios: cells untreated with any PIKK inhibitors, cells exposed to caffeine or those treated by Ku55933 and/or Nu7441 (Fig. 1A–C).
Fig 1.
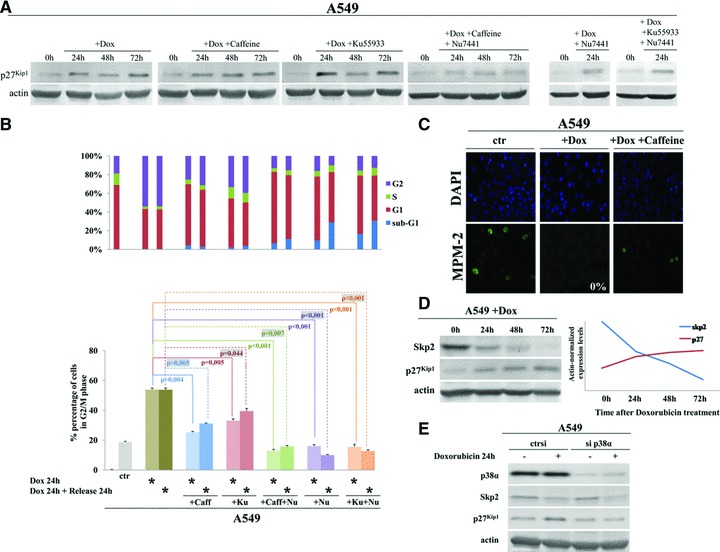
Analysis of Skp2, p27Kip1 and requirements for DDR kinases in doxorubicin-induced delayed cell-cycle checkpoint. (A) Representative immunoblots for p27Kip1 in A549 cells treated with 0.5 μM doxorubicin with or without inhibitors of ATM (Ku55933 [Merck, Athens, Greece] at a final concentration of 10 μM), ATM and ATR (Caffeine [Sigma, AntiSel, Athens, Greece] at a final concentration of 2 mM) and DNA-PK (Nu7441 [KuDOS Pharmaceuticals, Cambridge, UK] at a final concentration of 10 μM). All inhibitors were added to the medium 4 hrs before addition of doxorubicin. Cells were harvested prior to doxorubicin addition (control) and at 24, 48 and 72 hrs of treatment (actin = loading control). (B) Flow cytometry analysis of A549 cells treated as in (A) for 24 hrs and then released from doxorubicin treatment for another 24 hr culture in drug-free medium, and stained with propidium iodide. *, the applied treatment. Upper-panel bars: quantification of the percentage of cells in each cell cycle phase and apoptotic cells (see legend). Lower-panel bars: percentage of cells in G2/M phase for each treatment and time-point; and the statistical analysis. (Caff, caffeine; Ku, Ku55933; Nu for Nu7441). (C) Immunofluorescence visualization of the mitotic marker MPM-2 in A549 cell treated as in (B). (D) Representative immunoblots from a 3 day time course show an inverse correlation between Skp2 and p27Kip1 protein levels in A549 cells treated with 0.5 μM doxorubicin. Actin served as a loading control. (E) Representative immunoblots for p38α, Skp2 and p27Kip1 in A549 cells transfected with siRNA to p38α (sip38α) or control siRNA (ctrsi) and treated for 24 hrs with doxorubicin. Actin served as loading control.
The latter observation prompted us to further investigate the regulation of p27Kip1 protein level in the prolonged doxorubicin-evoked cell cycle arrest, another critical issue that was not mechanistically addressed by Cuadrado et al.[3] except for the finding that p27Kip1 protein abundance was dependent on signalling through the p38-MAPK kinase [3]. We particularly wished to assess the role of Skp2, a specificity factor in the SCF ubiquitin ligase complex, that promotes ubiquitin-dependent, proteasome-mediated degradation of p27Kip1 both in vivo and in vitro[5–7]. In addition, we have recently reported that in the A549 cells p27Kip1 protein levels are Skp2-dependent during unperturbed cell cycle progression [8]. Therefore, we recapitulated the experiments conducted by Cuadrado et al. by treating the A549 cells with doxorubicin and assessed protein levels of Skp2 in parallel with p27Kip1. As shown in (Fig. 1D), treatment with doxorubicin resulted in down-regulation of Skp2 and concomitant increase in p27Kip1 levels, a finding similar to that reported by Sugihara et al.[9]. This inverse correlation between Skp2 and p27Kip1 suggested the possibility that the lower abundance of Skp2 under conditions of prolonged genotoxic stress might limit the capacity of the cells to preserve the normally rapid turnover of p27Kip1 and thereby contribute to the observed increased abundance of p27Kip1 and the delayed ‘second-step checkpoint’. If true, Skp2 might operate either in a parallel mechanism, or in the same pathway with p38-MAPK [3]. To test these hypotheses, we knocked down p38α by RNA interference (sip38α) in doxorubicin-treated A549 cells. Depletion of p38α led to a concomitant decrease of Skp2 and p27Kip1 protein levels (Fig. 1E), indicating that Skp2 is unlikely to represent a major factor involved in p27Kip1 protein turnover under conditions of genotoxic stress. Our data also show that the observed decline of Skp2 is independent of p38-MAPK, and it might be attributed to a direct effect of doxorubicin as previously shown for several cell lines [9, 10]. These results with doxorubicin-treated A549 cells complement our previous findings [8] and suggest that p27Kip1 regulation is determined by the specific cellular environment, being dependent on Skp2-mediated proteolysis during unperturbed cell cycle progression, while during chemotherapy-induced genotoxic stress the activated p38-MAPK pathway plays a major role in inducing p27Kip1.
In conclusion, the novel findings from our present study can be summarized as follows: (i) p27Kip1 is not sufficient to maintain the durable (‘second-step’) cell-cycle arrest induced by doxorubicin; (ii) the activities of DDR-kinases are required for such prolonged cell-cycle arrest; (iii) ATM-, ATR- and DNA-PKcs-mediated signalling contribute to the doxorubicin-induced stabilization of p27Kip1 in a redundant manner and (iv) Skp2 is a regulator of p27Kip1 protein levels during unperturbed cell cycle progression, but under genotoxic stress induced by chemotherapeutic agents p27Kip1 levels are mainly determined by p38-MAPK signalling and the role of Skp2 is less apparent. Conceptually, our data with PIKK inhibitors being able to release the cells from the cell cycle block despite the maintained high p27Kip1 strongly indicate that PIKKs are not only jointly required for the enhanced p27Kip1 level, but they must regulate also p27Kip1-independent pathways that are critical for the G2 arrest, and these p27Kip1-independent mechanisms are more sensitive to PIKK silencing, since they are undermined by inhibition of even individual members of the PIKK family. Future studies are needed to elucidate potential cross-talks and mechanistic details of this intriguing aspect of cellular response to persistent DNA damage. Such studies should help better understand the effects of commonly used genotoxic anti-cancer drugs, and possibly identify some checkpoint components as targets to sensitize cancer cells to chemotherapy.
Acknowledgments
This work was supported by the Danish National Research Foundation, the Czech Minitsry of Education (MSMT6198959216) and the European Commission (projects GENICA, Infla-Care and CZ.1.05/2.1.00/01.0030). We thank KuDos pharmaceuticals for kindly providing DNA-PKcs inhibitor Nu7441.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 3.Cuadrado M, Gutierrez-Martinez P, Swat A, et al. p27kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 2009;69:8726–32. doi: 10.1158/0008-5472.CAN-09-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang H, Reinhardt HC, Bartkova J, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;15:1895–909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrano AC, Eytan E, Hershko A, et al. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biol. 1999;1:193–9. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 6.Sutterlüty H, Chatelain E, Marti A, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nature Cell Biol. 1999;1:207–14. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 7.Tsvetkov LM, Yeh KH, Lee SJ, et al. p27Kip1 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated Thr187 in p27. Curr. Biol. 1999;9:661–4. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 8.Pateras IS, Apostolopoulou K, Koutsami K, et al. Downregulation of the KIP family members p27KIP1 and p57KIP2 by SKP2 and the role of methylation in p57KIP2 inactivation in non small cell lung cancer. Int J Cancer. 2006;119:2546–56. doi: 10.1002/ijc.22214. [DOI] [PubMed] [Google Scholar]
- 9.Sugihara E, Kanai M, Saito S, et al. Suppression of centrosome amplification after DNA damage depends on p27 accumulation. Cancer Res. 2006;66:4020–9. doi: 10.1158/0008-5472.CAN-05-3250. [DOI] [PubMed] [Google Scholar]
- 10.Bar-On O, Shapira M, Hershko DD. Differential effects of doxorubicin treatment on cell cycle arrest and Skp2 expression in breast cells. Anti-Cancer Drugs. 2007;18:1113–21. doi: 10.1097/CAD.0b013e3282ef4571. [DOI] [PubMed] [Google Scholar]